
The Large srh Family of Chemoreceptor Genes in Caenorhabditis Nematodes Reveals Processes of Genome Evolution Involving Large Duplications and Deletions and Intron Gains and Losses
Abstract
The srh family of chemoreceptors in the nematodeCaenorhabditis elegans is very large, containing 214 genes and 90 pseudogenes. It is related to the str, stl, andsrd families of seven-transmembrane or serpentine receptors. Like these three families, most srh genes are concentrated on chromosome V, and mapping of their chromosomal locations on a phylogenetic tree reveals 27 different movements of genes to other chromosomes. Mapping of intron gains and losses onto the phylogenetic tree reveals that the last common ancestral gene of the family had five introns, which are inferred to have been lost 70 times independently during evolution of the family. In addition, seven intron gains are revealed, three of which are fairly recent. Comparisons with 20 family members in the C. briggsae genome confirms these patterns, including two intron losses in C. briggsae since the species split. There are 14 clear C. elegans orthologs for these 20 genes, whose average amino acid divergence of 68% allows estimation of 85 gene duplications in the C. elegans lineage since the species split. The absence of six orthologs in C. elegans also indicates that gene loss occurs; consideration of all deletions and terminal truncations of srh pseudogenes reveals that large deletions are common. Together these observations provide insight into the evolutionary dynamics of this compact animal genome.
[A truncated alignment of most annoted members of this protein family is available in Pfam v. 4.2 as family 7tm_5 (http://pfam.wustl.edu/); alignments of all translations are available as supplementary information at http://www.genome.org and can be opened with the program PAUP; alignments of all translations and genes are available athughrobe{at}uiuc.edu.]
Large gene families provide considerable insight into the evolutionary dynamics of genomes through analyses of the evolution of paralogous gene family members. I previously described the patterns of gene duplication, diversification, movement, and intron loss revealed by the large str and stlfamilies of chemoreceptor genes in the Caenorhabditis elegansand Caenorhabditis briggsae genomes (Robertson 1998). Thestr family contains the odr-10 gene encoding the ODR-10 chemoreceptor for diacetyl (Sengupta et al. 1996). ODR-10 is expressed in the AWA sensory neuron that mediates attraction to volatile chemicals (Bargmann and Mori 1997). Misexpression in sensory neuron AWB, which is known to mediate repulsion from diverse chemical stimuli (Bargmann and Mori 1997), led to repulsion from diacetyl, confirming the chemical specificity of the ODR-10 chemoreceptor and providing a simple mechanism for olfactory coding in nematodes (Troemel et al. 1997). Furthermore, this ODR-10 chemoreceptor mediates perception of diacetyl when expressed in mammalian cells (Zhang et al. 1997).
During analysis of the str and stl families, I encountered another very large family of related candidate chemoreceptors encoded by the C. elegans genome, here named the srh family (J. Spieth independently noted the size of this new family; pers. comm.). This family reveals very similar paralogous gene evolution, including frequent recent duplication of genes, their common degeneration to pseudogenes, and regular intron loss. Somewhat in contrast to the str and stl families, where only one intron gain was noted, seven intron gains are inferred for thesrh family. Comparison with orthologs in C. briggsaehelped to illuminate these processes, in particular indicating that many genes have duplicated in the C. elegans lineage since the two species lineages separated, whereas others have apparently been lost. Analysis of the sizes of deletions in pseudogenes supports the hypothesis that this small genome size is maintained by common large deletions, a process apparently shared with Drosophila fly genomes, but not the human genome.
RESULTS
The Large srh Chemoreceptor Gene Family
The C. elegans genome project is complete ( C. elegans Sequencing Consortium 1998; C. elegans Genome Consortium 1999) and searches for, and alignments of, genes were completed at the end of October 1999. All sequences in GenBank were employed, including a few from the HTGS database. Aligned reconstructions of these genes were communicated to those annotating the sequences, and have been employed in the annotations for many of the apparently functional genes. Some of the pseudogenes that I identify by their close similarity to other chemoreceptors can nevertheless be annotated as apparently reasonable genes by removal or truncation of exons with in-frame stop codons or frameshifting insertions/deletions (indels), and therefore their present annotations are questionable. Comparison with the closest functional gene in the phylogenetic tree below usually reveals their pseudogene status. Most of these clones have now been completed, annotated, and deposited in GenBank, and so the genes are identified herein by the gene numbers given in the annotations in the format Clone#.gene# (the remainder are identified by letters for gene numbers, particularly the C. briggsae genes). They have also been named in a srh series (see Fig. 3, below) with pseudogenes named if they encode more than half the amino acids of the closest intact relative. Smaller gene fragments were only employed below for analysis of truncation lengths.
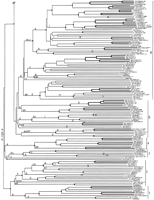

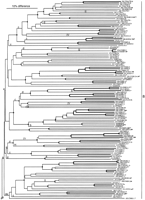
Phylogenetic tree relating members of the srh family of chemoreceptors. Subfamilies are indicated on the right by numbers. Bootstrap support >95% is indicated by a diamond on the relevant node, with a smaller circle indicating bootstrap support >75%. Inferred jumps of genes from chromosome V to another chromosome are indicated by roman numerals above the middle of the relevant branch. Lowercase letters above the base of the relevant branch indicate inferred intron loss, whereas uppercase letters indicate intron gain. Double-thickness lines connect genes that are inferred to have arisen by gene duplication since theelegans/briggsae species split. C. briggsae genes are indicated by bold type, all start with the letter G, and are not numbered. C. elegans genes are assigned gene numbers in asrh− series. Pseudogene status is indicated by symbols after each gene name. (#) Frameshift or large indel; (*) in-frame stop codon; (?) loss of start codon or questionable intron boundary.
Two hundred fourteen apparently functional C. elegans genes were identified in the srh family, defined somewhat arbitrarily as those with the amino acids ST (serine, threonine), or derivatives thereof, in the seventh transmembrane domain instead of the amino acids DP (aspartic acid, proline), or derivatives thereof, characteristic of the str, stl, and srdfamilies (residues 297 and 298 in ODR-10) (Fig. 1). Ninety certain or likely pseudogenes were also identified, which is 30% of the total, a frequency in agreement with the other three families (Robertson 1998). Many fragments of genes encoding <50% of the length of the closest intact receptor were excluded from the pseudogene set and not named. Many of the apparent pseudogenes in this family have multiple stop codons, frameshifts, or large indels that are unlikely to be sequencing errors. Therefore it is reasonable to conclude that even those with single base indels, single stop codons, and unacceptable intron splice junctions are pseudogenes. The sequencing accuracy rate of better than 99.99% for the nematode genome project makes it particularly unlikely that these apparent pseudogenes result from sequencing errors ( C. elegans Sequencing Consortium 1998; C. elegans Genome Consortium 1999). With the redundancy generated through sequencing of yeast artificial chromosome (YAC) clones spanning previously unclonable gaps, it was possible to re-examine many of these pseudogenes in YAC sequences and in each case, including several stop codons and single base indels, the same mutation was present in the YAC sequences.
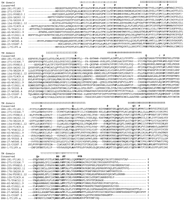
Alignment of the encoded amino acids of representative srhfamily chemoreceptors. One representative of each of the small basal subfamilies 1–4, two representatives each of subfamilies 5–7, and four representatives of subfamily 8 are shown, with the subfamily designations indicated at the end of the sequences. The seven transmembrane domains are indicated above the alignments following Fig.2. The alignments generally divide into blocks corresponding to these domains, with length variants between them, the exceptions being in the more variable TM4/5 region. The conserved amino acid positions that anchor the alignments are bold, and shown above the alignments, as are the inferred ancestral intron positions. Refined alignment of the highly variable amino and carboxyl termini is not shown.
Conceptual translations of these genes are readily alignable with each other for most of their length yet share as little as 12% amino acid identity with each other (Fig. 1)—the regions of less than certain alignment being the TM4/5 region, especially between subfamilies 1–4 and the remainder of the family (see below). The amino and carboxyl termini are also highly variable in sequence and length. A Kyte–Doolittle hydrophobicity plot (Fig. 2) for one of these, SRH-215/T20B3.3, shows how the seven transmembrane regions are usually readily identified.
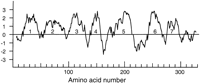
Kyte-Doolittle hydrophobicity plot for the SRH-215/T20B3.3 protein. Transmembrane regions are numbered.
Phylogenetic analysis of the 304 srh family members and 20 homologs in C. briggsae (see below) was performed on their conceptual translations using neighbor-joining. The “heuristic” algorithm of PAUP was then employed to examine more than three million rearrangements using tree-bisection-and-reconnection branch-swapping resulting in a minimum evolution tree 0.18% better, shown in Figure 3 and rooted by designating the subfamilies 1 and 2 as the outgroup (based on analyses of representatives of all four families using the srdfamily as the outgroup). Generally there is good bootstrap support for many terminal relationships and many small and large clades within subfamilies, however, within the large subfamilies there is usually little bootstrap support for the overall architecture of the relationships. Except for subfamilies 1–4 there is little support for the subfamilies themselves, and there is little support for the relationships of the subfamilies to each other.
Subfamilies are recognized to facilitate descriptions, however their definition by amino acid sequence and/or intron loss is not absolute, because several share features, and within otherwise well-defined subfamilies sometimes one of the defining sequences has changed in a subgroup. Unlike the str family where variations of the DP amino acid pair in TM7 were employed to distinguish the subfamilies, most members of the srh family have ST at these two positions, a defining feature of the family. Therefore, the eight subfamilies will simply be designated as 1–8, with additional definition by the two amino acids flanking the absolutely conserved arginine (R) near the end of TM3 (Fig. 1). The two small basal subfamilies 1 and 2 are characterized by the amino acids YRM and FRY, respectively. The small subfamily 3 is highly variable in this sequence, whereas subfamily 4 has (SN)R(IVL). Subfamily 5 is poorly defined, consisting of at least seven paraphyletic gene lineages with no bootstrap support for their relationships, and have the sequence NR(FQH). Basal members of subfamily 6 have NR(SN), but after gain of intron j (see below) they have SR(SA). Subfamily 7 is similarly diverse, with (NED)R(YR), whereas members of subfamily 8 have NR(LFY). Genes srh-24/R10D12.11 and srh-25/C54D10.6 branch near the base of subfamily 3 in Figure 3, however, they are highly divergent and this placement is tenuous, therefore they are not placed in a subfamily.
Chromosomal Location
The vast majority of genes in some large families, including the nuclear receptor superfamily (Sluder et al. 1999) and the str,stl, and srd families (J. Spieth, pers. comm.) are located on chromosome V. The same bias is overwhelmingly true for thesrh family, with 248 genes and pseudogenes on chromosome V, 34 on chromosome II, 15 on chromosome IV, 4 on chromosome X, 3 on chromosome I, and 0 on chromosome III. These genes are distributed roughly evenly along the lengths of these chromosomes (J. Spieth, pers. comm.), so there is little bias in location on a particular chromosome. To evaluate this enigmatic observation further, it is necessary to consider some additional details. These and other chemoreceptor genes commonly exist in large, tandemly repeated complexes that are presumably all derived by gene duplication from a single gene (Troemel et al. 1995; Robertson 1998). It is therefore best to evaluate this chromosomal location pattern further by employing the phylogenetic tree. It appears that the chromosome V location of the majority of these srh family genes is ancestral, and that movements to other chromosomes have occurred only rarely. Gene movements to other chromosomes were usually easily parsimoniously mapped on the tree, and the branches where such movements are inferred to have occurred are indicated with chromosome numbers (Fig. 3). Therefore, 17 moves to chromosome II are inferred, as are six moves to chromosome IV, two to chromosome X, one to chromosome I, and one back to chromosome V (17 of these 27 moves are so recent they involve only a single extant gene). The most convoluted of these gene movements is the old movement of a subfamily 6 gene to chromosome II, followed by a derived gene moving to chromosome IV, followed by gene srh-62/ZK6.9 returning to chromosome V. Relationships in this region of subfamily 6 are well supported by bootstrapping, making this by far the most parsimonious explanation of the chromosomal locations of this subset of its genes. The independence of most of these 27 moves to other chromosomes is well supported by bootstrapping. Two of these movements to other chromosomes are old enough that they have led to considerable gene duplication and diversification, and further movement within the new chromosome (the movements to chromosome II in subfamily 6 and to chromosome IV in subfamily 8). Preliminary analysis of mapping of each chromosome V gene onto the chromosome itself reveals abundant history of movement within the chromosome, but it is difficult to analyze whether these genes are more likely to move to new locations on chromosome V than to other chromosomes. It seems clear that the common ancestor of this family, and of this and the other three families, resided on chromosome V, and that the duplications leading to the four families and their early diversity occurred on this chromosome. The average size of srhgenes is ∼3 kb (1000 bp each of exons, introns, and flanking DNA), so this family alone accounts for about 1 Mb. Together with the amplification of the nuclear receptor superfamily (Sluder et al. 1999), the ancestry and amplification of these chemoreceptor families and perhaps other gene families on this chromosome might explain why chromosome V is the largest at 21 Mb.
Intron Evolution
The exon/intron structures of these genes were useful guides in their reconstruction, and an ancestral intron arrangement is easily established (Fig. 4; intron positions are indicated more precisely in Fig. 1). The common ancestor of this family appears to have had five introns, roughly evenly distributed along the length of the gene, although the fifth exon is rather short. One of these, intron d, is in precisely the same codon and phase as intron g of the related str and stl families, and can therefore be considered to be homologous and shared from a common ancestor of the three families. All of the others are apparently independent, more recent intron gains, and all are independent from those of thesrd family, which is considered to be ancestral to thesrh, str, and stl families (H. Robertson, unpubl.).
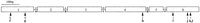
Reconstruction of the ancestral intron placements for the srhfamily of chemoreceptor genes. Exons are shown as open numbered boxes of roughly accurate length, whereas introns are shown as lettered lines. Phases of the introns are shown above them. (0) Between codons; (1) between the first and second bases of a codon; (2) between the second and third bases of a codon. Arrowheads indicate the positions of insertion of new introns f–l.
As was true for the str and stl families (Robertson 1998), the vast majority of intron changes involve loss, however it proved more difficult this time to map these losses parsimoniously on the phylogenetic tree (Fig. 3). Therefore the multiple losses of introns a and d near the base of subfamily 2, intron c at the bases of subfamilies 5 and 6, and intron e in subfamily 8 are questionable. Of these, the worst case involves intron e in subfamily 8, where it is present in only one gene, srh-210/D1065.4. An alternative evolutionary scenario for this intron might involve loss from the common ancestor of this subfamily (along with intron a) followed by regain in precisely the same codon and phase into this gene; however, this scenario seems unlikely and instead the poorly supported relationships of the basal lineages of subfamily 8 might be rearranged to yield only two losses of intron e. Conservatively then, 70 independent losses are inferred. Unfortunately, it is difficult to include intron losses as characters in deriving the tree because it is unclear how heavily intron losses should be weighted relative to single amino acid changes and inclusion of an intron presence/absence matrix greatly increases the computational complexity making analysis of this large dataset intractable. For these purposes, it is prudent not to include them in the data matrix, to allow quite independent mapping of the losses on the tree. Intron losses would nevertheless have considerable value as phylogenetic characters, particularly in that losses are likely to be irreversible, and depending on their weighting, would probably lead to minor rearrangements of the phylogenetic tree making the mapping of intron losses rather more parsimonious. This mapping nevertheless demonstrates how frequent these losses are, involving many independent losses of each intron in disparate lineages. The ancestral five-intron arrangement (Fig. 4) was maintained in two separate lineages of subfamilies 2 and 5 and persists in eight genes. As was true for the str and stl families, no gene has lost all of its introns, with only 13 reduced to a single intron, perhaps because at least one intron is necessary for expression of nematode genes (e.g., Okkema et al. 1993).
In contrast, there are seven instances of apparent intron gain within this family (Fig. 3). Remarkably, introns k and l appear to have been inserted into the same position of the same codon, however, this region of the carboxyl terminus of these proteins is highly divergent in amino acid sequence and so it is likely that they are simply independent insertions. The postulated gain of introns f and l in subfamilies 2 and 3 are the least certain of these because they could have been ancestral introns that were lost from the other subfamilies. Nevertheless, they are considered to be intron gains here. Introns j and k are also reasonably old gains within subfamilies 6 and 5. The remaining three intron gains—g, h, and i—were fairly recent, being found in only one or two genes in subfamilies 6 and 8. Logsdon et al. (1998) and Logsdon (1999) have reviewed the criteria required to establish recent origin of an intron, primarily that its novel appearance must on phylogenetic grounds greatly outweigh the likelihood of it being ancient but having suffered multiple independent losses, and that its origin can be determined from its sequence. With the possible exception of introns f, k, and l, the first criterion is convincingly satisfied here, with these seven introns (f–l) requiring postulation of 3, 12, 9, 13, 8, 5, and 5 independent losses, respectively, to be of ancient origin. Unfortunately, the second criterion cannot easily be established for nematode introns because they evolve in sequence so rapidly (see below for comparisons between C. elegans and C. briggsae). Therefore even the two introns shared by just two genes (h and i) have diverged completely in sequence and are unalignable between the pairs of genes, let alone with any other sequences in the C. elegansgenome. Intron g is 950 bp long and is present in only one gene,srh-240/F37B4.4b in subfamily 8, so appears to be the most recently acquired, nevertheless, its origin is unclear. The first ±170 bp are repeated ∼5 times elsewhere in the genome, including one repeat in the same cosmid, however, the remainder has no matches.
C. briggsae Homologs
The WashU GSC has generated 8 Mbp or ∼8% of the C. briggsae genome, providing 20 genes in 10 cosmids to compare with these C. elegans genes. These clones have not been annotated and deposited in GenBank; however, they are available from the WashU GSC database (pers. comm.). The phylogenetic relationships of these genes are shown in Figure 3, with the C. briggsae genes in bold type (the clone numbers all begin with G) and details of comparisons with orthologous genes in C. elegans are shown in Table 1. The levels of divergence between orthologous genes are comparable with those seen previously for the strand stl families (Robertson 1998) and a variety of other genes (summarized in de Bono and Hodgkin 1996).
Comparison of C. briggsae Chemoreceptor Genes with theirC. elegans Orthologs in thesrh Family
Convincing C. elegans orthologs are available for 14 of the 20C. briggsae genes, that is, those on clones that share several other genes or DNA sequences in reasonable, but not necessarily perfect, synteny (e.g., Kuwabara and Shah 1994; Robertson 1998). They generally encode proteins that are colinear with each other, except that sometimes the amino and commonly the carboxyl termini differ in length. The encoded amino acid sequences of these convincing orthologs retain 68% identity on average (Table 1). Simple inspection of Figure3 shows that large numbers of pairs, triples, quadruples, and even septuples of C. elegans genes are more similar than this, indicating that they originated by gene duplication after the species split; 85 gene duplications are inferred in the C. eleganslineage since the split (Fig. 3). The most extreme example involves a set of seven genes and pseudogenes on the overlapping cosmids F20E11 and F40D4 at the apex of subfamily 7, of which only one apparently remains functional, whereas three of another set of seven recently formed genes in this subfamily on cosmid K08G2 apparently remain functional. In contrast, six, or 30% of the C. briggsae genes do not have clear orthologs in C. elegans, so they must have been lost from the C. elegans genome since the split. The loss of G21D19.b, c, d, and e orthologs is the major contributor to this large number and may have involved a single large deletion in the C. elegans lineage.
Two other features of these interspecies comparisons are worth noting. First, as expected (e.g., de Bono and Hodgkin 1996; Robertson 1998;Thacker et al. 1999), the introns and the 5′ and 3′ flanking sequences have diverged so much they are unalignable. Consistent with this level of divergence, the frequency of synonymous changes,Ks , is very high (averaging 1.73, which is 7.7-fold higher than the average frequency of nonsynonymous changes) (Table 1). Even comparisons of pseudogenes between the species give highKs/Ka ratios, indicating that they became pseudogenes after the species split. Second, although most introns are still shared in particular positions in these genes, two or 2.4% [2/(41 × 2) = 0.024] have been lost since the species split, both from C. briggsae genes, a bias observed for other genes (e.g., de Bono and Hodgkin 1996;Robertson 1998; Thacker et al. 1999; Dufourcq et al. 1999).
Insertions and Deletions
The above comparisons with the C. briggsae genes strongly suggest that duplication of large regions of the genome including single or multiple genes are common, and that these are balanced by complete loss of genes, thereby maintaining a reasonably steady-state overall genome size. This dynamic equilibrium is in stark contrast to the situation of the human genome lineage, where large numbers of pseudogenes and enormous numbers of transposon insertions have persisted for a very long time with few large deletions removing them (e.g., Graur et al. 1989; Gu and Li 1995; Robertson and Martos 1997). The dynamics of the nematode genome appear more similar to those ofDrosophila species, where “junk” DNA is rapidly removed by large deletions (Petrov et al. 1996; Petrov and Hartl 1997, 1998). Large duplications are readily observable in the C. elegansgenome ( C. elegans Sequencing Consortium 1998; C. elegans Genome Consortium 1999; Semple and Wolfe 1999), however, identification of the balancing deletions is less obvious. In an attempt to quantify this deletion process, all of the pseudogenes and gene fragments in this dataset were examined for the length of indels when compared with their closest intact relative. Deletions were only included if they were within the gene so that their termini were clear, and only the lengths of exon sequence removed were counted as a conservative estimate. The results in Figure 5 show the expected high frequency of single base deletions, but in addition there are 21 deletions of longer than 20 bp, the longest being 786 bp (two very long insertions within genes were also observed, perhaps representing the type of event mediating the high frequencies of gene movement described above). Furthermore, some pseudogenes have truncations of one or other terminus, and there are many gene fragments with one or other terminus missing that were not formally included in the family. In these cases, the length of the truncation was determined as the exonic region of the gene that is missing, although of course this is a gross underestimate because the truncations may extend much further and again ignores introns that are commonly several hundreds of bases in the srh family. Offsetting this conservative measure is the likelihood that some of these gene fragments and truncated pseudogenes might actually be the ends of duplicated regions and not result from deletions at all. The average size of 31 such truncations was 514 ± 56 bp (s.e.).
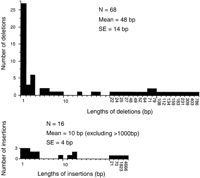
Frequency distributions of lengths of deletions and insertions insrh chemoreceptor family pseudogenes and fragments. After lengths of 20 bp, only lengths of indels actually present are shown.
DISCUSSION
This is one of the largest families of genes reported in the C. elegans genome, with the 214 genes encoding apparently functional chemoreceptors constituting ±1% of its functional gene complement or proteome, estimated at ±19,000 by the C. elegansSequencing Consortium (1998). Many authors have defined protein/gene families as those sharing at least 45% amino acid identity, however, such a definition is inappropriate for families of proteins in C. elegans where rates of molecular evolution appear to be particularly high (note that even orthologous chemoreceptors in the congeners C. elegans and C. briggsae on average share only 68% identity). Such a definition would lead to splitting thissrh family into at least 100 families. Extending the family to include subfamilies 1–4, whose members share as little as 12% amino acid identity with the rest of the family, is justified on the basis of maintaining the cohesiveness of the family. It can also be justified on the basis of their shared ancestral intron arrangement, which is quite different from the related families. In preliminary large-scale analyses including the srd, str, and stlfamilies, the srh family as defined here is monophyletic and a sister group to the str and stl families. There is every reason to believe that these are all chemoreceptors given the sister relationship of the srh family to the strfamily, which contains the ODR-10 diacetyl receptor (Sengupta et al. 1996; Troemel et al. 1997; Zhang et al. 1997). Certainly it is difficult to imagine what other function such a large family of transmembrane receptors might serve. The str, stl, and srd families consist of about 200, 40, and 60 functional chemoreceptor genes, respectively (Sonnhammer and Durbin 1997;Robertson 1998; updated in November 1999), so together these three families encode at least 300 chemoreceptors. In addition, there are now ∼200 genes (Bargmann 1998) in the sra, srb,sre, and srg families originally identified byTroemel et al. (1995), and there are several smaller, undescribed families of likely chemoreceptors in the genome bringing the total of functional chemoreceptor genes to ∼800 encoding ±4% of theC. elegans proteome.
Presumably, these hundreds of receptor proteins are involved in detection of the many water-soluble and volatile chemicals that this nematode can perceive (Bargmann and Mori 1997). Troemel et al. (1995)demonstrated that representatives of their five families are probably primarily expressed in the chemosensory neurons by examining expression of fusion reporter genes under control of their promoter regions. Similar tests have been performed for 16 representative genes in thesrh family. Six were expressed only in sensory neurons, five in sensory neurons plus elsewhere, four only elsewhere, and one was not expressed (Y. Zhang and C. Bargmann, pers. comm.). There are just 16 pairs of chemosensory neurons (Bargmann and Mori 1997), and generally receptor genes are only expressed in one, or at most two, pairs of neurons (Troemel et al. 1995); therefore, on average, 40–50 different genes must be expressed in each pair of neurons. They are not expressed at high levels because among the ±73,000 C. eleganssequences in dbEST there is only one expressed sequence tag (EST) from a cDNA clone from a srh family member (the 3′ read from clone yk446b1 matches gene srh-2/C05E4.a in subfamily 1). The identification of at least 14 Gα proteins expressed exclusively in subsets of these and related sensory neurons suggests that there are multiple transduction pathways for diverse chemoreceptors expressed in the same neuron (Jansen et al. 1999).
The patterns of molecular evolution of these srh family genes abundantly confirm and extend those described previously for thestr and stl families (Robertson 1998). For example, in each case there is abundant evidence for recent duplications of genes; within the srh family 85 gene duplications can be inferred since the split from the congener C. briggsae. Whereas many of these duplicated genes have apparently retained functionality while presumably diverging to new odorant specificities, 44 have become pseudogenes. Simple inspection of the phylogenetic relationships of these and other pseudogenes in Figure 3 shows that they are all young pseudogenes. Presumably most pseudogenes are rapidly removed by random large deletions of hundreds of bases in the genome, which are revealed here by comparison of pseudogenes and gene fragments with their most closely related apparently functional gene. In addition, several orthologs of genes in C. briggsae are missing in C. elegans, presumably by deletion. Semple and Wolfe (1999) recently described similar large deletions when comparing three large recently duplicated regions of the C. elegansgenome. These results show how frequent large random deletions can explain the small size of the C. elegans genome in the face of clearly frequent gene duplications. In this regard,Caenorhabditis nematodes are more like Drosophilaflies (Petrov et al. 1996; Petrov and Hartl 1997, 1998) than humans (Graur et al. 1989; Gu and Li 1995; Robertson and Martos 1997).
As was the case for the str and stl families, intron losses prevailed numerically over intron gains (70 to 7), although the bias was not as extreme as in the str and stlfamilies (165 to 1) (Robertson 1998). In part, this is because the ancestral intron arrangement for the family appears to have been five introns, versus eight each for the str and stlfamilies, thereby providing fewer opportunities for intron losses, and in addition several introns were lost early in large subfamilies, further reducing the opportunities for subsequent independent intron losses. Most of these losses are of single introns at a time, with just four convincing cases of adjacent introns being lost on single branches of the tree. I suggested previously that these independent losses of single introns might best be explained by precise in-frame deletion through nonhomologous recombination between short direct repeats at the 5′ and 3′ splice junctions (Robertson 1998). A similar mechanism might also be responsible for the frequent large deletions discussed above.
The gain of at least five and probably seven new introns within thesrh family strongly supports the notion of introns as recent acquisitions of eukaryotic genes (e.g., Logsdon et al. 1998). Logsdon (1999) was able to find only four such convincing examples in recent literature, and this srh family provides at least five. Given this clearcut evidence for recent intron acquisition, it is reasonable to infer that all of the ancestral introns in the srh,str, and stl families are also older intron gains, especially because none are shared with the more distantly relatedsrd family. Gotoh (1998) examined intron gains and losses in the 60 p450 genes then available in the C. elegans genome, and in these older and more conserved gene lineages that can be traced to common ancestries with vertebrates and insects, found similarly high rates of intron losses and gains. These results therefore strongly support the “introns-late” view of intron evolution (e.g., Logsdon et al. 1998; Logsdon 1999).
One of the most remarkable aspects of this family is the concentration of genes on chromosome V. The same is true for the relatedstr, stl, and srd families (J. Spieth, pers. comm.) suggesting that the common ancestor of all of them resided on this chromosome, and most of the expansion of these large families has occurred on this chromosome. A similar pattern has been reported bySluder et al. (1998) for the large nuclear receptor superfamily. Mapping of the srh gene locations on the phylogenetic tree shows that only occasional gene movements to other chromosomes have occurred, and only a few of those have led to expansion of groups of genes on other chromosomes. For some reason, most movements off chromosome V in this family have been to chromosome II, which might be taken as evidence for some kind of physical association of these two chromosomes that predisposes such movement; however, examination of the other families (J. Spieth, pers. comm.) shows that they have most frequent movements to chromosomes IV and X. Most of these movements to other chromosomes are very recent, because they involve single genes. A possible inference is that most genes that moved to other chromosomes have subsequently been lost. Perhaps genes on chromosome V are more likely to persist evolutionarily because of increased frequencies of gene conversion maintaining their integrity. Semple and Wolfe (1999)report that although frequencies of gene conversions between members of gene families in C. elegans are relatively low, they do occur more commonly between genes on the same chromosome. Examination of other large gene families on chromosome V and other chromosomes may help illuminate this puzzle.
METHODS
Searches of the nonredundant and HTGS DNA databases at NCBI (Benson et al. 1998) were conducted using TBLASTN v2.05 (Altschul et al. 1997) to recover the intron/exon arrangements of these genes, which were then aligned by eye in the editor of PAUP for the Macintosh (Swofford 1998). This process was repeated iteratively until most members of thesrh family had been identified. The divergent subfamilies 3 and 4 were discovered using a PSI-BLAST search (Altschul et al. 1997). Shared subsets of five introns at exactly the same positions were useful landmarks, especially for alignment of pseudogenes, and the NSPL program of GeneFinder from the Baylor College of Medicine WWW site (http://dot.imgen.bcm.tmc.edu:9331/gene-finder/gf.html) was also used to help identify intron boundaries. The encoded translations were initially similarly aligned by eye in the PAUP editor. Alignments of transmembrane (TM) regions 1, 2, 3, 6, and 7 are unambiguous, being easily anchored by several highly conserved amino acids (see Fig. 1). The boundaries of TM4 and TM5 were sometimes difficult to align confidently between subfamilies 1–4 and the rest of the family, therefore for the phylogenetic analysis an alignment obtained using ClustalX at default settings was employed (Jeanmougin et al. 1998). This yielded the same blocks of aligned amino acids for the transmembrane domains, differing only in minor points regarding placement of gaps between them, but manual adjustment was necessary for more appropriate alignment of many of deletions in pseudogenes. All amino acid positions were employed for the phylogenetic analyses to provide the maximum possible information within subfamilies. Phylogenetic analysis was performed using neighbor-joining followed by tree-bisection-and-reconnection branch-swapping as implemented by PAUP v4.0b2a for the Macintosh (Swofford 1998). Bootstrap analysis employed 1000 neighbor-joining replications. Molecular evolution of pairs of genes was assessed by computing the frequencies of synonymous (Ks ) and nonsynonymous (Ka ) changes following Nei and Gojobori (1986), using the Macintosh programKsKa Calc (H. Akashi, pers. comm.).
Acknowledgments
I thank the Genome Sequencing Centers at Washington University, St. Louis, USA, and the Sanger Center, Cambridge, UK, for communication of DNA sequence data before publication; John Spieth, Steve Jones, and Richard Durbin for their assistance in annotating these nematode genes; John Spieth for communication of chromosomal locations of these and other chemoreceptor genes; and Yongmei Zhang and Cori Bargmann for communicating their unpublished expression studies. This work was supported by NSF grant IBN 96-04095.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
E-MAIL hughrobe{at}uiuc.edu; FAX (217) 244-3499.
-
- Received June 4, 1999.
- Accepted December 17, 1999.
- Cold Spring Harbor Laboratory Press








