
The Mouse Clock Locus: Sequence and Comparative Analysis of 204 Kb from Mouse Chromosome 5
Abstract
The Clock gene encodes a basic helix-loop-helix (bHLH)–PAS transcription factor that regulates circadian rhythms in mice. We previously cloned Clock in mouse and human using a battery of behavioral and molecular techniques, including shotgun sequencing of two bacterial artificial chromosome (BAC) clones. Here we report the finished sequence of a 204-kb region from mouse chromosome 5. This region contains the complete loci for the Clock andTpardl (pFT27) genes, as well as the 3′ partial locus of the Neuromedin U gene; sequence analysis also suggests the presence of two previously unidentified genes. In addition, we provide a comparative genomic sequence analysis with the syntenic region from human chromosome 4. Finally, a new BAC transgenic line indicates that the genomic region that is sufficient for rescue of the Clock mutant phenotype is no greater than 120 kb and tightly flanks the 3′ end of the Clockgene.
[The sequence data reported in this paper have been submitted to the GenBank data library under accession no. AF146793.]
The last three years have witnessed remarkable progress in our understanding of the molecular basis of circadian rhythms (Wilsbacher and Takahashi 1998; Dunlap 1999; Strayer and Kay 1999; King and Takahashi 2000). In most if not all living systems, a conserved transcription-translation-based negative autoregulatory feedback loop appears to form the basis for circadian mechanisms. In mammals, the transcription factors CLOCK and BMAL (also known as MOP3) activate expression of the Period (Per) andCryptochrome (Cry) genes; the steady-state mRNA levels of these genes express circadian rhythms of abundance in the suprachiasmatic nucleus (SCN), which is the site of the master circadian pacemaker in mammals (Gekakis et al. 1998; Hogenesch et al. 1998; King and Takahashi 2000; Shearman et al. 2000). Through mechanisms that are still being defined, the PER and CRY proteins translocate to the nucleus and inhibit the activity of CLOCK and BMAL acting upon the promoter regions of the Per and Crygenes (Gekakis et al. 1998; Sangoram et al. 1998; Griffin et al. 1999;Kume et al. 1999). Finally, turnover of the inhibitory complex leads to the release of CLOCK/BMAL inhibition, and CLOCK/BMAL-driven transcription begins anew.
Because transcriptional control plays such an important role in the circadian model, it will be essential to define the regulatory elements that control circadian gene expression. Genomic sequence of circadian loci will provide extremely useful information for the study of transcriptional regulation. For example, in the Drosophila period (dper) gene, an E box element (5′-CACGTG-3′) lies within a 69-bp enhancer that is required for high-amplitude cycling of the per transcript (Hao et al. 1997). The dper E box element and those found in the promoter regions of mouse Period1 (mPer1), mousevasopressin, and Drosophila timeless (dtim) are known to mediate CLOCK/BMAL-activated transcription of those genes (Darlington et al. 1998; Gekakis et al. 1998; Jin et al. 1999;Ripperger et al. 2000). Genomic sequence also provides an important tool for circadian gene discovery, as shown with the identification of human Per1 (Sun et al. 1997). Here we report the complete sequence of two overlappingmouse BAC clones from mouse chromosome 5 and describe the structures of the Clock locus and four other genes within this 204-kb interval. In addition, we provide a comparative genomic sequence analysis with the syntenic region from human chromosome 4. Finally, we present new BAC transgenic rescue results that reduce the genomic interval that is necessary for properClock expression.
RESULTS
Shotgun Sequencing and Finishing of Two Bacterial Artificial Chromosome Clones
Twelve bacterial artificial chromosome (BAC) clones were isolated in a library screen for clones spanning the Clock nonrecombinant region (King et al. 1997a). Based on genetic and physical mapping near the Clock locus, the 140-kb clone BAC 54 and the 160-kb clone BAC 52 were chosen for sequencing (King et al. 1997a; King et al. 1997b). These two BACs were also selected for transgenic rescue experiments (Antoch et al. 1997), as was BAC 49 (described in this paper). Previous results showed that the full-length BAC 54 clone rescued the phenotypic effects of the Clock mutation (Fig.1; Antoch et al. 1997).
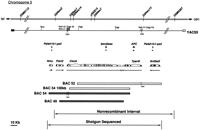
Physical map of the 204 kb flanking the Clock locus. TheClock locus lies at the midportion of mouse chromosome 5. The genomic locus is updated from Antoch et al. (1997); genetic markers and restriction sites are indicated on the chromosomal and YAC55 maps. The positions of pseudogenes Pafah1b1-ps2 (2 copies),bendless, and APC are shown relative to Nmu,Pdcl2, Clock, Tpardl, and Srd5a2l. The bacterial artificial chromosome (BAC) clones that rescue theClock mutation, BAC54 and BAC49, are shaded, whereas the nonrescuing BAC clones 52 and 54 (100 kb) are shown as open boxes. BAC 54 and BAC 52 were sequenced to generate the data presented here.
We used the shotgun sequencing approach (Sulston et al. 1992;Ansari-Lari et al. 1998) to obtain genomic sequence near theClock locus. Specifically, we adapted the high-throughput shotgun sequencing protocols available at the Washington University Genome Sequencing Center (http://genome.wustl.edu/gsc/index.shtml) and the Whitehead Institute for Biomedical Research (http://www-genome.wi.mit.edu/) for use in an individual laboratory. BAC 52 and BAC 54 were subcloned into M13 shotgun libraries; approximately 1200 BAC 52 and 1400 BAC 54 M13 subclones were purified and sequenced. This first phase of sequencing generated many small contigs (2 kb–30 kb), most of which were joined via reverse strand sequencing of M13 clones at the ends of the contigs. The prefinished project consisted of five contigs (10 kb–70 kb); gaps were filled using PCR and primer walking. The complete, finished sequence encompasses a region of 204 kb in the midportion of mouse chromosome 5 (Figs. 1, 2). Figure 2, a percentage identity plot (PIP; see below) of the mouse sequence against two unfinished human BAC clones (GenBank accession nos. AC069200 andAC024243), displays the organization of genes and their exons, repetitive elements, CpG islands, predicted promoter elements, pseudogenes, and genetic markers within the region. The complete loci of two previously identified genes, Clock (GenBank accession no. AF000998) and Tpardl (pFT27; GenBank accession no. M23568), map to this region. The 3′ end of another previously identified gene, Neuromedin U (Nmu) precursor (GenBankAF203444), maps to the telomeric end of BAC 54. Two Unigene clusters lie within this interval, one at the centromeric end of BAC 52 and the other between Nmu and Clock. Finally, pseudogenes for platelet-activating factor acetylhydrolase Ib alpha subunit (Pafah1b1-ps2), adenopolyposis coli (APC), and ubiquitin-conjugating enzyme homolog (bendless) exist in the 204-kb region (Joslyn et al. 1991; Su et al. 1992; Yamaguchi et al. 1996; Peterfy et al. 1998). The Pafah1b1-ps2 andbendless pseudogenes are full-length, while the APCpseudogene represents 1240 bases near the 3′ end of the functionalAPC gene. All three pseudogenes appear to have arisen via fusion with a member of the long-terminal-repeat retrotransposon family followed by insertion at this locus of study (Miki et al. 1992; Peterfy et al. 1998).
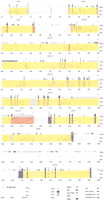
Percentage identity plot (PIP) of 204-kb mouse sequence against unfinished human sequence. The PIP plots the mouse genomic sequence on the x-axis and the percentage identity to the human sequence on the y-axis. Annotation of the mouse sequence is indicated along the top of each plot: gene name, gene orientation, exons, repetitive elements, and CpG islands. Gene regions are shaded in yellow, and the exons of each gene are marked as full-length bands in corresponding colors: Nmu, light green; Pdcl2, orange; Clock, pink; Tpardl, purple; andSrd5a2l, blue. Human Clock exon 1 is shown in red at 54.0 kb. Pseudogenes are shown as light gray full-length bands:Pafah1b1-ps2, 10 kb; bendless, 106 kb; APC, 142 kb; and Pafah1b1-ps2, 187 kb. Predicted promoter elements are shown as dark green half-length bands: promoter 1 (P1), 16.2 kb; P2, 35.0 kb; P3, 36.2 kb; P4, 154.3 kb; P5, 154.8 kb; and P6, 198.5 kb. SSLP markers are shown as dark gray half-bands: D5Nwu 1, 35.9 kb; D5Nwu14, 48.0 kb; D5Nwu7, 90.9 kb; and D5Nwu13, 190.4 kb. See key for annotation of exons, UTRs, repetitive elements, and CpG islands.
Neuromedin U Locus
BLASTN homology revealed the presence of the three 3′-most exons of mouse Nmu precursor mRNA (GenBank accession no. AF203444). Exon/intron boundary locations and exon and intron lengths are listed in Table1. Nmu codes for the Nmuprecursor peptide and is orthologous to rat Nmu (Lo et al. 1992). The three mouse Nmu exons (named exon a–c) lie at the telomeric end of BAC 54 and encompass bases 506–827 of the gene (Fig.2). A consensus polyadenylation signal occurs 22 bases upstream of the 3′ end. Neither GRAIL norGenScan gene prediction programs detected anyNmu exons.
Gene Exon/Intron Characteristics
A region with BLASTX homology to F0membrane domain ATP synthase subunit g from cow, mouse, and human (GenBank accession nos. AAB31108, CAA76699, and AAC61597, respectively) was detected within intron b of Nmu (data not shown; Collinson et al. 1994). Due to low similarity and the lack of ESTs with which to characterize the area, we did not further investigate this similarity.
Unigene Cluster: Phosducin-Like-2 (Pdcl2) Locus
A PowerBLAST (Zhang and Madden 1997) search revealed a family of >50 mouse RIKEN mouse EST clones immediately centromeric ofNmu. The ESTs showed a clear exon/intron structure of 5 exons that spanned 13 kb; the 5′ end of the alignment arose precisely at an intron/exon junction (Fig. 2). A 717-bp open reading frame, devoid of an initiating methionine, lies at the 5′ end of the 1038-bp contiguous alignment of the EST family. The putative translation displays similarity to human Phosducin-Like Protein (PhLP; GenBank accession no. AAD01930). Due to this similarity, we named this putative gene Phosducin-like protein similar 1 (Pdcl2). Because we did not determine the 5′ end of this gene, we annotate it as exon a to exon e. At the 3′ end, a polyadenylation signal occurs at bases 1020–1027. GRAIL detected exon b with inaccuracies at the 5′ and 3′ ends and accurately predicted exons c and d. GenScan accurately predicted all five exons, as well as a potential 87-bp initiator exon 1682 bases upstream of exon a.
Clock Locus
The Clock gene spans 90 kb and contains 23 conventional exons, as well as one alternatively spliced exon (exon 1a; in Fig. 2, exon 1a is not included; King et al. 1997a). Exon/intron boundary locations, exon lengths, and intron lengths are listed in Table 1. The ATG initiator codon occurs in exon 4 and agrees well with the Kozak consensus sequence (Kozak 1996; King et al. 1997a). The region encoding the basic helix-loop-helix regulatory motif spans exons 5 and 6; the PAS A-encoding region lies in exons 7, 8, and 9; the PAS B-encoding region lies in exons 12 and 13; and the region coding for a glutamine rich domain is found in exons 19–23. GRAILand GenScan differed remarkably in their abilities to detect Clock exons (Table 1).GRAIL identified only 13 of the 24 exons; of the 13 Clock exons GRAIL found, 7 were assigned an incorrect splice site. In contrast,GenScan detected 17 Clock exons, 16 of which were predicted with accurate splice sites. Exons 1a, 1b, 2, 8, and 10, which range in size from 70 bp to 181 bp, were not detected by either program. Single nucleotide polymorphisms at 34.5 kb and 124.0 kb were detected between C57BL/6J, the strain used in circadian behavior experiments and that carries the Clock mutation (Jackson Laboratory Stock no. 002923), and 129S1/Sv- + p +Tyr - c MgfSl-J / + (Jackson Laboratory Stock no. 00090), the 129 strain from which the CJ7 ES cell line was derived and used for the Research Genetics BAC library construction (Swiatek and Gridley 1993). Finally, 12 simple sequence repeats were detected in the Clock locus, some of which are known to be polymorphic between mouse strains (dark gray half-length bands in Fig. 2; GenBank accession nos. AF146780, AF146792, andAF146786).
We previously reported the presence of two Clock transcripts (8 kb and 11 kb) in all tissues that express Clock (King et al. 1997a). The smaller transcript was found to be 7490 bp in length (Genbank accession no. AF000998; King et al. 1997a), but the longer transcript was not detected in any of the mouse cDNA libraries we screened. A BLASTN search of the genomic sequence against the EST database (Altschul et al. 1990) revealed several expressed sequence tags (ESTs) just downstream of the 3′ end of the 7490 bp transcript. These ESTs, most of which are 3′ reads in the correct orientation with respect to Clock, may represent the terminal end of the larger Clock transcript. However, the EST sequences are not contiguous with the knownClock transcript. One family of ESTs (960 bp in length) begins 37 bases downstream from Clock. This family appears to be primed off of a poly(A) tract in the genomic sequence and likely does not represent a true expressed sequence. A second EST family of 710 bp arises 370 bases downstream from the first family, and this cluster contains a polyadenylation signal in an appropriate position for an expressed sequence (EST family GenBank accession nos. AA607824,AW492507, AA164040, AI843957, AI451178, and AV367325). If this 710-bp EST family does indeed represent Clock, it predicts a 9.6-kb transcript.
Tpardll Locus
Tpardl, also known as pFT27, encodes a putative transmembrane protein of unknown function (Fig. 2; Akagi et al. 1988). The 5′ UTR, complete coding sequence, and 3′ UTR comprise six exons over 25 kb. Exon/intron boundary locations, exon lengths, and intron lengths are listed in Table 1. The 3′ end of Tpardllies 2.9 kb away from the Clock polyadenylation signal which generates the 7490-bp transcript and merely 624 bp away from the 710 bp EST family downstream from Clock.GenScan detected all six exons; exons 1–5 were predicted accurately, but the 3′ end of exon 6 was underestimated.
Unigene Cluster: Steroid 5-alpha Reductase Type 2-Like(Srd5a2l) Locus
The PowerBLAST search revealed a second family of >50 mouse and human ESTs centromeric of Clock. When aligned to the genomic sequence, the mouse ESTs represent a gene that spans 15 kb and includes six exons (Fig. 2). The 1746-bp contiguous EST alignment contains a large open reading frame that encodes a putative protein of at least 330 amino acids. This hypothetical protein shares similarity withCaenorhabditis elegans hypothetical protein Z71178 (GenBank accession no. CAA94885), as well as human and rat 3-oxo-5-alpha-steroid 4-dehydrogenase 2 (GenBank accession nos. P31213 and P31214, respectively; Andersson et al. 1991; Normington and Russell 1992;Wilson et al. 1994). Due to these protein similarities, we named this gene Steroid 5-alpha reductase type 2–like(Srd5a2l). Sequencing of the 3′-most EST clone (IMAGE 949802, GenBank accession no. AA537506) revealed two polyadenylation signals, one at bases 1483–1490 and the other at bases 1706–1713 of the contiguous EST alignment. IMAGE clone 949802 was used to probe a Clontech mouse multiple tissue Northern blot (data not shown). Transcripts of ∼1.6 kb and ∼1.8 kb in length were detected, which confirmed the existence of Srd5a2l and is consistent with the two polyadenylation sites. Srd5a2l is highly expressed in liver and kidney; moderately expressed in heart, spleen, and lung; minimally expressed in brain and testis; and not expressed in skeletal muscle. Although the transcript sizes agree reasonably well with the length of the contiguous EST alignment, no conclusive data confirms the 5′ end of Srd5a2l. Therefore, we consider this gene partial and annotate the gene as exon a to exon f.GenScan detected all of the proposed exons except exon e, which GRAIL accurately predicted.
Comparative Genomic Sequence Analysis
Previously, the human CLOCK exon structure was determined via sequencing of a human BAC using CLOCK cDNA-derived primers (Steeves et al. 1999). The exon structure of mouse Clock and human CLOCK is perfectly conserved between mouse coding exons 4–23 and human coding exons 3–22 (human exons were named arbitrarily, as the BAC from which they were sequenced does not contain the 5′ end of the gene; Steeves et al. 1999). Human CLOCK 5′ UTR exons 1 and 2 do not appear to be conserved with mouse Clock5′ UTR exons 1, 2, or 3 (data not shown). We searched the HTGS database with the human CLOCK (GenBank accession no. AB002332;Nagase et al. 1997) and the mouse Pdcl2 cDNA sequences and identified two nonoverlapping human chromosome 4 BAC clones. Both clones are in HGTS Phase 1 of sequencing at the Washington University Genome Sequencing Center. BAC RP11–528I4 (GenBank accession no.AC024243), which contains sequence similarities to Pdcl2, contains 164070 bp in 18 unordered pieces. BAC clone RP11–177J6 (GenBank accession no. AC069200), which contains the humanCLOCK transcription unit, contains 148205 bp in 19 unordered pieces. All CLOCK exons map with 100% identity to RP11–177J6. Of note, human exons 1, 2, and 3 align within a single contig; as in mouse, a large intron (20.4 kb) lies between human exons 1 and 2. Human exons 11–22 align within the largest contig, while exons 4–10 are interspersed among four small contigs.
We used PipMaker(http://nog.cse.psu.edu/pipmaker) to create a PIP for comparison of the finished 204-kb mouse sequence to the unfinished human sequences (Fig.2; Schwartz et al. 2000). The PIP indicates extensive similarity between the human RP11–528I4 (Pdcl2-containing) sequence and the 0 kb–40 kb subregion of mouse sequence, as well as a small region of similarity at 48 kb of the mouse sequence. The region at 48 kb has no significant similarity to sequences in theBLAST nonredundant nucleotide database. The PIP also displays similarity between the human RP11–177J6 (CLOCK-containing) sequence and the 53.0 kb–185.2 kb subregion of the mouse sequence. The PIP, therefore, suggests that the gap between the two human clones corresponds to an ∼13-kb region in mouse (40 kb–53 kb), all of which lies in the 29-kb mClockintron 2. The coding exons of all genes in the 204-kb mouse sequence show >75% identity with the human sequence. Significant similarity exists within the unmasked portions of introns as well (mouse repetitive elements were masked usingRepeatMasker; see Methods) and may represent conserved regulatory regions.
Promoter Analysis
Transgenic Rescue of the Clock Mutant Phenotype Using BAC 49
Previously, we showed that the 140-kb transgene BAC 54 could rescue the phenotypic effects of the Clock mutation (Antoch et al. 1997). BAC 54 contains the entire Clock gene, 34 kb of upstream sequence and 12 kb of downstream sequence. The 120-kb BAC 49 flanks the Clock gene more tightly than BAC 54, as it contains the complete Clock coding region, 25 kb of upstream sequence and only 4 kb of downstream sequence. Given this information, we generated a transgenic line via pronuclear injection of BAC 49 to confirm functionally the extent of the Clock gene regulatory region.
Due to the breeding scheme, all BAC 49 transgenic founder progeny were heterozygous for the Clock mutation. Representative activity records show that BAC 49 transgene-positive progeny show wild-type phenotypic effects: The free-running period is shorter than 24 h (mean = 23.51 ± 0.052 h), and a saturating light pulse administered at approximately circadian time 17 results in a 3.77 ± 0.453-h phase delay (Fig. 3). In contrast, nontransgenic progeny from this founder express all the phenotypic characteristics of Clock/+ animals, including longer circadian period (mean = 24.04 ± 0.06 h) and 5.645 ± 0.706-h phase delay in response to a light pulse at circadian time 17 (Fig. 3). These results indicate that the genomic interval that is sufficient for Clock expression in the circadian context is no larger than 120 kb. Most, if not all, of the circadian regulatory elements are likely to be found either in the 25-kb upstream region or within the exon/intron structure, as the downstream region encompasses only 4 kb and contains the 3′ end of the Tpardl gene.
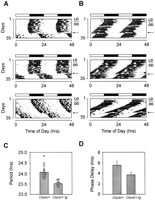
The BAC 49 transgene rescues Clock/+ mutant phenotype. Wheel-running activity records and period estimates of Clock/+ F1 transgenic mice and their nontransgenic littermates are shown. A total of 19 control and 15 transgenic animals were tested for their circadian phenotype. All animals were maintained on an LD12:12 cycle for 1 wk (LD) and were transferred into constant darkness (DD) on the day indicated by the bar to the right of each actogram. All animals received a saturating 6-h light-pulse at CT17 after a 21-d exposure to DD (indicated by an arrow). (A) Activity records of three individual Clock/+ mice without the BAC 49 transgene. Light pulse results in an average phase delay of 5.645 ± 0.706 h. (B) Activity records of three individual Clock/+ mice with the BAC 49 transgene. Light pulse results in an average phase delay of 3.77 ± 0.453 h. (C) Period estimate inClock/+ nontransgenic mice and Clock/+ transgenic (tg) mice. Average period length in Clock/+ mice is 24.04 ± 0.06 h (n = 19), while average period length inClock/+ tg mice is 23.51 ± 0.052 h (n = 15). The difference in average period is significant by t-test analysis (P <0.00001). (D) Phase delay response to a light pulse in Clock/+ nontransgenic mice and Clock/+ transgenic (tg) mice. Average phase delay in Clock/+ mice is 5.645 ± 0.706 h (n = 10), while average phase delay in Clock/+ tg mice is 3.77 ± 0.453 h (n = 11). The difference in average phase delay is significant byt-test analysis (P <0.03).
Promoter Prediction Programs
To facilitate the analysis of regulatory regions, particularly those of Clock, we utilized the TSSGpromoter prediction program and MatInspectortranscription factor binding site analysis program (Quandt et al. 1995). Taking the error rate of TSSG into account (at 50%–55% true promoter recognition, one false-positive promoter per 5000 bp will be found on average), we disregarded predicted promoters with a score less than 5.0. Thirteen promoters were predicted throughout the 204-kb region, six of which had a score >7.5. Of these six predicted promoters, promoter 1 (P1) lies withinPdcl2, P2 and P3 flank exon 2 of the Clock gene, P4 and P5 flank exon 1 of the Tpardl gene, and P6 lies just centromeric of Srd5a2l (dark green half-length bands in Fig.2). Groups of lower-scoring promoters flank the beginning of theClock and Tpardl genes (data not shown), and, in general, high scores occurred near regions of high GC content in the genomic DNA.
MatInspector identified several interesting binding sites in the regions upstream of Clock,Tpardl, and Srd5a2l. The Clock 10-kb upstream region contains four E boxes (two 5′-CACGTG-3′ sequences and two 5′-CAGCTG-3′ sequences), three activator protein 1 (AP-1) sites, seven consensus GATA sites, six fork-head transcription factor (HNF3β) sites, three NF-κB sites, and eight core-consensus Sp1 sites (5′-GGGCGG-3′). To date, all E boxes that confer circadian rhythm regulation have the 5′-CACGTG-3′ sequence; whether additional bases outside of the E box are required for circadian rhythm specificity is unknown (Gekakis et al. 1995;Darlington et al. 1998; Hogenesch et al. 1998; Sangoram et al. 1998;Jin et al. 1999). A 7-kb region comprising both predicted promoters ofTpardl includes four E boxes (three subtypes of the 5′-CANNGT-3′ consensus), two fork-head transcription factor sites, a cyclic AMP response element binding (CREB) site, and four Sp1 sites near the P4 promoter. An AP-1 site and nuclear factor Y (NFY) site lie near the P5 promoter. The 3.1-kb region upstream ofSrd5a2l contains 6 E boxes (5′-CACGTG-3′ and 5′-CAGCTG-3′), 4 Sp1 sites, an AP-1 site, and an NFY/CAAT binding site. We interpret all promoter and binding-site predictions with caution, as no prediction program is perfect; however, these results will aid in the design of experiments concerning the regulation of these genes.
DISCUSSION
The results of the completed yeast, C. elegans andDrosophila genome sequencing projects, as well as the growing human and mouse genome sequencing projects, will generate a genomic structure/function framework on which gene expression models can be built across eukaryotic families. The field of circadian rhythms will certainly benefit from the complete sequencing of the mouse andDrosophila genomes, as these organisms constitute two of the four well-understood, genetically amenable circadian model systems. The current circadian model indicates that the regulation of circadian gene expression is a key component of rhythm generation. The genomic sequence of circadian loci will provide much information toward understanding this regulation. To this end, we sequenced two BAC clones that encompass the mouse circadian Clock locus. In addition toClock, this 204-kb region contains the Tpardl(pFT27) gene, the 3′ end of the Nmu gene, and two Unigene clusters.
Each Unigene cluster shows similarity to known proteins. The PDCL2 putative product encodes a protein of at least 239 amino acids. The conceptual translation shows similarity throughout the coding sequence to predicted proteins from Drosophila melanogaster(GenBank accession no. AAF49974.1), Schizosaccharomyces pombe (GenBank accession nos. CAB39851.2 and T40100),Arabidopsis thaliana (GenBank accession no. CAB87763.1), andSaccharomyces cerevisiae (GenBank accession no. S67183). Lower similarity is found within PDCL2 putative amino acids 35–179 to the carboxy-terminal portion of several orthologs of Phosducin-Like Protein (PHLP), including human (GenBank accession no. AAC78849.1) and rat (long and short forms; GenBank accession nos. AAB00333.1 andAAB00334.1, respectively). In the retina, phosducin down-regulates the phototransduction cascade by binding the G-protein βγ subunit and preventing reaggregation with the Gα transducin subunit (Lee et al. 1992). Rat phosducin-like protein potentially regulates G-protein function, and the PhLP1 phosducin family isoform was shown to bind Gβγ (Miles et al. 1993; Craft et al. 1998). The exclusively testicular expression of Pdcl2 suggests a possible G-protein regulator in testicular function. The SRD5A2L putative product encodes a protein of at least 331 amino acids; this conceptual translation shows highest similarity throughout the coding region with unknown proteins in Arabidopsis thaliana (GenBank accession nos.AC007195_5 and AC010926_21), CG7840 gene product in Drosophila melanogaster (GenBank accession nos. AAF52635.1), and hypothetical protein B0024.13 in C. elegans (GenBank accession no. T18648) with BLASTX-nr E values from 8e-20to 2e-14 (Altschul et al. 1990). SRD5A2L putative amino acids 238–349 shows lower similarity with members of the 3-oxo-5 alpha-steroid 4-dehydrogenase type 2 (steroid 5-alpha-reductase 2) family, with a BLASTX-nr E value of 5e-10 to the human ortholog (GenBank accession no. P31213;Labrie et al. 1992). Steroid 5-alpha-reductase 2 is a critical enzyme in androgen physiology and is expressed both in androgen-responsive tissues such as prostate as well as in hair follicles, cerebral cortex, hepatocytes, bile duct cells, and fat cells (Andersson et al. 1991;Eicheler et al. 1994). Srd5a2l is widely expressed, with highest expression in liver and kidney (Fig. 3), and its function remains to be determined.
The function of Tpardl remains unknown, even 11 years after it was cloned from the pluripotent mouse F9 teratocarcinoma cell line (Akagi et al. 1988). Its gene product encodes a 323–amino acid protein with six putative transmembrane domains, and the gene is down-regulated following treatment with retinoic acid, cAMP, or the phorbol ester tetradecanoyl phorbol acetate (TPA) (Miyata et al. 1987; Akagi et al. 1988). A CpG island exists upstream of the Tpardl gene, and the TSSG program predicted two promoters near this island. The MatInspector binding-site prediction program (Quandt et al. 1995) identified binding sites for E box binding proteins, fork-head transcription factors, NFY, AP-1, and CREB. In fact, this gene contains the only consensus CRE of any of the genes reported here; it may play a role in the regulatory effects of cAMP on Tpardl. Tpardl is a candidate for the tumorigenic effects of TPA (Akagi et al. 1988); knowledge of theTpardl promoter sequence may provide information about the relationship between TPA, Tpardl down-regulation, and tumorigenesis.
In contrast to the other genes, the function of Clock is relatively well understood. The Clock mutation was discovered in mice through a mutagenesis screen for altered locomotor activity (Vitaterna et al. 1994). In constant darkness, wild-type mice (strain C57BL/6J) typically complete one circadian cycle with a period ranging from 23.3 to 23.8 h (Vitaterna et al. 1994). In contrast, heterozygousClock mutant mice show an ∼1-h increase in period length over wild-type, and homozygous Clock mutants show an ∼4-h increase in period length, followed by a loss of rhythmicity (Vitaterna et al. 1994). Therefore, the Clock mutation affects both period length and stability (Vitaterna et al. 1994). TheClock gene was cloned using a combination of techniques: (1) rescue of the Clock mutant phenotype by transgenic BAC clones, (2) transcription unit analysis via cDNA subtraction and an SCN cDNA library screen, (3) elucidation of gene structure using genomic survey sequencing data, and (4) mutation detection through comparison of genomic sequence from Clock mutant animals and wild-type animals (Antoch et al. 1997; King et al. 1997a). Clock encodes a bHLH-PAS transcription factor that is a positive activator within the circadian autoregulatory negative feedback loop (King and Takahashi 2000). However, little information exists about the regulation ofClock itself. The transcriptional regulation of Clockmay not seem important initially; only the circadian activity of the CLOCK protein, which appears to be regulated by the PER, CRY, and possibly TIM proteins, is required for proper rhythm generation (Gekakis et al. 1998; Sangoram et al. 1998; Griffin et al. 1999;Kume et al. 1999). Nonetheless, the regulated expression ofClock should still play a critical role in the circadian cycle. For example, the Clock expression profile may be different in the SCN, as compared to the rest of the body. Indeed, although no rhythm in mClock expression has been reported in the mouse SCN, a low-amplitude rClock rhythm was detected in rat retina (Namihira et al. 1999).
The organization of mClock exons 1 through 3 is intriguing: exons 1a, 1b, and 2 fall within a 1.5-kb region and then a 29-kb intron precedes exon 3. Excluding repeat elements, this intron shows no similarity to the nonredundant nucleotide, nonredundant protein, and EST databases. No promoter elements were predicted within the repeat-masked region, and the interval contains a binding element profile similar to a nonpromoter region (i.e., a cDNA). We cannot rule out an enhancer function within the intron; even though the nonrescuing BAC clones contain this intron (Fig. 1), it is possible that position effects could mask a regulatory element near the 5′ end of the intron.
The transgenic rescue experiments with BAC 49 reduce the region that is sufficient for proper Clock expression in the circadian context from 140 kb to 120 kb. BAC 54 is 135 kb in length; 35 kb of potential promoter sequence and 12 kb of potential 3′ enhancer sequence exist in this construct. BAC 49 contains 25 kb of upstream sequence and only 2–4 kb of downstream sequence (depending on the identity of the 3′ EST family flanking the 3′ end of the knownClock gene). The presence of the 3′ end of Tpardlwithin 3 kb of the Clock gene argues against a 3′ enhancer sequence in either BAC 49 or BAC 54. Together, these data indicate that most if not all the circadian regulatory elements lie within theClock locus itself and a 25-kb upstream interval.
The human Clock transcript shares a high level of structural homology with the mouse Clock transcript: From the initiator ATG-containing exon in mouse and in human, each exon/intron boundary is identical between the two species (Steeves et al. 1999). Such conservation of gene structure appears to be a common theme (Hardison et al. 1997; Ansari-Lari et al. 1998; Blechschmidt et al. 1999). Indeed, significant similarity was detected between the mouse sequence and the unfinished human chromosome 4–derived BAC sequence. Three regions of similarity stand out: region A, 28.5 kb–29.6 kb; region B, 32.3 kb–38.7 kb; and region C, 126.7 kb–129 kb (Fig. 2). Region A contains sequence present in BAC 49 but absent from the nonrescuing BAC 54 100-kb clone (Fig. 1). This area could represent a Clockpromoter region that is required for proper expression of theClock gene. In addition, MatInspectordetected putative AP-1 and E box binding sites within the 27 kb–34 kb region of the mouse sequence. The AP-1 sites may represent targets for the input pathways that mediate entrainment of circadian rhythms (Kornhauser et al. 1992; Takeuchi et al. 1993). The presence of E box elements in the Clock promoter may suggest, but does not dictate, a circadian regulation of Clock expression. In region B, the 32.3–34.0-kb portion is excluded from the BAC 54 100-kb construct, but the remainder of this 6.4-kb region is included in the nonrescuing clone. Therefore, region B may be critically important for appropriate expression of Clock as a complete, uninterrupted unit. Recent advances in BAC engineering should make it possible to identify any regulatory elements via deletion scanning mutagenesis through regions A and B (Jessen et al. 1998). Region C represents the putative 3′ UTR of the ∼10-kb mouse Clock transcript described above and provides additional evidence in support of our hypothesis concerning the long Clock transcript. Finally, the PIP shows differences between the mouse and human genomic regions, particularly in the large Clock intron from each species. Human CLOCK exon 1, which shows no similarity to themClock 5′ UTR exons, aligns at 54.0 kb of the mouse sequence (61% identity; red band in Fig. 2), hCLOCK exon 2 does not align with the mouse sequence, and hCLOCK exon 3 aligns with mClock exon 4 at 72.1 kb (93% identity). The mouse-human alignment suggests an 18.1-kb distance betweenhCLOCK exons 1 and 3; however, the hCLOCK alignment to human genomic sequence clearly shows a 27-kb distance between human exons 1 and 3 (data not shown). These analyses suggest that rearrangements have occurred in human genomic sequence relative to mouse genomic sequence.
We anticipate that comparative sequence analyses will be of great use in the study of human circadian diseases such as delayed sleep phase syndrome and advanced phase sleep syndrome (Weitzman et al. 1981;Jones et al. 1999). Furthermore, the genes that make up the core circadian autoregulatory feedback loop appear to be well-conserved between Drosophila and mammals (Wilsbacher and Takahashi 1998;Dunlap 1999; King and Takahashi 2000). Combining the forward genetic approach of gene discovery with the resources of genomic sequence will lead to rapid, parallel identification of novel circadian genes among many organisms. These genetic tools will undoubtedly accelerate our understanding of the circadian clock, including input pathways to the clock, output pathways from the clock, and the core circadian oscillator itself.
METHODS
Shotgun Sequencing and Finishing of BAC 52 and BAC 54
BAC DNA Isolation and Purification
BAC DNA was isolated from 250 mL cultures via an alkaline lysis method using 6 mL of Solution I (50 mM glucose; 10 mM EDTA at pH 8.0; 25 mM Tris at pH 8.0), 8 mL Solution II (1% SDS, 0.2 N NaOH), and 6 mL Solution III (60 mL of 5M potassium acetate, 11.5 mL of glacial acetic acid, 28.5 mL water). After addition of Solution II, the cultures were mixed gently and incubated on ice for 7.5 min; after addition of Solution III, the lysates were gently mixed and cleared of cellular debris by two 25-min centrifugations at 14,000×g, 4°C. The supernatant was precipitated with 0.7 volumes of isopropanol and centrifuged at 2000×g for 10 min at room temperature. The air-dried DNA pellet was resuspended in 2.8 mL TE at pH 8.0 that contained 10 μg/μL RNase A, and the solution was incubated for 30 min at 37°C. BAC DNA was purified via double cesium chloride centrifugation; 3.25 g of CsCl (Gallard-Schlesinger Industries) and 280 μL of 10 mg/mL ethidium bromide (EtBr) were added to the resuspended BAC DNA. This solution was centrifuged at 3000×g for 10 min at room temperature; the density of the clear bottom layer was adjusted to 1.55 g/mL if necessary. DNA was loaded into a 3.9-mL Quick-Seal polyallomer tube (Beckman) and centrifuged in an Optima TLX ultracentrifuge (Beckman) at 80,000 rpm for 16 h at room temperature in a TLN100 rotor. The lower (plasmid) band was pulled using an 18-gauge needle, transferred to a new Quick-Seal tube, filled with 1.55 mg/mL CsCl in TE, and centrifuged at 100,000 rpm for 4 h at room temperature. This second CsCl centrifugation step was required to purify the BAC DNA from contaminating Escherchia coli DNA. The lower band from the second spin was pulled, diluted with 3 volumes of water, and EtBr was extracted three times with butanol. DNA was precipitated with 2 volumes of ethanol and stored overnight at −20°C to maximize BAC DNA recovery. Following centrifugation and a 70% ethanol wash, the BAC DNA pellet was air-dried and resuspended in 10–20 μL TE. Concentration was determined using fluorometry. This prep typically yields about 10–20 μg of supercoiled BAC DNA.
M13 Shotgun Library Construction
For most steps, M13 vector preparation and construction of M13 libraries were completed according to the Washington University Genome Sequencing Center protocols (available athttp://genome.wustl.edu/gsc/Protocols/protocols.shtml).
M13 Vector Preparation
M13mp19 RFI (Pharmacia) was digested with SmaI (NEB) and then precipitated using 1/50 volume of 5 M NaCl and 2.5 volumes of 95% ethanol. The resultant DNA pellet was treated with 5 units/μg DNA of calf intestinal alkaline phosphatase for 1 h at 37°C. M13 DNA was phenol extracted once and then purified using a two-gel procedure: DNA was run on a 0.8% agarose gel in 1× TAE for 4 h at 60 volts and then the linear M13 band was excised and run again in a new 0.8% low-melting-point agarose gel for 3 h at 60 volts. The linear M13 band was gel-purified and resuspended in water.
Size Selection of BAC DNA
BAC DNA was sonicated using a Heat Systems Ultrasonics sonicator (model W-225R) with a chilled-cup horn probe. 2.5 μg BAC DNA was diluted in 1× mung bean nuclease buffer (Pharmacia) to a final volume of 60 μL and sonicated for 7–8 sec at power level 6. DNA fragments were blunt-ended using 1 unit mung bean nuclease (Pharmacia) for 10 min at 30°C. Proteins were phenol-chloroform-isoamyl extracted and DNA was precipitated. The resultant DNA pellet was resuspended in 10 μL of water. Size selection was performed by running the DNA on a 0.8% SeaPlaque GTG agarose (FMC) gel in 1× TAE (no ethidium bromide) for 3.5 h at 60 volts. Size marker lanes run on the same gel were cut out and stained; these markers were used to estimate and excise the 1.3 kb–1.7 kb region in the unstained sheared BAC DNA lane. The size-selected DNA was gel-purified using β-agarase (NEB) according to the quick-equilibration protocol, and 2 units β-agarase per 100 mg of agarose were used at 42°C in an overnight digestion. DNA was precipitated and resuspended in 7 μL of water. DNA concentration was measured using a Hoefer TKO 100 fluorometer (Pharmacia).
Ligation and Transformation
Twenty nanograms of prepared vector and 100–200 ng sheared, size-selected BAC DNA (in 1–3 μL) were added to 1 μL of 10× blunt-end ligation buffer (0.5M Tris-Cl, 0.1M MgCl2, 0.1M DTT, 0.5mM ATP, 0.5 mg/ml BSA at pH 7.8) and 0.3 μL of T4 high concentration ligase (New England Biolabs) in a final volume of 8 μL. The ligation reaction was incubated overnight at 15°C. For transformation of the M13 library, XL1-Blue MRF' cells were grown in YT broth +12.5 μg/mL tetracycline to O.D. 600 = 0.8. Top YT agar was freshly prepared, aliquoted into 6-mL samples, maintained at 45°C in a water bath, and supplemented with 50 μL of 25 mg/mL IPTG and 50 μL of 25 mg/mL of X-gal. The ligation reaction was diluted 25-fold in water, and 2 μL were electroporated into 25 μL of electrocompetent XL1-Blue MRF'. Electroporator (BTX 600) settings were voltage = 1.7 kV and resistance = 129 ohm. The electroporated cells were mixed with 400 μL of prewarmed SOC and 100 μL of XL1-Blue MRF' lawn cells. This solution was mixed with the top YT and plated onto prewarmed YT +12.5 μg/mL tetracycline plates. Plates were incubated for 12–16 h at 37°C. Individual M13 plaques were picked with a sterile toothpick, eluted into 400 μL of SM buffer and stored at 4°C.
Ninety-six-Well M13 Template Purification
Preparation of single-stranded M13 DNA was performed using the Solid Phase Reversible Immobilization (SPRI) technique for M13 (Hawkins et al. 1994). All steps were carried out in 96-tube format using Fisher-racked 1.1 mL tubes (P/N 1184294); 650 μL of XL1 Blue MRF' starter culture was inoculated with 40 μL of the M13-containing SM stock and grown for 16 h with shaking at 37°C. Cultures were centrifuged at 2000×g for 15 min; 450 μL of lysate was added to 45 μL of 7.5% SDS and incubated for 10 min in an 85°C water bath to lyse the phage coats. Magnetic beads (Bangs Estaphor number MN0009200CN) were washed three times in 0.5 M EDTA at pH 8.0 and resuspended at 20 μg/μL in the same EDTA buffer. DNA was hybridized to 50 μL (1 mg) of washed beads in 545 μL of hybridization solution (26% PEG 8000/20mM MgCl2) for 30 min at room temperature. Because the tubes were full to capacity, they were centrifuged for 3 min at 1000×g to collect the beads initially. The supernatant was discarded by inverting the tube box and magnetic stand (Promega) together so as to keep the beads and DNA in the tubes. The beads were washed twice in 200 μL of 70% ethanol and collected using the magnetic stand. The beads were air-dried for 10 to 20 min and then DNA was eluted in 30 μL of water for 2 min. Using this protocol, 192 M13 templates could be prepared in about 5 h with an average yield of 1 μg of sequence-ready DNA. A similar protocol is currently available athttp://www-seq.wi.mit.edu/protocols/M13SPRI.html.
Sequencing of M13 Templates
M13 sequencing reactions were run in an ABI PRISM Catalyst Turbo 877 Molecular Biology LabStation using the Dye Primer Cycle Sequencing Ready Reaction-21 M13 Kit (Perkin Elmer) at half-chemistry concentrations: 2.0 μL of A and C premixes and 4.0 μL of G and T premixes were used for each reaction; 1.5 μL and 3.0 μL of template were delivered to the A/C and G/T reactions, respectively. Thermocycling conditions were modified as recommended for use of FS Dye Primer chemistry with single-stranded template, and 15 μg glycogen (Boehringer Mannheim) per reaction was used as carrier after precipitation. Sequences were run on a 4% Long Ranger acrylamide gel (FMC) using an ABI PRISM 377 DNA Sequencer. Electrophoresis run time was 7.5 h, and read length was truncated at 650 bases due to peak expansion and subsequent base-calling errors past ∼650 bases. If necessary, ends were trimmed using Sequencher 3.0 (Gene Codes, Inc.) with the following criteria: (1) at the 5′ end, sequence was trimmed until the first 25 bases contained no more than three consecutive ambiguities; (2) 100 bases after the 5′ trim, sequence was trimmed after the first 25 bases that contained more than three consecutive ambiguities; and (3) sequence was trimmed from the 3′ end until the last 25 bases contained no more than three consecutive ambiguities.
Reverse Sequencing
Double-stranded M13 DNA was generated using PCR with M13 Universal and M13 reverse primers on 0.5 μL of the M13 SM stock as template; cycling conditions were 94°C for five minutes, 26 cycles of 94°C for one minute, 55°C for one minutes, and 72°C for one minute; 72°C for 10 minutes; and 4°C hold. PCR products were purified from mineral oil by pipeting the reaction onto a strip of Parafilm (3M), rolling the droplet along 5–10 cm of Parafilm, and collecting the aqueous droplet. Primers and unincorporated nucleotides were removed from the PCR products using the PCR SPRI technique (T. Hawkins, unpubl.). For a 50 μL PCR, 10 μL of washed magnetic beads (see “High throughput M13 prep” above) and 50 μL of PCR hybridization buffer (2.5M NaCl/20% PEG8000) were added to the PCR product, mixed, and incubated for 10 min at room temperature. Beads were magnetically collected, the supernatant was discarded, and the beads were washed two times with 150 μL of 70% ethanol. Beads were air-dried for 10 min and then resuspended in 20 μL of 10 mM Tris-Acetate at pH 7.8 for 5 min. Reverse-sequencing reactions were performed on 50 ng purified PCR product using the Catalyst 877 Turbo robot with Dye Primer M13 Reverse Ready Reaction Mix (Perkin Elmer) as for the M13 forward shotgun samples, and products were run on an ABI 377. The PCR purification protocol is currently available athttp://www-seq.wi.mit.edu/protocols/PCRSPRI.html.
Direct BAC Sequencing
BAC DNA was used as sequencing template for finishing purposes. Template was prepared either by CsCl centrifugation or using the Qiagen Midi Plasmid Prep Kit (Qiagen). One to 1.5 μg BAC DNA, 20 pmole primer, and 8 μL Big Dye Terminator Ready Reaction Mix were used in 20-μL reactions. Reactions were run in a Perkin Elmer 9700 thermal cycler using conditions of 95° for 5 minutes; 34 cycles of 95°C for 30 seconds, 56°C for 1 minute, 60°C for 4 minutes; and 4°C hold.
Gap PCR on BAC Template
The Stratagene TaqPlus Long PCR system was used to generate PCR products across four large (3.5 kb, 5.8 kb, 6.5 kb, 7.8 kb) gapsin genomic sequence. Primers were designed from unique genomic sequence flanking the gaps. The TaqPlus low-salt buffer was used in the reaction that produced the 6.8-kb product, while the high-salt buffer was used in all other reactions. The reactions for the three largest products used thermal cycling conditions of 94°C for 5 minutes; 16 cycles of 94°C for 15 seconds, 60°C for 1 minute, and 72°C for 10 minutes; 12 cycles of 94°C for 15 seconds, 60°C for 1 minute, and 72°C for 10 minutes with an increasing increment of 15 seconds per cycle; 72°C for 10 minutes, and 4°C hold. The reaction conditions for the 3.5-kb product were identical except for a 6-min extension time. For the 6.5-kb and 6.8-kb products, ten independent reactions were performed, pooled, and sequenced; for the 3.5 kb and 5.8 kb products, three independent reactions were performed and sequenced individually (to resolve Taq polymerase incorporation errors).
A relatively short (250-bp) gap was amplified using AmpliTaq polymerase (Perkin Elmer) with cycling conditions of 94°C for 2 minutes; 26 cycles of 94°C for 30 seconds, 55°C for 1 minute, and 72°C for 1 minute; 72°C for 20 minutes, and 4°C hold. Four independent reactions were performed and sequenced separately. All gap PCR products were sequenced by primer walking across both strands for the entire length of the product. Reactions were run using the Big Dye Terminator Ready Reaction Mix (Perkin Elmer).
Analysis of Genomic Sequences
All alignments were performed using Sequencher 3.0 (Gene Codes Corp.). Individual M13 sequences were BLASTsearched using BLASTN against the nr nucleotide and dbEST databases, BLASTX against the nr protein database, and TBLASTX against the nr protein database (Altschul et al. 1990). For the complete, finished BAC 54 and BAC 52 sequence, repeat elements were masked usingRepeatMasker against the rodent Repbase database (A.F.A. Smit and P. Green, unpubl.; available athttp://www.genome.washington.edu/UWGC/analysistools/repeatmask.htm). Masked output was BLAST searched inPowerBLAST (Zhang and Madden 1997) against the nr nucleotide, EST, and STS databases forBLASTN (parameters of M = 1, N = −3, S = 40, S2 = 40) and the nr protein database forBLASTX (parameters of S = 90, S2 = 90, -filter = seg). Masked sequence was also searched againstGRAIL (Xu et al. 1994) andGenScan (Burge and Karlin 1997) for exon prediction. Promoter analysis was performed usingTSSG (V.V. Solovyev et al., unpubl.; available at http://dot.imgen.bcm.tmc.edu:9331/seq-search/gene-search.html) and MatInspector (Quandt et al. 1995). The PIP was created using Advanced PipMaker(http://nog.cse.psu.edu/pipmaker; Schwartz et al. 2000) using the “Search both strands” and “Show all matches” options with the following files: sequence 1, GenBank accession no. AF146793 (mouse genomic); sequence 2, GenBank accession no. AC069200 and AC024243(human genomic); mask file, RepeatMaskeragainst the rodent Repbase database (as above); exon file, manually generated; first sequence underlay file, manually generated.
Generation of BAC Transgenic Mice
Circular BAC DNA was purified using the CsCl method described above, resuspended in injection buffer (10 mM Tris-HCl at pH 7.5, 0.1 mM EDTA, 100 mM NaCl, 30 μM spermine, 70 μM spermidine) to a concentration of 1 ng/μL, and injected into the oocytes of wild-type CD1 females. Injections were done at the Northwestern University/CMIER Transgenic facility as previously described (Hogan et al. 1994). Transgenic mice were identified using both PCR and Southern blot analysis of genomic DNA prepared from tail biopsies as described in Antoch et al. (1997). A 240-bp fragment corresponding to the Sp6-end of BAC vector-insert junction was amplified using primers 5′-GACCATGATTACGCCAAGCTATTTAG and 5′-ACTGTAGCAGTCATCACCTTAACCC. Out of 14 mice tested, two were positive for the transgene by PCR analysis and only one (no. 5633) by both PCR and Southern hybridization. This transgenic founder (female) was transferred to the Northwestern University Center for Experimental Animal Resources and crossed to a Clock/Clock (BALB/cJ X C57BL/6J) F3 male. The progeny (all heterozygous for the Clock mutation) were analyzed for transgene presence using PCR. Both transgenic animals and their nontransgenic littermates were analyzed for their locomotor activity.
Locomotor Activity Rhythm Monitoring and Analysis
Mice were housed in LD 12:12 (lights on at 0500, Central Standard Time) and behaviorally tested after reaching 8 wk of age. Locomotor activity was performed as described in Vitaterna et al. (1994). Mice were individually housed in the running-wheel cages for 1 wk in LD 12:12 and then transferred to constant darkness for 3 wk. To confirm Clock/+ phenotype, a 6-h saturating light pulse at circadian time 17 was given at day 21 in constant darkness. Activity was monitored with an on-line PC computer system (Stanford Software Systems, Chronobiology Kit).
Period calculations were performed for a 20-d interval in constant darkness (1–20 d) by χ2 periodogram analysis as previously described (Antoch et al. 1997) using ClockLab Software (Circadian Systems). Statistical analyses were performed using one-way Generalized Linear Models Analysis of Variance using Number Cruncher Statistical Systems (NCSS 97).
WWW RESOURCES
dot.imgen.bcm.tmc.edu:9331/seq-search.html.
genome.wustl.edu/gsc/index.shtml.
genome.wustl.edu/Protocols/protocols.shtml.
www.genome.washington.edu/UWGC/analysistools/repeatmask.htm.
Acknowledgments
This work was supported by the NSF Center for Biological Timing, an Unrestricted Grant in Neuroscience, from Bristol-Myers Squibb, NIMH grant R37 MH39592 and Northwestern University (J.S.T.). We thank Stephanie Chissoe, Cecelia Boysen, Eric Lander, Trevor Hawkins, and Richard Gibbs for advice on genomic sequencing; Lynn T. Doglio and NU/CMIER (Childrens Memorial Institute for Education and Research) Transgenic Facility for producing BAC transgenic mice; Adrian Smit for advice on BLAST searching with rodent sequences; and Jani Kushla and Andrew Whiteley for technical assistance. We also thank the Genome Sequencing Center, Washington University, St. Louis, MO, for communication of DNA sequence (GenBank accession nos. AC069200 and AC024243) data prior to publication. J.S.T. is an Investigator at the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL j-takahashi{at}northwestern.edu; FAX (847) 491-4600.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.155400.
-
- Received July 13, 2000.
- Accepted October 24, 2000.
- Cold Spring Harbor Laboratory Press








