
Abstract
With sequence analysis of the human genome well underway, there is an increasingly urgent challenge to understand the fundamental function and interplay of genes that build and maintain an organism. Several approaches will be critical for interpreting gene function, including random cDNA sequencing, expression profiling in different tissues, genetic analysis of human or model organism phenotypes, and creation of transgenic or “knockout” animals. Traditional gene-trapping approaches, in which genes are randomly disrupted with DNA elements inserted throughout the genome, have been used to generate large numbers of mutant organisms for genetic analysis. Recent modifications of gene-trapping methods and their increased use in mammalian systems are likely to result in a wealth of new information on gene function. Various trapping strategies allow genes to be segregated based on criteria like the specific subcellular location of an encoded protein, the tissue expression profile, or responsiveness to specific stimuli. Genome-wide gene-trapping strategies, which integrate gene discovery and expression profiling, can be applied in a massively parallel format to produce living assays for drug discovery.
Classical Genetics, Genomics, and Analysis of Gene Function
The term “genomics” was proposed in 1986 by Thomas Roderick (McKusick 1997a) to describe the study of a genome by molecular means distinct from traditional genetic approaches. Genomics evolved from a much older word, genome (McKusick 1997a). A fusion ofgene and chromosome, a genome is the complete collection of genes possessed by an organism. Living creatures result from the delicate interplay between a “functional” genome and environmental factors. With complete sequence of many genomes, including biological workhorses like Escherichia coli(Blattner et al. 1997), Saccharomyces cerevisiae (Mewes et al. 1997), and Caenorhabditis elegans (The C. elegansSequencing Consortium 1998), and with the Human Genome Project “ahead of schedule and under budget” (Collins 1995; Collins et al. 1998), the scientific focus is shifting towards development of methods that effectively use this wealth of structural genomic information (Collins et al. 1998).
The ultimate goal of the Human Genome Project is to understand how the genome builds, maintains, and operates an organism. Multiple strategies at the DNA, RNA, protein, cellular, and organism level will be key in achieving this goal. Hybridization-based techniques, like oligonucleotide chips (Fodor et al. 1991) or cDNA arrays (Schena et al. 1995), can reveal the differential expression profiles of numerous genes in response to a stimulus (DeRisi et al. 1997; Wodicka et al. 1997). Gene expression profiles can also be elucidated through random sequencing of cDNA libraries derived from specific tissues or cell types (Okubo et al. 1992; Fannon 1996; Okubo and Matsubara 1997). Analysis of these expressed sequence tags (ESTs) can provide an “in-silico” gene expression landscape. Shifting towards the gene products, large two-dimensional polyacrylamide gels can be used to monitor the expression of at least a subset of proteins (Geisow 1998;Persidis 1998). In addition, various methods are available for elucidating physical interactions between proteins in live cells (Fields and Song 1989; Miyawaki et al. 1997).
Classical and molecular genetics provide a variety of powerful tools useful for understanding gene function and studying complex developmental events. Many structure–function relationships have been elucidated using elegant genetic approaches and by cloning disease associated genes. However, these strategies are too time consuming for efficient genome-wide investigation. This review will focus on the use of random gene-trapping techniques to gain insights into gene function. These methods are producing valuable raw material for current and future bioinformatics databases that will catalog many biological processes. Ultimately, this information should help researchers predict gene function in much the same way sequence databases are currently used in structural genomics efforts.
Gene Capturing and Mutagenesis
Conceptually, one straightforward way to discern the function of a gene is to observe the effects of an impaired or mutated gene on an organism over a desired number of generations. Many gene mutations have consequently been implicated in a variety of human medical conditions. In addition, chromosomal markers identified through arduous structural genomics approaches have led to remarkable successes in positional cloning of disease related genes. The important functional insights gained through time-consuming map-based discovery of genes like the cystic fibrosis transmembrane conductance regulator (Riordan et al. 1989; Rommens et al. 1989) or BRCA-l (Miki et al. 1994) have resulted in the mapping of many phenotypic variations to a specific gene. Much of the genetic basis of disease work has culminated in the Online Mendelian Inheritance in Man (OMIM) database provided on the National Center for Biotechnology Information (NCBI) Web site (http://www.ncbi.nlm.nih.gov/Omim/), which currently lists ∼500 human genes that have disease-producing mutations (McKusick 1997b).
Nonhuman organisms have been exploited to gather additional information about gene function. Each organism offers its own unique advantages and drawbacks. Yeast, as the simplest eukaryote, is a logical place to start when searching for basic understanding of cell biology. Studies with C. elegans, Drosophila, zebrafish, and other model systems have revealed the functions of many genes during embryonic development or complex intercellular signaling. These organisms, however, are very distantly related to humans; so mammalian systems are required to expand the knowledge base and ultimately for pharmacological evaluations. The combined use of embryonic stem (ES) cells with homologous recombination in mice has created a very useful system for functional genomic study, allowing researchers to modify or eliminate any known gene and analyze its function (Capecchi 1989;Joyner 1993).
In contrast to the rational “one gene at a time” approaches, enhancer-trapping methods evolved to probe the entire genome simultaneously. Originally described in bacteria, enhancer trapping was first demonstrated using bacteriophage transposable elements to insert a reporter gene at scattered sites throughout the E. coligenome (Casadaban and Cohen 1979; Bellofatto et al. 1984). Chromosomal integration of a transposable element tagged the integration site and often mutated the gene into which it inserted. Mutants resulting from this approach could be selected using genetic screens. Because the insertion site was tagged, the mutated genes could be readily identified, which made this approach more efficient than traditional chemical mutagenesis (Meneely and Herman 1979; Rinchik 1991). The success and conceptual simplicity of the enhancer trap method was quickly adapted for use in other model systems including plants (Schell 1987), C. elegans (Hope 1991), and Drosophila(O'Kane and Gehring 1987). When the enhancer-trapping element consists of a cDNA encoding an easily monitored reporter gene like β-galactosidase, the expression pattern of the trapped gene can be visualized. In Drosophila, the P-transposon system has been used with great success to create transformed animals carrying enhancer detectors (Rubin and Spradling 1982; O'Kane and Gehring 1987). Large numbers of enhancer trap lines have been established and evaluated using both phenotypic and expression analysis (Bier et al. 1989;Spradling et al. 1995). Specific mutant lines can then be chosen for further study when there is a correlation between expression and phenotype.
Although enhancer traps are widely used, certain gene traps were developed as an alternative to enhancer traps to capture open reading frame information. The identification of target genes using enhancer trapping was sometimes problematic because the site of reporter insertion could be as much as 100 kb from the target gene. This would require extensive characterization of the genomic insertion site to identify candidate target genes. Gene trapping varies from enhancer trapping in that, instead of using a minimal promoter, gene trap vectors provide specific sequences that generate fusion RNA transcripts when inserted into a gene (See Fig. 1). This feature makes gene trapping (vs. enhancer trapping) especially advantageous in mammalian cells that have complex genomic organization, including large introns and small exons, because the trapped gene can be identified by mRNA sequence.
Schematic overview of enhancer and gene trapping. Normally a wild-type gene produces an unspliced RNA transcript that is processed by linking splice acceptor (SA) sites to splice donor (SD) sites. Transcription initiates at the promoter (Pro) and is regulated by enhancer sequences that can be located thousands of base pairs away from the promoter. (AAAAAAA) Poly-(A)-tail. (A) An enhancer-trapping element contains a minimal promoter and a translation start site. When the element integrates near an endogenous enhancer, that enhancer controls expression of the reporter gene. (B) In the basic gene-trapping element, a reporter gene sequence is flanked by splice acceptor (SA) and polyadenylation (pA) sequences. When the trapping element integrates within an intron, a fusion transcript is generated as a result of splicing of the endogenous splice donor with the vector-provided acceptor. As with enhancer trapping, the result is expression of the reporter gene that is faithfully controlled by the regulatory elements of the trapped gene locus. (C) The downstream sequences of the trapped gene can also be captured in a fusion transcript by providing a splice donor in the trapping element.

It is believed that the use of random “knockout” mutagenesis by gene trapping in mice will result in further enhancements for functional analysis of the genome. Typical vectors used for knockout gene trapping contain an acceptor site for RNA splicing followed by a reporter gene and then a transcript-terminating polyadenylation sequence (Brenner et al. 1989; Gossler et al. 1989; Friedrich and Soriano 1991; Skarnes et al. 1992). These gene-trapping vectors can be introduced into the genome of murine ES cells by electroporation or replication-deficient self-inactivating retroviruses. Insertion of this vector into an intron results in premature termination of the captured allele in which the splice donor at the 3′ end of an endogenous exon is “trapped” into splicing with the splice acceptor from the gene trap vector. The result is a fusion mRNA in which the reporter gene from the vector becomes transcriptionally regulated by the trapped promoter. Expression of the reporter gene can be used to monitor the spatial and developmental transcription profile of the trapped locus (See Fig. 2).
Overview of gene trapping applications. Depending on the model system, gene-trapping elements can be introduced throughout a genome using traditional plasmid vectors, transposons, or modified retroviruses. When the trapping element contains a reporter gene, the expression pattern of the tagged gene can be observed throughout the organism at various stages of development. If the tagging element disrupts the tagged gene, animals bred to homozygosity can be useful gene knockout models. The expression of tagged genes in individual cells can be monitored with the CCF2 fluorescent substrate of β-lactamase. Fluorescent activated cell sorting can be used to isolate cells in which the β-lactamase reporter gene has trapped a gene that responds to a stimulus of interest. Whether gene trapping is applied to whole organisms or individual cells, the trapping element facilitates rapid identification of the trapped gene.
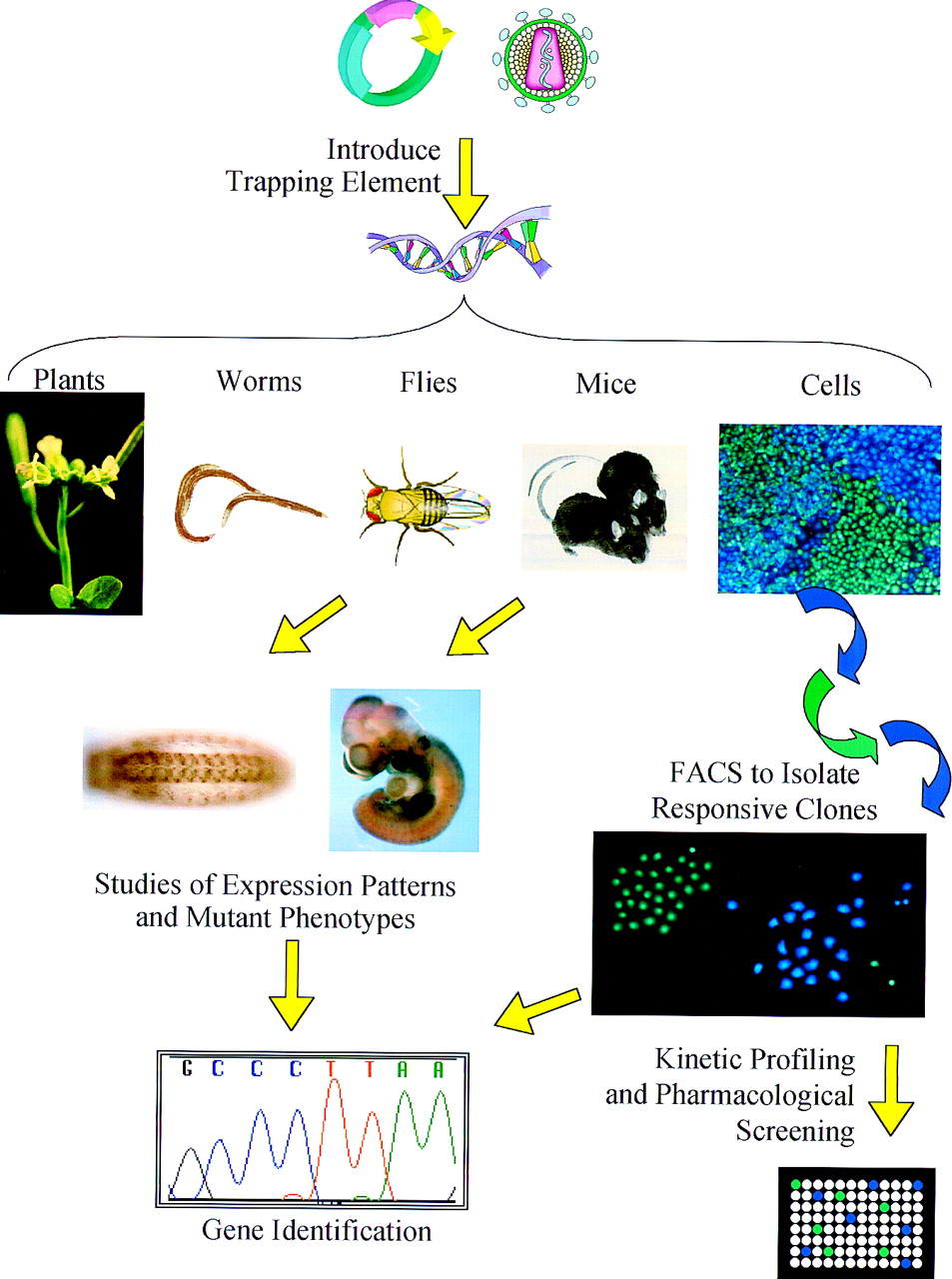
Sequence Acquisition: Identification of Trapped Genes
Recent reports have demonstrated the feasibility of using knockout gene-trapping approaches to create libraries of murine ES cells in which each individual clone has one gene disrupted and tagged. In gene-trapping approaches, a fusion RNA is generated so the trapped endogenous gene from each isolated clone of interest can be identified using rapid amplification of cDNA ends (RACE), a method pioneered by Michael Frohman (Frohman et al. 1988). With this approach, PCR is used to amplify the unknown 5′ or 3′ end of the fusion mRNA. The design of the vector dictates whether 5′ and/or 3′ fusions with endogenous gene transcripts are produced.
Several reports have demonstrated that a gene trap method can effectively knock out genes and that a 5′ RACE protocol can be used to obtain sequence information about the disrupted genes (Skarnes et al. 1992, 1995; Forrester et al. 1996; Chowdhury et al. 1997). In the largest study reported to date using 5′ RACE, 115 sequences were successfully recovered from 153 cell lines (Townley et al. 1997). Sequence information from some of these murine ES cell clones is available on the Web (http://socrates.berkeley.edu/∼skarnes/), and this set currently includes >250 characterized lines. In addition, details on a large number of other academic mouse ES cell tagging efforts have also recently been reported (Chowdhury et al. 1997; Hicks et al. 1997; Couldrey et al. 1998; Voss et al. 1998).
Researchers at Lexicon Genetics (Woodlands, TX) have reported sequence information of 2000 genes in murine ES cells (Zambrowicz et al. 1998). The vector design used in this study provides an internal constitutive promoter that drives expression of a selectable marker sequence with a 3′ splice donor in place of a poly(A) signal. This vector should only confer resistance to ES cells where the vector traps an endogenous splice acceptor and polyadenylation signal. The higher rate of “novel” sequence recovery reported in this study, compared with that obtained previously (Skarnes et al. 1992; Townley et al. 1997), may suggest that these vectors tag genes that are under-represented in the EST databases or that tagging occurs in genes that are not normally transcribed (pseudogenes, line elements, interspersed repeats). Because these poly(A) trap vectors do not require integration into an actively transcribed gene for selection, they can be useful for identifying genes that are inactive in undifferentiated ES cells and become actively transcribed during differentiation (Salminen et al. 1998).
Although it may be possible to generate a complete set of murine gene trap ES cell lines, that would serve only as a starting point for functional analysis of the genome. The benefits of this approach include the potential to discover novel phenotypes and create useful in vivo model systems for the study of disease. The strategy is also well suited for studying embryonic development (Friedrich and Soriano 1991;Skarnes et al. 1992). Roadblocks for broad application in understanding all mouse genes include the effort and expense of generating mouse lines because generation of homozygous mutant animals requires breeding of heterozygous lines that carry the gene trap mutation. In addition, once the animals are generated, one must search for phenotypes that may be masked by partial redundancy of closely related genes. One additional limitation to using the gene trap approach for generating knockout mice results from the fact that a gene trap vector may “tag” a gene, reliably tracking its expression with a reporter gene, without completely disrupting it. In some cases the full-length trapped gene may still be expressed as a result of differential or inefficient splicing, or an active fragment could be expressed when the trapping element inserts in the 3′ region of a gene.
Genome-Wide Functional Analysis in Mammalian Cells
Molecular expression profiling by hybridization and mutational analysis via traditional or gene trap methods are providing valuable information that will help index functions of many genes. The information gained may ultimately lead to better medicines and treatments. However, if one hopes to rapidly move from the vast amount of genomic information to therapeutic intervention, additional technologies are required.
Modern drug discovery efforts require bioassays to sort through the large number of potentially bioactive compounds generated by conventional and combinatorial chemistry. Large-scale mutagenesis in mice can provide functional information about genes and generate model systems for the study of some diseases, but mice are not well suited for high-throughput screening. The hybridization DNA array technologies can observe changes in gene expression on a genome-wide basis, but they are limited to only those genes that have already been cloned. To assess the gene expression effects of numerous test compounds using DNA arrays would require RNA isolation from pools of treated cells, probe generation from this material, and then hybridization to a chip for each assay. Such an approach is not currently cost effective for large-scale screening. The need for an integrated technology platform to discover genes, pathways, and corresponding lead drug candidates drove development of novel approaches, termed “gene-to-screen” genomics. Gene-“tagging” strategies have been described that may provide a link between trapping methods and drug discovery (Whitney et al. 1998).
The first step in this gene-to-screen approach is creation of a reporter gene-tagged library in a particular cell line. A library consists of millions of cells, each carrying the same tagging element integrated into a different site within the genome. The gene-tagging element typically contains a splice acceptor and reporter gene. As with knockout approaches in ES cells, the gene-tagging element can be introduced into the genome using physical methods such as transfection and electroporation (Skarnes et al. 1992) or with viral vectors (Friedrich and Soriano 1991; Gogos et al. 1997; Hicks et al. 1997;Zambrowicz et al. 1998). The result is reporter gene expression in those cells in which the tagging element has inserted into an actively transcribed gene.
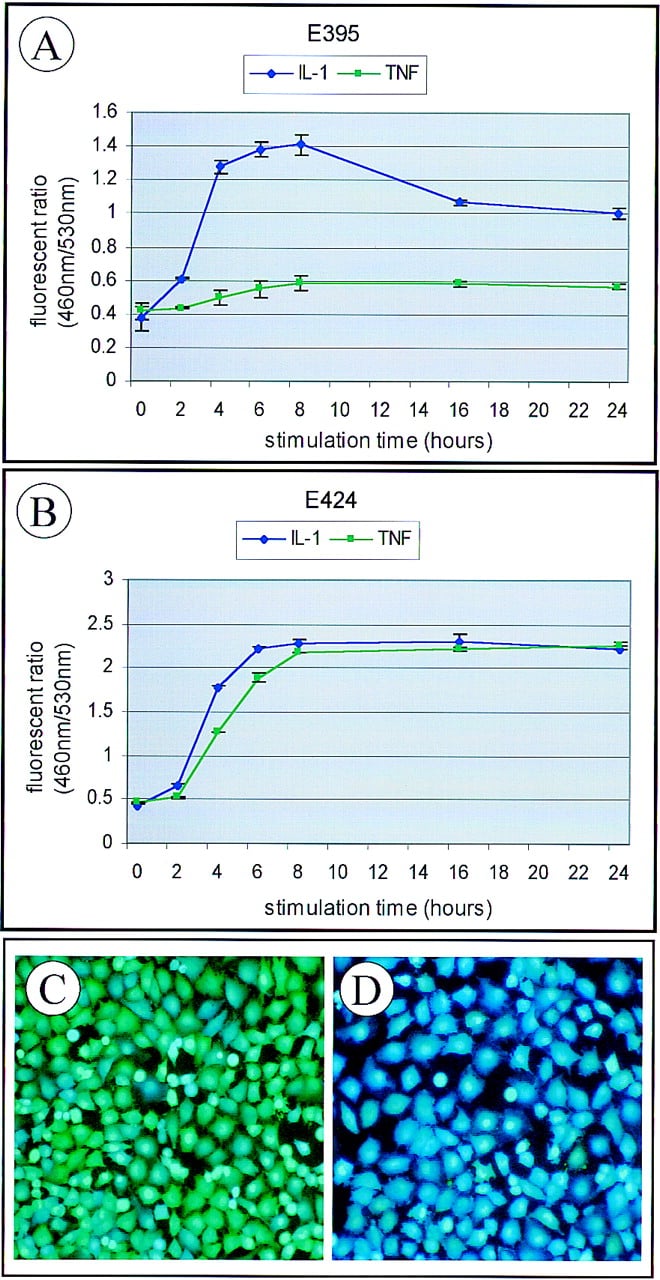
A new and highly sensitive reporter system has recently been described and is well suited for gene tagging. A nontoxic fluorescent substrate of β-lactamase, CCF2-AM, enables real-time and sensitive monitoring of transcription in live cells (Zlokarnik et al. 1998). A β-lactamase tagged library can be used to clone genes with a variety of interesting expression characteristics (Whitney et al. 1998). Any β-lactamase tagged cell library can be subjected to fluorescent activated cell sorting to rapidly separate clones where the tagged gene is constitutively expressed. In addition, sequential rounds of sorting can be used to identify cell clones whose genes are induced or repressed by different agents including receptor ligands, drug candidates, and viruses (Rao 1998) (See Fig. 3).
β-Lactamase tagged clones faithfully report real-time transcriptional responses in living cells. A β-lactamase tagged library was created in human endothelial cells. Fluorescent activated cell sorting (FACS) was used to isolate cell clones that responded to stimulation by a proinflammatory cytokine with an increase in β-lactamase expression. Kinetic profiling plots are shown for two of the cell lines isolated in which response is the ratio of blue fluorescent emission (460 nm) compared with green fluorescent emission (530 nm). Cells were stimulated with either interleukin-1β (IL-1β) or tumor necrosis factor-α (TNFα) at a concentration of 10 ng/ml. Results plotted are the average fluorescent ratio plus or minus s.d. for four parallel experiments. Digital fluorescent images are shown of clone E424 either unstimulated (C) or stimulated with IL-1β for 6 hr (D) and loaded with the CCF2 β-lactamase substrate. The genes tagged in these two clones were identified by RACE as CCA3 (GenBank no. AB000216;A) for E395 and RelB (GenBank acession no. M83221, B) for E424.
The Future of the Trapping Trade
Gene-trapping methods are already proving to be indispensable for functional analysis of the genome. Versatility is one strength of this family of molecular tools, which is helpful when attempting to analyze complex genomes. With this goal in mind, gene-trapping schemes that can act as a filter and allow discovery of a functionally related set of genes are useful. One example of a fairly “broad-pass” filter would be a technique to trap only genes that encode secreted or membrane proteins (Skarnes et al. 1995). Such a “secretory-trap” approach can ultimately reveal the proteins important for communication between cells. Another strategy that has been used in mice is the “induction-trap” in which ES cells are selected based on responsiveness of the trapped gene to growth and differentiation stimuli like retinoic acid or nerve growth factor (Forrester et al. 1996; Bonaldo et al. 1998; Salminen et al. 1998). Techniques that can specifically trap and monitor genes expressed at low levels will be valuable for “genome closure” because these genes are currently under-represented in compiled EST databases. Reporter cell lines generated by some trapping methods will be useful tools for investigation of the signal transduction pathways that regulate expression of the trapped genes. This will ultimately provide another layer of functional genomic information.
We wish to acknowledge the many fine contributions to the study of genome function that could not be included in this review because of space constraints. We wish to thank Drs. D. Nelson, M. Liyanage, M. Whitney, P. England, P. Negulescu, and T. Rink for critical review of this article. K.G.X. wishes to thank Dr. F. Craig for early inspirational discussions.
Notes
[1] Corresponding author.
Notes
[2] E-MAIL [email protected]; FAX (619) 404-6719.
REFERENCES
- ↵V. BellofattoL. ShapiroD.A. Hodgson(1984) Generation of a Tn5 promoter probe and its use in the study of gene expression in Caulobacter crescentus. Proc. Natl. Acad. Sci. 81:1035–1039.
- ↵E. BierH. VaessinS. ShepherdK. LeeK. McCallS. BarbelL. AckermanR. CarrettoT. UemuraE. Grell(1989) Searching for pattern and mutation in the Drosophila genome with a P–lacZ vector. Genes & Dev. 3:1273–1287.
- ↵F.R. BlattnerG. Plunkett IIIC.A. BlochN.T. PernaV. BurlandM. RileyJ. Collado-VidesJ.D. GlasnerC.K. RodeG.F. MayhewJ. GregorN.W. DavisH.A. KirkpatrickM.A. GoedenD.J. RoseB. MauY. Shao(1997) The complete genome sequence of Escherichia coli K-12. Science 277:1453–1474.
- ↵P. BonaldoK. ChowdhuryA. StoykovaM. TorresP. Gruss(1998) Efficient gene trap screening for novel developmental genes using IRES beta geo vector and in vitro preselection. Exp. Cell Res. 244:125–136.
- ↵D.G. BrennerS. Lin-ChaoS.N. Cohen(1989) Analysis of mammalian cell genetic regulation in situ by using retrovirus-derived “portable exons” carrying the Escherichia coli lacZ gene. Proc. Natl. Acad. Sci. 86:5517–5521.
- ↵M.R. Capecchi(1989) Altering the genome by homologous recombination. Science 244:1288–1292.
- ↵M.J. CasadabanS.N. Cohen(1979) Lactose genes fused to exogenous promoters in one step using a Mu-lac bacteriophage: In vivo probe for transcriptional control sequences. Proc. Natl. Acad. Sci. 76:4530–4533.
- ↵K. ChowdhuryP. BonaldoM. TorresA. StoykovaP. Gruss(1997) Evidence for the stochastic integration of gene trap vectors into the mouse germline. Nucleic Acids Res. 25:1531–1536.
- ↵F.S. Collins(1995) Ahead of schedule and under budget: The Genome Project passes its fifth birthday. Proc. Natl. Acad. Sci. 92:10821–10823.
- ↵F.S. CollinsA. PatrinosE. JordanA. ChakravartiR. GestelandL. Walters(1998) New goals for the U.S. Human Genome Project: 1998-2003. Science 282:682–689.
- ↵The C. elegans Sequencing Consortium (1998) Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 282:2012–2018.
- ↵C. CouldreyM.B. CarltonJ. FerrierW.H. ColledgeM.J. Evans(1998) Disruption of murine alpha-enolase by a retroviral gene trap results in early embryonic lethality. Dev. Dyn. 212:284–292.
- ↵J.L. DeRisiV.R. IyerP.O. Brown(1997) Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278:680–686.
- ↵M.R. Fannon(1996) Gene expression in normal and disease states—identification of therapeutic targets. Trends Biotechnol. 14:294–298.
- ↵S. FieldsO. Song(1989) A novel genetic system to detect protein-protein interactions. Nature 340:245–246.
- ↵S.P. FodorJ.L. ReadM.C. PirrungL. StryerA.T. LuD. Solas(1991) Light-directed, spatially addressable parallel chemical synthesis. Science 251:767–773.
- ↵L.M. ForresterA. NagyM. SamA. WattL. StevensonA. BernsteinA.L. JoynerW. Wurst(1996) An induction gene trap screen in embryonic stem cells: Identification of genes that respond to retinoic acid in vitro. Proc. Natl. Acad. Sci. 93:1677–1682.
- ↵G. FriedrichP. Soriano(1991) Promoter traps in embryonic stem cells: A genetic screen to identify and mutate developmental genes in mice. Genes & Dev. 5:1513–1523.
- ↵M.A. FrohmanM.K. DushG.R. Martin(1988) Rapid production of full-length cDNAs from rare transcripts: Amplification using a single gene-specific oligonucleotide primer. Proc. Natl. Acad. Sci. 85:8998–9002.
- ↵M.J. Geisow(1998) Proteomics: One small step for a digital computer, one giant leap for humankind. Nat. Biotechnol. 16:206.
- ↵J.A. GogosW. LowryM. Karayiorgou(1997) Selection for retroviral insertions into regulated genes. J. Virol. 71:1644–1650.
- ↵A. GosslerA.L. JoynerJ. RossantW.C. Skarnes(1989) Mouse embryonic stem cells and reporter constructs to detect developmentally regulated genes. Science 244:463–465.
- ↵G.G. HicksE.G. ShiX.M. LiC.H. LiM. PawlakH.E. Ruley(1997) Functional genomics in mice by tagged sequence mutagenesis. Nat. Genet. 16:338–344.
- ↵I.A. Hope(1991) “Promoter trapping” in Caenorhabditis elegans. Development 113:399–408.
- ↵A. Joyner(1993) Gene targeting: A practical approach. (Oxford University Press, UK).
- ↵V.A. McKusick(1997a) Genomics: Structural and functional studies of genomes. Genomics 45:244–249.
- ↵(1997b) Mendelian inheritence in man. Catalog of human genes and genetic disorders (Johns Hopkins University Press, Baltimore, MD), 12th ed. ibid.
- ↵P.M. MeneelyR.K. Herman(1979) Lethals, steriles and deficiencies in a region of the X chromosome of Caenorhabditis elegans. Genetics 92:99–115.
- ↵H.W. MewesK. AlbermannM. BahrD. FrishmanA. GleissnerJ. HaniK. HeumannK. KleineA. MaierlS.G. OliverF. PfeifferA. Zollner(1997) Overview of the yeast genome. Nature 387:7–65.
- ↵Y. MikiJ. SwensenD. Shattuck-EidensP.A. FutrealK. HarshmanS. TavtigianQ. LiuC. CochranL.M. BennettW. Ding(1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266:66–71.
- ↵A. MiyawakiJ. LlopisR. HeimJ.M. McCafferyJ.A. AdamsM. IkuraR.Y. Tsien(1997) Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388:882–887.
- ↵C.J. O'KaneW.J. Gehring(1987) Detection in situ of genomic regulatory elements in Drosophila. Proc. Natl. Acad. Sci. 84:9123–9127.
- ↵K. OkuboK. Matsubara(1997) Complementary DNA sequence (EST) collections and the expression information of the human genome. FEBS Lett. 403:225–229.
- ↵K. OkuboN. HoriR. MatobaT. NiiyamaA. FukushimaY. KojimaK. Matsubara(1992) Large scale cDNA sequencing for analysis of quantitative and qualitative aspects of gene expression. Nat. Genet. 2:173–179.
- ↵A. Persidis(1998) Proteomics. Nat. Biotechnol. 16:393–394.
- ↵A. Rao(1998) Sampling the universe of gene expression. Nat. Biotechnol. 16:1311–1312.
- ↵E.M. Rinchik(1991) Chemical mutagenesis and fine-structure functional analysis of the mouse genome. Trends Genet. 7:15–21.
- ↵J.R. RiordanJ.M. RommensB. KeremN. AlonR. RozmahelZ. GrzelczakJ. ZielenskiS. LokN. PlavsicJ.L. Chou(1989) Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 245:1066–1073.
- ↵J.M. RommensM.C. IannuzziB. KeremM.L. DrummG. MelmerM. DeanR. RozmahelJ.L. ColeD. KennedyN. Hidaka(1989) Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 245:1059–1065.
- ↵G.M. RubinA.C. Spradling(1982) Genetic transformation of Drosophila with transposable element vectors. Science 218:348–353.
- ↵M. SalminenB.I. MeyerP. Gruss(1998) Efficient poly A trap approach allows the capture of genes specifically active in differentiated embryonic stem cells and in mouse embryos. Dev. Dyn. 212:326–333.
- ↵J. Schell(1987) Transgenic plants as tools to study the molecular organization of plant genes. Science 237:1176–1183.
- ↵M. SchenaD. ShalonR.W. DavisP.O. Brown(1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270:467–470.
- ↵W.C. SkarnesB.A. AuerbachA.L. Joyner(1992) A gene trap approach in mouse embryonic stem cells: The lacZ reported is activated by splicing, reflects endogenous gene expression, and is mutagenic in mice. Genes & Dev. 6:903–918.
- ↵W.C. SkarnesJ.E. MossS.M. HurtleyR.S. Beddington(1995) Capturing genes encoding membrane and secreted proteins important for mouse development. Proc. Natl. Acad. Sci. 92:6592–6596.
- ↵A.C. SpradlingD.M. SternI. KissJ. RooteT. LavertyG.M. Rubin(1995) Gene disruptions using P transposable elements: An integral component of the Drosophila genome project. Proc. Natl. Acad. Sci. 92:10824–10830.
- ↵D.J. TownleyB.J. AveryB. RosenW.C. Skarnes(1997) Rapid sequence analysis of gene trap integrations to generate a resource of insertional mutations in mice. Genome Res. 7:293–298.
- ↵A.K. VossT. ThomasP. Gruss(1998) Efficiency assessment of the gene trap approach. Dev. Dyn. 212:171–180.
- ↵M. WhitneyE. RockensteinG. CantinT. KnappG. ZlokarnikP. SandersK. DurickF.F. CraigP.A. Negulescu(1998) A genome-wide functional assay of signal transduction in living mammalian cells. Nat. Biotechnol. 16:1329–1333.
- ↵L. WodickaH. DongM. MittmannM.H. HoD.J. Lockhart(1997) Genome-wide expression monitoring in Saccharomyces cerevisiae. Nat. Biotechnol. 15:1359–1367.
- ↵B.P. ZambrowiczG.A. FriedrichE.C. BuxtonS.L. LillebergC. PersonA.T. Sands(1998) Disruption and sequence identification of 2,000 genes in mouse embryonic stem cells. Nature 392:608–611.
- ↵G. ZlokarnikP.A. NegulescuT.E. KnappL. MereN. BurresL. FengM. WhitneyK. RoemerR.Y. Tsien(1998) Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science 279:84–88.