
High-Resolution BAC-Based Map of the Central Portion of Mouse Chromosome 5
Abstract
The current strategy for sequencing the mouse genome involves the combination of a whole-genome shotgun approach with clone-based sequencing. High-resolution physical maps will provide a foundation for assembling contiguous segments of sequence. We have established a bacterial artificial chromosome (BAC)-based map of a 5-Mb region on mouse Chromosome 5, encompassing three gene families: receptor tyrosine kinases (PdgfraKit-Kdr), nonreceptor protein-tyrosine type kinases (Tec–Txk), and type-A receptors for the neurotransmitter GABA (Gabra2, Gabrb1, Gabrg1, and Gabra4). The construction of a BAC contig was initiated by hybridization screening the C57BL/6J (RPCI-23) BAC library, using known genes and sequence tagged sites (STSs). Additional overlapping clones were identified by searching the database of available restriction fingerprints for the RPCI-23 and RPCI-24 libraries. This effort resulted in the selection of >600 BAC clones, 251 kb of BAC-end sequences, and the placement of 40 known and/or predicted genes within this 5-Mb region. We use this high-resolution map to illustrate the integration of the BAC fingerprint map with a radiation-hybrid map via assembled expressed sequence tags (ESTs). From annotation of three representative BAC clones we demonstrate that up to 98% of the draft sequence for each contig could be ordered and oriented using known genes, BAC ends, consensus sequences for transcript assemblies, and comparisons with orthologous human sequence. For functional studies, annotation of sequence fragments as they are assembled into 50–200-kb stretches will be remarkably valuable.
With the recent publication of the human genome sequence by the International Human Genome Project and Celera Genomics (International Human Genome Sequencing Consortium 2001; Venter et al. 2001), the mouse genome is the next major goal of large-scale genomic sequencing efforts. According to the current strategy, a draft sequence will be obtained by a combination of a “whole-genome shotgun” approach and “clone-by-clone”-based sequencing (Battey et al. 1999; Pennisi 2000; Marshall 2000, 2001). Systematic restriction fragment analysis or “fingerprinting” and bacterial artificial chromosome (BAC)-end sequencing of all clones in the selected C57BL/6J (RPCI-23 and RPCI-24) BAC libraries will permit long-range association of sequence contigs. In the second phase, this effort will “generate the complete sequence coverage and assemble the entire sequence into a finished, highly accurate form,” that is, the sequence will be contiguous with <1 error per 10,000 bases (http://www.nih.gov/science/models/mouse/mouseseq/index.html). A challenge confronted by this approach is that in the initial phase a large portion of the mouse genome will be available in draft form only. Therefore, it will be important to integrate available functional data and add biological information to this sequence while it is still in the form of collections of noncontiguous sequence segments that correspond to individual BAC clones. For researchers who are using the mouse as a model organism, annotation of genomic fragments as they are assembled to 50–200-kb stretches will be remarkably valuable. Several powerful experimental approaches, particularly transgenesis, depend on the availability of cloned fragments that span large genomic regions. Moreover, interspecies sequence comparisons can be initiated on noncontiguous mouse and human sequence (Bouck et al. 2000).
We have initiated an effort to obtain the genomic sequence of a 5-Mb region in the central portion of mouse Chromosome 5. This region encompasses at least three gene clusters of biological significance — a cluster of three receptor tyrosine kinases: Kit, Pdgfra,and Kdr (formerly Flk1), two related cytoplasmic kinases: Tec and Txk, and a cluster of at least four genes encoding gamma-aminobutyric acid (GABA) receptor subunits: Gabra2, Gabrb1, Gabrg1, andGabra4 (Kozak and Stephenson 2000). Orthologous clusters are located in the centromeric region of human chromosome 4, with theGABRA2, GABRB1, and GABRG1 loci cytogenetically localized to the short arm (4p12–p14), and thePDGFRA–KIT–KDR cluster to the long arm (4q12–q13) (http://www.ncbi.nlm.nih.gov/Omim/Homology/), indicating that this contiguous region in the mouse is interrupted by a centromere in the human genome. There is great interest in the genomic sequencing and comparative sequence analysis of this region. Of particular importance is the analysis of regulatory elements involved in the complex pattern of expression of these genes, which are members of large gene families, during development and in the adult brain.
In this paper, we report a BAC-based physical map encompassing the two gene clusters Pdgfra–Kit–Kdr and Tec–Txk in the central portion of mouse Chromosome 5. This map, together with a physical map of the Gabr gene cluster generated previously (Lengeling et al. 1999), provides a template for systematic, clone-by-clone sequencing of a 5-Mb genomic segment. In anticipation of the contiguous sequence of the entire region, we have selected three BAC clones for which draft sequence has been generated to illustrate the current status of genomic resources in the mouse, specifically the databases of fingerprinted BAC clone and BAC-end sequences. In addition, we present our annotation pipeline, which consists of public tools and resources, some developed by our groups. Our study shows that with the genomic resources and annotation tools currently available, working draft sequence can be organized into an intermediate collection of ordered and oriented sequence contigs, which can be annotated to capture biologically relevant information, such as the positions of transcribed sequences and highly conserved regions. With this information, functional studies can be initiated while awaiting contiguous, high-quality whole-genome sequence.
RESULTS
Isolation of BAC Clones and Contig Development
The region in the central portion of mouse Chromosome 5, flanked by the Gabr gene cluster on the proximal end (Lengeling et al. 1999), and the Clock gene on the distal end (Wilsbacher et al. 2000), was selected to develop a sequence-ready BAC contig. To initiate the construction of a BAC-based physical map for the remaining region, hybridization probes were designed from six genes known to map to this region (Txk, Tec, Pdgfra, Kit,Kdr, and Clock) (Table 1, below), 12 markers placed on the genetic map previously (D5Mit83, D5Mit113,D5Mit134, D5Mit305, D5Mit336,D5Mit201, D5Mit202, and D5Mit203) or physical map (Nwu8, Nwu12, Nwu13, andNwu1), and three sequence tag site (STS) markers (D5Ber1, D5Buc2, and D5Buc4) derived from yeast artificial chromosome (YAC)-end clones (Nagle et al. 1994, 1995;Brunkow et al. 1995) (Table 2, available as supplementary material athttp://www.genome.org). Based on the previously reported PFGE map (Nagle et al. 1995) we estimate that this region covers 5 Mb. The mouse RPCI-23 BAC library was screened with these probes, and the selected BAC clones were confirmed by dot-blot colony hybridization assays and PCR STS content-mapping. In the second phase, chromosome walks were initiated from the BAC islands, BACs from these islands were end-sequenced and the sequence data were used to develop new hybridization probes for the library screen. STS content mapping allowed ordering and orientation of 200 identified BAC clones within four contigs: the previously described Gabr cluster (Lengeling et al. 1999), a contig encompassing the Tec–Txk cluster, a short contig around the D5Mnl25e locus, and a large contig encompassing the Pdgfra–Kit–Kdr cluster and a segment located distal to Clock (Fig. 1). The ordering of four BAC islands in this region was based initially on the mouse Chromosome 5 genetic linkage map, YAC-based map, and PFGE map (Dietrich et al. 1992; Brunkow et al. 1995; Nagle et al. 1995; King et al. 1997a,b).
Gene/EST/Locus List
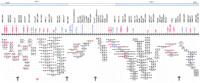
A BAC-based STS/EST-content map of theGabrg1–Gabra2–Gabrb1–Tec–Txk–Pdgfra–Kit–Kdr–Clockregion on mouse Chromosome 5 (oriented with centromeric endleftward and telomeric end rightward). The orthologous regions in the human genome are indicated on thetop (blue and open boxes). The relative positions of mapped STSs and ESTs are indicated on the top of the map, including the corresponding loci names (e.g., D5Buc); the isolated BAC clones are shown as horizontal lines below with circles along the lines indicating positive STS hits. The STSs were developed from BAC insert ends (black); BAC-end STSs corresponding to the original BAC clone (yellow); known genes (red); and SSLP markers (blue). BAC clones were isolated from the C57BL/6J library (RPCI-23). The map is displayed with equal spacing between STS/EST markers and the depicted clones together span a distance of ∼5 Mb. The ordering of five BAC islands in this region was based on the mouse Chromosome 5 genetic linkage, YAC-based, PFGE and RH map (Dietrich et al. 1992; Brunkow et al. 1995; Nagle et al. 1995; King et al. 1997a,b; Tarantino et al. 2000). Purple arrows indicate gaps in the STS-content map, and the red asterisk indicates the gap that was closed by examining the BAC fingerprint cluster shown in Figure 2.
To extend and link these BAC-contigs and to increase the pool of BAC-ends that could be used for the annotation of draft sequence in this 5-Mb region, we searched the Mouse Fingerprint Database (http://www.bcgsc.bc.ca/projects/mouse_mapping, March 15, 2001 release) for additional clones, redundant or partially overlapping with those placed in the contig based on the STS content mapping. This search identified 22 fingerprint clusters encompassing 200 originally identified and 414 additional BAC clones. Each one of these 22 clusters contained more than one clone from the “original list.” An additional 20 clusters, however, contained only one BAC clone originally placed in the contig based on the STS mapping, and because of the uncertainty about their map position, they were not included in any further analysis. The overall collection of fingerprinted BACs significantly increased the depth of the physical map, and in some cases the generated “fingerprint cluster” closed the gap between the originally defined BAC islands (Fig.2). We anticipate that with the rapid increase in the number of RPCI-23 BACs characterized by restriction fingerprinting, the majority of the existing gaps in the BAC-based map will be closed, allowing the selection of a minimal tiling path across the entire region.

A representative cluster of BAC clones obtained by the analysis of FPC fingerprints for the RPCI-23 library (http://www.bcgsc.bc.ca/projects/mouse-mapping). A query with the clones RPCI-23–294A15 (D5Mit305 region) and RPCI-23–65I8 (the Tec-Txk region) identified 148 overlapping clones, 17 of which were previously placed on the contig by hybridization screen and STS mapping. This fingerprint cluster provided a link between two contigs identified originally by STS content mapping (shown by the red asterisk in Fig. 1). The sequenced and annotated clones RPCI-23–294A15 and RPCI-23–65I8 are shaded, and the color-coded triangles indicate BAC-ends identified through sequence annotation.
Integration of Radiation Hybrid and BAC-Based Maps Through DoTS Assemblies
A distinguishing aspect of the analysis and annotation of BAC clones presented in this paper is the use of a data warehouse, the Genomics Unified Schema (GUS). GUS is a relational database that organizes biological sequences and integrates the associated sequence annotation based on the central dogma of biology (DNA→RNA→protein) (Davidson et al. 2001). GUS is also a data warehouse that unifies data from public resources such as GenBank/EMBL/DDBJ and SWISS-PROT. A major component of GUS is the clustering and assembly of expressed sequence tags (ESTs) and mRNAs to generate consensus sequences. This process serves to extend the available sequence for each EST, integrate annotation associated with each input sequence (e.g., tissue source, radiation hybrid map location, predicted gene function), and generate a nonredundant set of sequences (gene index) for further annotation and analysis. These transcript assemblies for human and mouse were originally developed as the Database of Transcribed Sequences (DoTS) and are referred to as humDoTS and musDoTS, respectively. DoTS is now incorporated into GUS and can be accessed at http://www.allgenes.org.
DoTS assemblies provide the ability to integrate annotations associated with distinct sequences, including ESTs. For example, an EST that has been placed on the radiation hybrid (RH) map can be linked with one that has been mapped on the fingerprint map if both belong to the same DoTS assembly (and therefore presumably represent the same gene transcript) (Fig. 3, below). Known genes, whose chromosomal location is usually also known, are included in DoTS assemblies through mRNAs. The currently available database of fingerprinted RPCI-23 and RPCI-24 BAC clones contains information about EST content, obtained by hybridizing the BAC libraries with 14,000 mouse ESTs (J. McPherson and M. Marra, unpubl., http://www.bcgsc.bc.ca/projects/mouse_mapping). Also, >10,000 mouse ESTs have been mapped on the T31 RH mapping panel (McCarthy et al. 1997; Van Etten et al. 1999, T. Hudson and P. Denny, pers. comm.). DoTS serves to integrate this information and link it with known and predicted genes.

Integration of the RH and BAC fingerprint map through DoTS assemblies. using the DoTS assembly for the Kit gene as an example. This DoTS assembly contains two ESTs that can be linked to the same contig in the fingerprint map (left). E-PCR links the assembly, and hence the fingerprint contig, to the central portion of mouse Chromosome 5 on the MIT/Whitehead radiation hybrid map (right).
Warehousing this RH and fingerprint data in GUS enabled us to develop several searches and database queries of use in analyzing the mouse genome. The first query displays all RH markers for a chosen chromosome (obtained from the MIT/Whitehead Mouse RH Mapping Project) and corresponding DoTS genes. A second search allows an investigator to enter a list of RPCI-23 and RPCI-24 BAC clone addresses and obtain all fingerprint contigs that contain the submitted BACs along with DoTS genes and RH markers, mapped by hybridization to this contig. A summary list of DoTS transcripts is given highlighting ESTs that hybridize to multiple contigs. Also, the fingerprint database can be searched with a list of known genes/EST accession numbers or DoTS assembly IDs. These database queries are available on a supplemental “Mouse Chromosome 5” site (http://www.cbil.upenn.edu/mouse/chromosome5). Note that despite using a single region of Chromosome 5 as a test case, all the queries discussed can be applied to the entire mouse genome.
Using the fingerprint search query, we examined the 22 BAC fingerprint clusters described above spanning our region of interest and identified 70 DoTS assemblies therein (http://www.cbil.upenn.edu/mouse/chromosome5/fpc-search.php3). To evaluate the likelihood that these DoTS assemblies represent transcribed sequences in this region, we used the following lines of evidence: (1) correspondence to previously described genes (Table1); (2) correspondence to mouse ESTs mapped to the central portion of mouse Chromosome 5 based on the RH and/or genetic map; and (3) BLAT alignment with an mRNA or ESTs in the corresponding region of human chromosome 4 (http://genome.ucsc.edu; December 12, 2000 freeze). Only 10 of the 70 DoTS assemblies found on the fingerprint map were validated with at least one additional line of evidence. A large number of the ESTs (20%) found on the fingerprints map to multiple contigs and therefore must be taken with caution.
BAC-End Sequence Annotation
Forty-seven out of 90 markers on this BAC-based physical map were generated from BAC-end sequences produced in the course of contig construction. In the initial phase of contig construction, we determined the end sequences of a subset of the BACs in the contig, but then used the database of BAC-end sequences for RPCI-23 and RPCI-24 BACs generated by The Institute for Genome Research (TIGR) (http://www.tigr.org/tdb/bac_ends/mouse/bac_end_intro.html). We searched this database for BAC-end sequences for all 614 BAC clones. From these 614 BAC clones, 427 mouse BAC-end sequences (mBESs) were identified. The average read size is ∼477 bp, giving a total of 251 kb from the contig. Comparing this set of BAC ends with the whole mBES dataset allowed us to characterize aspects of the genomic organization of this region with respect to the entire genome. We annotated the mBES sequences for repeat content, mouse shotgun reads, ESTs, and human draft sequences. Compared with the whole genome dataset (366,024 sequences and 170 Mb), the Chromosome 5 contig subset has a lower overall repeat content: 31% versus 36%. Although the GC contents are similar, the subset has higher SINE content and lower L1 content, suggesting that the subset sequences are from a gene-rich region. Further evidence that the contig is from a gene-rich region is provided by matches to ESTs and mRNAs, represented by the TIGR mouse gene index. A higher fraction of mBESs (5.5% for the subset and 2% for the whole set) was found to match the TIGR mouse gene index. TheBLAST searches identified 13 BAC ends with significant homology to known or novel genes and ESTs (Table 1). We later confirmed these assignments by RH analysis (data not shown) or by examining draft sequence in the orthologous portion of the human genome. For example, the REST gene, detected by two independent BAC ends, maps distal to CLOCK in the human draft sequence assembly (http://genome.ucsc.edu; December 12, 2000 freeze).
The contig mBESs were also compared with the mouse whole-genome shotgun reads from the Trace Archive athttp://www.ncbi.nlm.nih.gov/blast/mmtrace.html, and matches with identity ≥99%, match length ≥100 bp, and unmatched bases on each end of mBES (overhang) ≤50 bp were selected. About 50% of the mBESs (40% of the bases) were found to match the shotgun reads with an average match length of 378 bp and an average identity of 99.2%. The current shotgun data are 8,107 Mb total (∼2.7× genome) and this result suggests that at least 50% of the mouse genome is represented in the whole-genome shotgun reads.
Annotation of Draft Sequence
We have annotated genomic sequence for three representative BAC clones from the established physical map. Annotation of draft sequence was performed on the BAC clone that encompasses the known genesTec and Txk (RPCI-23–65I8). We also chose a BAC clone which, based on STS mapping, did not contain any known gene (RPCI-23–294A15), and a clone that encompasses the 5′ portion of theKit gene and 150 kb of its upstream region (RPCI-23–232H18).
The analysis of these BAC sequences consisted of ordering and orienting the draft sequence contigs, performing framework annotation (identifying repeats, genes, BAC ends, etc.), and comparing them with orthologous human sequence. Working draft sequence for each BAC clone consists of a collection of contigs sequenced at threefold redundancy, with an average size of ∼10 kb and ranging in size from 1 kb to >60 kb. The true order of the pieces is not known and their order in each GenBank sequence record is arbitrary. The contigs are first filtered to block repeat sequences using RepeatMasker. The sequence is then analyzed using a combination of BLAST searches against GenBank (nonredundant protein, GSS, HTG) and GUS (humDoTS and musDoTS). Genes (from GenBank and/or GUS) that align with multiple draft sequence contigs are used to infer the relative order and orientation of the relevant contigs. BLAST hits are displayed with AnnotView (Fischer et al. 1999) for manual inspection to arrange the contigs based on these hits. Unlike comparative sequence annotation programs such as PIPMaker(Schwartz et al. 2000), or Vista (Dubchak et al. 2000), which each combine a sequence alignment algorithm with a specialized display, AnnotView is an algorithm-independent interactive display tool that can be used to display various types of annotation. Information about the extent of overlap between BAC clones generated during the course of BAC fingerprinting, the sizes of individual BAC clones, and the BAC-end sequences all provide valuable information for positioning sequence contigs. In the second step of the annotation protocol, a string of provisionally ordered and oriented contigs (a GUS “virtual” sequence) is further annotated with repeats, gene predictions, EST homologies, potential matrix attachment regions, CpG islands, transcription factor-binding sites, conserved regions, etc. Figure 4illustrates the final annotation of three representative BAC clones (not all data shown).
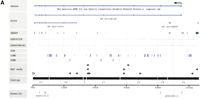

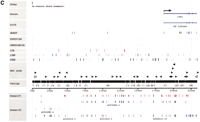
(A–C) Sequence annotation of three BAC clones:D5Mit305/Corin (A), Tec–Txk (B), and the Kit upstream region (C). Sequence: The labeled horizontal axis in the middle of each figure indicates the total amount of draft sequence that has been successfully ordered and oriented. Numbers indicate the order and orientation of the individual draft sequence fragments with respect to the original GenBank entry. For example, a fragment labeled “(21)” indicates that draft sequence fragment 21 has been placed in that location in the reverse complement orientation (with respect to the GenBank entry, in which the orientation of the individual fragments is typically arbitrary). Known genes: Shown at the top of each figure, this includes both the annotation of known genes present in GenBank and also the sim4 alignment of any known mRNAs against the mouse genomic sequence. Arrows are used to indicate the start of transcription where appropriate.BLAST-sim4 versus DoTS (EST assemblies). Indicates the (fully-automated) alignment of predicted transcripts from the Database of Transcribed Sequences (see http://www.allgenes.org) with the mouse genomic sequence. Similar transcripts are first identified by aBLASTN search. Those exceeding a user-defined similarity threshold are then sim4-aligned with the genomic sequence. A post-processing step we have implemented eliminates those sim4 alignments that are not “consistent” (with the hypothesis that the transcript represents a “real” gene at that position). The post-processing eliminates numerous low-quality alignments and uses heuristics to identify predicted transcripts that are deemed likely to represent artifacts in the EST databases. EST similarity (dbEST): The results of a BLASTN search against the dbEST database of ESTs. Hits are filtered to show only those alignments that are at least 50 bp long and that show at least 85% identity. GENSCAN: The results of running GENSCAN on both masked and unmasked sequence are shown in gray. Thin lines represent predicted introns and thick lines represent predicted exons. ECRs (Evolutionarily conserved regions): An additional line (Kit only) indicates regions more highly conserved with human finished sequence. These regions were identified with cross_match, using the available finished human sequence for the Kit upstream region (GenBank accession no.AC006552.) Only regions >100 bp and with at least 70% identity are displayed. RepeatMasker: The positions of individual repeat elements are indicated in threeRepeatMasker classes. Repeat elements were identified with the April 4, 1999 version of RepeatMasker using the default parameters (except for the use of “–mus” to indicate rodent sequence). Other features: Indicates any other sequence features of note (Kit only). Human draft sequence: The presence of orthologous human draft sequence on chromosome 4 is indicated below the sequence “axis.” In the interactive version of the display it is possible to click on individual sequences to obtain information on the level of sequence conservation and links to the actual human sequence. The human draft sequence was identified by a BLASTN search of HTG, filtered to show only those meeting the same thresholds as the ECRs (at least 100 bp and no less than 70 % identity.) The GenBank accession numbers of these sequences are shown in the figures. Human sequences labeled as being from chromosome 4 are shown in blue. Those from any other chromosome are red, and those with no chromosomal assignment specified in the GenBank record are gray. TIGR RPCI-23 and RPCI-24 BAC end sequences: We retrieve BAC-end sequences on a daily basis from TIGR, annotating them and entering them into our database. The sequences were searched using BLASTN and those likely to represent true positives are indicated.
BAC Containing the D5Mit305 Marker
The BAC clone RPCI-23–294A15 was originally isolated in a chromosome walk from the Gabrb1 gene (Figs. 1 and 4A). This BAC clone was shown to contain several STSs corresponding to ends of overlapping BAC clones, as well as the polymorphic markerD5Mit305. We performed sequence analysis and annotation of RPCI-23–294A15 (AC036146.2) as an example of a clone that was not shown previously to contain any known gene. BLAST searches (against GUS), however, identified homology to Corin(low-density lipoprotein receptor related protein 4, DT.60104108, and DT.40171971) at 97%–100% identity. This finding was supported further by the identification of 10 BLAST hits to a GenBank protein (NCBI accession NP_05856.1) that correspond to 10 exons of the DoTS transcript assembly. It is striking that all 11 ESTs in the corin DoTS assemblies are derived from testis cDNA libraries, although this gene was described in the original publication as expressed “almost exclusively in heart in mouse and human” (Yan et al. 1999). Interestingly, the second DoTS assembly, DT.40171971, appears to represent an alternatively spliced form of the gene. Sequence annotation revealed 13 BAC ends located within the 100 kb of ordered and oriented sequence, and 10 of these BAC ends are corroborated by the fingerprint map (Fig. 2).
BAC Containing the Tec–Txk Region
Annotation of the RPCI-23–65I8 BAC clone (AC013623.3) showed that the Tec and Txk genes span ∼155 kb: 105 and 55 kb, respectively (Fig. 4B). This analysis confirmed the previously reported small intergenic distance between these two genes (in human) of ∼1.5 kb and that the two genes are arranged in the same transcriptional orientation (Ohta et al. 1996). Tec and Txk have 18 and 15 exons, respectively. In addition to two known genes, at least one additional gene, and perhaps others, were found in the distal part of the RPCI-23–65I8 BAC clone. Three independent lines of evidence support this finding: (1) hits to GUS (DT.40185452/EST C89493, DT.55237874/EST AI875966); (2) exon predictions byGENSCAN, and (3) hits to the nonredundant protein database (GenBank accession no. AB041581).
The highest density of sequences conserved with human was found at the 5′ portion of the Tec gene. Thirty-five BAC-ends were identified in the 200 kb of annotated sequence, and 27 are corroborated by the fingerprint map. To illustrate the utility and high accuracy of the currently available fingerprint map, we show the positions of these BAC ends on the annotated sequence (Figs. 2 and 4B; and supplemental Fig. 5B available on-line at http://www.genome org).
BAC Containing the Upstream Region of Kit
STS content mapping showed that the RPCI-23–232H18 BAC clone encompasses the 5′ end of the Kit gene at one end and sequences shown previously to contain the distal breakpoint of theRw mutation, which are located 150 kb upstream ofKit. BLAST searches with 260 kb of RPCI-23–232H18 draft sequence (AC013622.2), composed of 26 contigs, confirmed matches with the first six exons of Kit, theRw breakpoint region, and 31 BAC ends (Fig. 4C). The positions of the majority of these BAC ends (25/31) are corroborated by the fingerprint map (data not shown). With this information we were able to align only six of the 26 contigs (or 32% of the available sequence) located at the two ends of the BAC clone. Annotation of the draft sequence of this clone, however, was facilitated with the availability of high-quality finished sequence for the orthologous region of the human genome (GenBank accession no. AC006552.7). The use of finished human sequence has been shown recently to allow the ordering and orientation of contigs covering approximately half of the region contained in 2.2× draft sequence (Pletcher et al. 2001). The comparison of RPCI-23–232H18 with the human clone identified >60 evolutionary conserved regions (ECRs), exceeding a defined threshold of sequence identity (>50 bp, >60% identity). No evidence was found that any of these regions represent new genes. These segments of homology allowed the ordering and orientation of additional contigs that did not contain homology to known genes or ESTs. In summary, the homology against human finished sequence, together with the BAC-end sequences, permitted the ordering and orientation of 23 of the 26 contigs, equal to 98% of the available draft sequence.
DISCUSSION
The work described in this paper illustrates the generation of a sequence-ready map of a 5-Mb region in the mouse genome known to encompass three gene clusters (Gabrg1–Gabra2–Gabrb1, Tec–Txk andPdgfra–Kit–Kdr). The BAC contig was generated using the C57BL/6J BAC (RPCI-23) library, which has been designated by the community and mouse sequencing centers as a source of BACs for “clone-by-clone” sequencing (http://www.nih.gov/science/models/mouse/mouseseq/index.html). This paper takes, as an example, genomic analysis of 5 Mb of the mouse genome and sequence annotation of three representative BAC clones to illustrate three major points: (1) the current status of genomic resources, such as a well-characterized large insert (BAC) library for which >270,000 clones have already been fingerprinted, and for which BAC-end sequences for >170,000 clones are available; (2) the utility of annotated draft sequence for functional studies; and (3) first insights into novel features of intra- and intergenic regions gained through comparative sequence analysis of this chromosomal region. The corresponding chromosomal region in the human genome encompasses the centromeric portion of chromosome 4 and the human draft sequence in this region contains several gaps. For example, the segment betweenTec and Pdgfra on our map is missing in the human sequence (http://genome.ucsc.edu; December 12, 2000 freeze).
Ten known genes and >20 STS markers were used to initiate the construction of a BAC-based physical map around three gene clusters in the central portion of mouse Chromosome 5. The recently established databases of RPCI-23 and RPCI-24 clone fingerprints and BAC-end sequences were used to increase the density of BACs in the region of interest. Using this combined approach, we were able to identify >600 BAC clones classified into four megabase-length contigs. The high degree of redundancy and accuracy of these maps will eventually allow efficient selection of overlapping clones for sequencing. Systematic sequencing of megabase lengths of contiguous sequence in a genetically well-characterized portion of a chromosome may provide a useful pilot project. The biologists interested in developmental pathways in which these gene clusters participate will benefit from sequence annotation and identification of BAC clones that encompass regions of biological significance. Similarly, these contiguous sequences, encompassing known and predicted genes, provide a useful experimental sample for assessing existing and new approaches to genomic sequence annotation and analysis. During the course of fly genome sequencing, a 2.9-Mb region encompassing the Adh gene provided “a valuable test of the longer-term strategy of sequencing and annotating the entire genome of this fly” (Ashburner 2000).
The established BAC-based physical map provides positions for 30 known genes and ESTs, in addition to at least 10 novel DoTS assemblies that were found by integrating the fingerprint map and an established database of assembled ESTs. The annotation of BAC clones described in this paper demonstrates that we were able to identify novel genes, even in a historically well-characterized chromosomal region. This collection of >40 known and/or potential genes will facilitate annotation of genomic sequence in this 5-Mb region. Although this report describes sequence annotation for a small sample of three BAC clones, it illustrates different levels of confidence from the various approaches used for gene annotation. GENSCAN is highly sensitive in predicting exons, however, it is known to have a high false positive rate. Without further evidence, these predictions are suspect. The alignment of either DoTS assemblies or protein sequence at high stringency to genomic sequence provides stronger evidence for the presence of a gene, although the gene may be fragmented or a pseudogene. When all three methods agree (GENSCAN, DoTS, GenBank protein), the assignment of a gene can be made with high confidence. For example, the identification of a novel gene with 97%–100% identity to corin (Lrp4) was supported by all three lines of evidence. On the other hand, in the region upstream ofKit, no GUS or GenBank database matches were found, despite the presence of GENSCAN exon predictions throughout the entire region.
The sample annotation of three BAC clones also illustrates the potential utility of BAC ends for the assembly of low coverage (1–3×) clone-based sequence and whole-genome shotgun sequence. In the case of these clones, homologous BAC-end sequences predicted from the fingerprint map were identified on average every 6–8 kb along the length of the assembled BAC sequences. Whereas a dense map of ordered and oriented BAC ends may in some regions allow assembly of whole-genome shotgun reads, and therefore eliminate the need for clone-based sequencing, because of the high repeat content of a mammalian genome, BAC-based sequencing will expedite assembly. Clone-based sequencing will ultimately be required in the final stages of sequencing and gap closure and will provide the high-quality sequence necessary for functional genomic efforts.
In summary, mouse draft genomic sequence can be annotated using a variety of sources, including orthologous human sequence, leading to the identification of new genes and the localization of known genes. It is the ability to integrate whole-genome BAC-based maps with genetic maps that will allow draft sequence annotation to be understood in terms of gene function and phenotype.
METHODS
BAC Library Screening
BAC clones were isolated by hybridization of probes to high-density library filters from the mouse BAC (RPCI-23) library (Osoegawa et al. 2000). Initial screenings used PCR-product probes designed from cDNAs and MIT markers that had been assigned to this region previously. In subsequent screenings, the probes used were primarily PCR products generated from BAC-end sequence data (Table 2), but also ESTs and other STS markers (YAC and cosmid-end sequence). Probe DNA was labeled in agarose with [α32P]dCTP by random primer extension (Feinberg and Vogelstein 1984).
Clone Analysis and Contig Construction
All BACs isolated were arrayed as colony dot blots in a 96-well format. BACs were grown overnight in 100 μL of LB/chloramphenicol, replicated onto nylon filters and grown for 8 h on LB agar plates. Filters were processed using alkaline lysis and Proteinase K/Sarkosyl treatment (http://www.resgen.com/depts/rnd/rapid.html).
BAC DNA was prepared from 5-mL overnight cultures using a standard alkaline lysis procedure and the DNA pellet was resuspended in 40-μL H2O. NotI digests of mini-prep BAC DNA were used to determine clone insert size. Twenty-microliter reactions containing 5 μL DNA were digested with 5 U of NotI enzyme for 2 h and subsequently run on a Pulsed Field Gel (16-h run time, switch time 5 sec to 15 sec). EcoRI and HindIII digests of mini-prep DNA were used to fingerprint clones (Marra et al. 1997). Clone ordering was performed manually using FPC (http://www.sanger.ac.uk).
BAC DNA for end-sequencing was prepared from 200-mL overnight cultures according to the modified protocol for BACs using P100 midi-prep columns (QIAGEN). Automated didoxy-terminator cycle sequencing was carried out with SP6 and T7 primers on BAC DNA (2 μg of DNA in a 20- μL reaction) using ABI Big Dye Terminator sequencing chemistry withTaq FS polymerase (Applied Biosystems). Reaction products were purified through G-50 spin columns and analyzed on ABI 377 sequencers (DNA Sequencing Facility, Department of Genetics, University of Pennsylvania, Philadelphia, PA).
All BAC-end sequences were assessed for development of new STS markers, and analyzed for sequence similarities using the BLASTnetwork (http://www.ncbi.nlm.nih.gov/cgi-bin/BLAST/nph-blast?Jform=0). Sequence data was also analyzed using RepeatMasker(http://ftp.genome.washington.edu/cgi-bin/RepeatMasker) to determine the presence of repeat sequences and Primer 3.0 (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3.cgi) for selection of PCR primers. Primers and respective PCR conditions used in this work are listed in Table 2. PCR was performed with diluted mini-prep BAC DNA in 12.5-μL reactions consisting of 1× buffer (20 mM Tris-HCl at pH 8.3, 50 mM KCl, and 1.5 mM MgCl2), 0.2 mM each dNTP, 0.4 μM each STS primer, and 0.1 U of Taq polymerase (Roche) with the following conditions: 94°C for 4 min, 94°C for 30 sec, annealing for 30 sec (temperatures listed in Table 2), 72°C for 30 sec, for 35 cycles.
Integration of Fingerprint Map with Radiation Hybrid Map and DoTS
The flat file version of the mouse fingerprint data was converted into relational form and loaded into the GUS database, which also contains the mouse radiation hybrid mapping data available from the Whitehead Institute. Several searches of the fingerprint data were then implemented. The first accepts a list of clones and displays all the fingerprint contigs that contain one or more of the specified clones. It also displays the markers from the fingerprint map, many of which can be linked to ESTs through the Washington University clone IDs in GUS. These markers can be linked in turn with DoTS genes either by e-PCR (for STS markers) or through an associated EST identifier, by determining which DoTS assembly contains the EST. Many of the DoTS assemblies have been placed on the RH map by e-PCR, providing additional information on the relative positions of the fingerprint clusters. This search page was used to probe the March 15, 2000 release of the fingerprint database with 200 known RPCI-23 clones, yielding four singleton fingerprint contigs, 22 nonsingleton contigs, and 70 DoTS assemblies localized to the region. About 10% (19) of the 200 clones had not yet been fingerprinted. The remaining searches display more detailed information about fingerprint contigs and link the RH data for each chromosome to DoTS (http://www.cbil.upenn.edu/mouse/chromosome5/).
Sequence Analysis
BAC-End Sequence Analysis
The TIGR database of RPCI-23 BAC-end sequences is downloaded on a nightly basis (ftp://ftp.tigr.org/pub/data/m_musculus/bac_end_sequences). Indices are built on the flat files for rapid access. A local file contains the names of those BACs that have been localized to the region of interest on Chromosome 5 (either during the original physical map construction or as a result of the subsequent fingerprint analysis.) This file and the TIGR database are used to update a local web site that shows the status of the BAC-end sequencing project with respect to this restricted set of Chromosome 5 BACs.
On a weekly basis, all new BAC-end sequences are loaded into the GUS data warehouse (Davidson et al. 2001). BAC-end sequences are masked for repeats and low complexity sequence with the April 4, 1999 release ofRepeatMasker (A. Smit and P. Green, unpubl.) using the default parameters, except for the use of the “–mus” option for rodent sequence. Using the “–s” option for increased sensitivity had a negligible effect (data not shown.) Masked BAC-end sequences from the Chromosome 5 set were then used to search the following subsets of GenBank: dbEST (EST sequences), GSS (Genome SurveySequence, including BAC-end sequences), and HTG (High Throughput Genomic sequences, including human and mouse draft sequence from genome sequencing projects). Masked BAC-end sequences were also searched against the NCBI nonredundant nucleic acid (nt) and protein (nr) databases and the clustered and assembled ESTs in the DoTS subset of the GUS database (http://www.allgenes.org).
The nr and DoTS search results were stored in GUS, all other search results were stored in flat files. The above searches were performed with Washington University BLASTX 2.0 (nr) andBLASTN 2.0 (all other databases) using the default parameters (Altschul et al. 1990). All of these steps are performed automatically by Perl scripts. Tasks repeated on a periodic basis are run automatically from crontab entries.
Draft Sequence Analysis
Each of the three draft BAC sequence entries was retrieved from GenBank and a Perl script was used to split the sequence into its component contigs, removing the stretches of Ns (if any) used to separate them. The script also entered each contig into the GUS database. The individual draft sequence contigs were masked for repeats using RepeatMasker (as described above) and then searched against RPCI-23 BAC-end sequences from Chromosome 5, dbEST, DoTS, and the nonredundant nucleotide and protein databases (also as described above). The resulting annotation was examined in the bioWidgetAnnotView application. For each BAC, the order and orientation of as many of the contigs as possible was determined manually, using matching BAC-end pairs (where present) and any similarity to known genes or other sequence landmarks. For the BAC containing the 5′ end of c-Kit (RPCI-23–232H18), similarity to orthologous human finished sequence (accession no. AC006552.7) was used to order and orient draft contigs further (under the assumption that this region of the mouse genome contains no small-scale rearrangements or inversions relative to human).
For each BAC, a “Virtual Sequence” was created in the GUS database to reflect the tentative arrangement of sequence fragments. Each virtual sequence was then subjected to the framework annotation process described below and the bioWidget application AnnotViewwas used to view and verify the consistency of the results and produce PostScript figures for publication (Fig. 4; supplemental Fig. 5 available on-line at http://www.genome.org).
Comparative Sequence Analysis
The cross_match program was used to identify regions of high sequence similarity between the human and mouse Kit upstream regions.
Framework Annotation
GUS is a data warehouse of sequence and annotation obtained from a variety of public sources. GUS is a relational database designed to represent data from multiple organisms and biological systems. Currently, data from mouse, human (http://www.allgenes.org), andPlasmodium falciparum (Plasmodium Genome Consortium,Nucleic Acids Res. 2001; http://plasmodb.org) are stored in GUS.
The virtual sequences for the three BAC clones were subjected to framework sequence annotation. The first step is to mask repetitive and low-complexity DNA with RepeatMasker(http://repeatmasker.genome.washington.edu/cgi-bin/RM2_req.pl). As with the BAC-end sequences, Washington University BLASTN 2.0 was used to search NCBI's nonredundant protein database (nr) and Washington University (http://blast.wustl.edu) was used to search dbEST, HTG, and the nonredundant nucleotide database.BLASTN was also used to search the TIGR RPCI-23 BAC-end sequence database (http://www.tigr.org/tdb/bac_ends/mouse/bac_end_intro.html). All searches used the masked sequence. The searches against HTG were further post-processed to identify hits against homologous human draft sequences, in particular those mapped to chromosome 4.
mRNA sequences for the genes known previously to be in each BAC were aligned with the sequence using sim4 (Florea et al. 1998).GENSCAN (Burge and Karlin 1997) was used to predict genes on both the masked and unmasked sequences, using the parameter file for human/vertebrates (HumanIso.smat). Finally, potential coding regions were also identified by a BLAST-sim4 search of the DoTS database — BLASTN was used to search DoTS, and high-scoring DoTS assemblies were then aligned with the genomic sequence using sim4. A set of consistency rules was then applied to the resulting alignments to eliminate those deemed likely to represent false positives (e.g., attributable to spurious or incomplete sequence similarity, genomic contamination in the ESTs, or errors in the DoTS assemblies).
Acknowledgments
This paper is dedicated to the memory of Chris Overton. We thank the Whitehead/MIT Genome Sequencing Center for the BAC sequence; J. Lehoczky for the BAC-fingerprint analysis; L. Tarantino, A. Lengeling, and S. Kanes for their contribution in the early stages of this project; C. Otmani, O. Valladares, and B. Dong for technical assistance; and K. Dewar, B. Birren, and H. Riethman for helpful discussions and comments on the manuscript. These studies were supported by grants from the National Institutes of Health (HD 28410 to M.B.), (RO1HG01539 to C.S.), Department of Energy (DE-FG02-DOE00ER62893 to C.S.), and from the PENN Genomics Institute Pilot project.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Present address: Genomics Institute of the Novartis Research Foundation, San Diego, CA 92121, USA.
-
↵5 These authors contributed equally to this work.
-
↵6 Corresponding author.
-
E-MAIL bucan{at}pobox.upenn.edu; FAX (215) 573-2041.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.195101.
-
- Received May 3, 2001.
- Accepted July 25, 2001.
- Cold Spring Harbor Laboratory Press








