
Abstract
The evolutionary history of a gene helps predict its function and relationship to phenotypic traits. Although sequence conservation is commonly used to decipher gene function and assess medical relevance, methods for functional inference from comparative expression data are lacking. Here, we use RNA-seq across seven tissues from 17 mammalian species to show that expression evolution across mammals is accurately modeled by the Ornstein–Uhlenbeck process, a commonly proposed model of continuous trait evolution. We apply this model to identify expression pathways under neutral, stabilizing, and directional selection. We further demonstrate novel applications of this model to quantify the extent of stabilizing selection on a gene's expression, parameterize the distribution of each gene's optimal expression level, and detect deleterious expression levels in expression data from individual patients. Our work provides a statistical framework for interpreting expression data across species and in disease.
Comparative genomics has identified and annotated functional genetic elements by their evolutionary patterns across species (Rubin et al. 2000; Kellis et al. 2003; Siepel et al. 2005; Pollard et al. 2006; Lindblad-Toh et al. 2011). Current comparative studies focus primarily on analysis of genomic sequences, relying on a well-established theoretical framework developed from observations that neutral sequence diverges linearly across time (Harris 1966; Lewontin and Hubby 1966; Kimura 1968; Jukes and King 1971). These methods allow for detection of sequence elements that evolve slower (e.g., due to purifying selection) or faster (e.g., due to positive selection or relaxed selective constraints) than expected under the null model of neutral evolution.
It has long been accepted that divergence of gene regulation, manifested by phenotypic changes in gene expression, also plays a key role in evolution (King and Wilson 1975; Wang et al. 1996; Pierce and Crawford 1997; Ferea et al. 1999; Fraser et al. 2010). An evolutionary analysis of gene expression should help interpret gene function and evolutionary processes in ways that cannot be addressed by sequence alone: The extent of stabilizing selection on a gene's expression level in different tissues could reveal the one(s) in which the gene plays the most important role; the strength of evolutionary constraint on a gene's expression level could help interpret expression levels observed in clinical samples; and identifying genes whose expression level is under directional (positive) selection can help assess the basis of lineage- and species-specific phenotypes.
Multiple studies have analyzed expression data collected across mammalian species using various heuristic methods for defining conserved and divergent expression levels (Chan et al. 2009; Brawand et al. 2011; Merkin et al. 2012; Perry et al. 2012). However, there is currently no consensus on a quantitative framework for addressing the functional questions related to evolution of expression levels, due in part to a lack of agreement for how to best model expression evolution in mammals. In Drosophila, studies have found that unlike sequence evolution, divergence of gene expression levels is not continuously linear across evolutionary time. Instead, it reaches saturation due to stabilizing selective pressures, requiring more sophisticated models than standard neutral drift models (Bedford and Hartl 2009; Kalinka et al. 2010). In contrast, initial gene expression studies in mammals have been hampered by small data sets leading to inconsistent reports on the relative contribution of neutral drift and stabilizing selection within the mammalian lineage (Khaitovich et al. 2004; Yanai et al. 2004; Blekhman et al. 2008; Brawand et al. 2011). Early microarray-based studies observed a linear relationship between expression differences and divergence time across primates, suggesting neutral evolution (Enard et al. 2002; Khaitovich et al. 2004, 2005). Subsequent analysis, however, suggested that these observations were confounded by microarrays containing only human DNA probes (Gilad et al. 2006) which, once accounted for, left few differences in primate expression levels, highlighting stabilizing selection as the dominant mode of expression evolution. A more recent large-scale study of expression evolution across nine mammals profiled by RNA-seq (Brawand et al. 2011)—alleviating the limitations of hybridization technology—noted that more closely related species indeed have more similar expression levels (supporting a neutral model), but also that rates of expression evolution appear to be slow (supporting a major role for purifying selection). This lack of clarity on the correct model for expression evolution has resulted in conflicting usage of both pure neutral drift models (Perry et al. 2012) and those that incorporate stabilizing selection (Brawand et al. 2011) in comparative analyses across mammals, rendering different studies difficult to compare or interpret. Moreover, it has not been substantially explored how to use such models, once fit, to draw conclusions on gene function beyond theoretical inferences about fitness gains and selective effects (Bedford and Hartl 2009; Nourmohammad et al. 2017). The applicability of evolutionary models of gene expression to gain insight about transcriptional pathways and their relationships to healthy and disease processes has yet to be widely explored.
Here, we use a comprehensive RNA-seq data set from 17 mammalian species and seven different tissues to characterize the pattern of expression evolution across the mammalian lineage. We find that an evolutionary model that incorporates stabilizing selection is most appropriate for describing mammalian expression evolution. We further develop a framework, based on the previously proposed Ornstein–Uhlenbeck (OU) model, to parameterize the distribution of evolutionarily optimal gene expression, and we use this distribution to quantify the extent of stabilizing selection on a gene's expression, identify deleterious expression levels in patient expression data, and detect directional selection in lineage-specific expression programs.
Results
Expression differences between mammalian species saturate with increasing evolutionary time
To systematically explore mammalian expression evolution, we compiled a data set across the mammalian phylogeny spanning 17 species and seven different tissues (brain, heart, muscle, lung, kidney, liver, testis) (Fig. 1A; Supplemental Table S1). The data set combines published data for 12 species (Harr and Turner 2010; Brawand et al. 2011; Merkin et al. 2012; Pipes et al. 2013; Cortez et al. 2014; Wong et al. 2015) with data for five additional species we newly collected here to improve phylogenetic coverage (Methods). We focused on the 10,899 Ensembl-annotated mammalian one-to-one orthologs (Aken et al. 2017). We confirmed the quality of gene annotations by realigning transcriptomes across species and finding that 95%–99% of Ensembl's one-to-one orthologs were also identified as reciprocal-best alignments by our procedure; moreover, the mean sequence identity between Ensembl-annotated orthologs and their human counterpart decreases linearly with evolutionary time (Supplemental Fig. S1). Additionally, as expected, expression profiles first cluster by tissue and then by species, and their hierarchical clustering closely matches the phylogenetic tree (Supplemental Figs. S2, S3).
Expression evolution across mammalian lineages is accurately modeled by the OU process. (A) Data overview. Phylogenetic tree of all 17 mammals (left) marked by tissue types (colored dots) for which profiles are included. (*) Newly generated data. (B) Expression divergence is not linear. Shown is the pairwise mean squared expression distances (y-axis) between mammals and human for liver samples across evolutionary time, as estimated by substitutions per 100 bp (x-axis). (Error bars) standard deviation of the mean across replicates; (solid line) nonlinear (y = axk) regression fit. (C) OU model. Equation describing OU model (top): (σ) rate of genetic drift; [dB(t)] Brownian motion; (θ) optimal expression level; (α) strength of selection. (Left) Simulated trajectories of expression (y-axis) over evolutionary time (x-axis) under a Brownian motion (top) and OU (bottom) process. Ten example trajectories are shown. (Right) Mean squared distance to initial value (y-axis) across time (x-axis) from 1000 simulated trajectories. (D) Distribution of optimal expression. (Top) Illustration of the change in probability distribution of expression (y-axis) across time (x-axis) under an OU process. The distribution stabilizes as time approaches infinity. (Bottom) Scatter plot of log10TPM values (y-axis) across all liver samples (x-axis) of two example genes with low (NRBP1) and high (APOA4) variance. (Solid and dotted red lines) Estimated mean and variance, respectively, of the asymptotic (optimal) distribution of each gene's expression value estimated using the OU process. Note that mean and variance are calculated in log space.
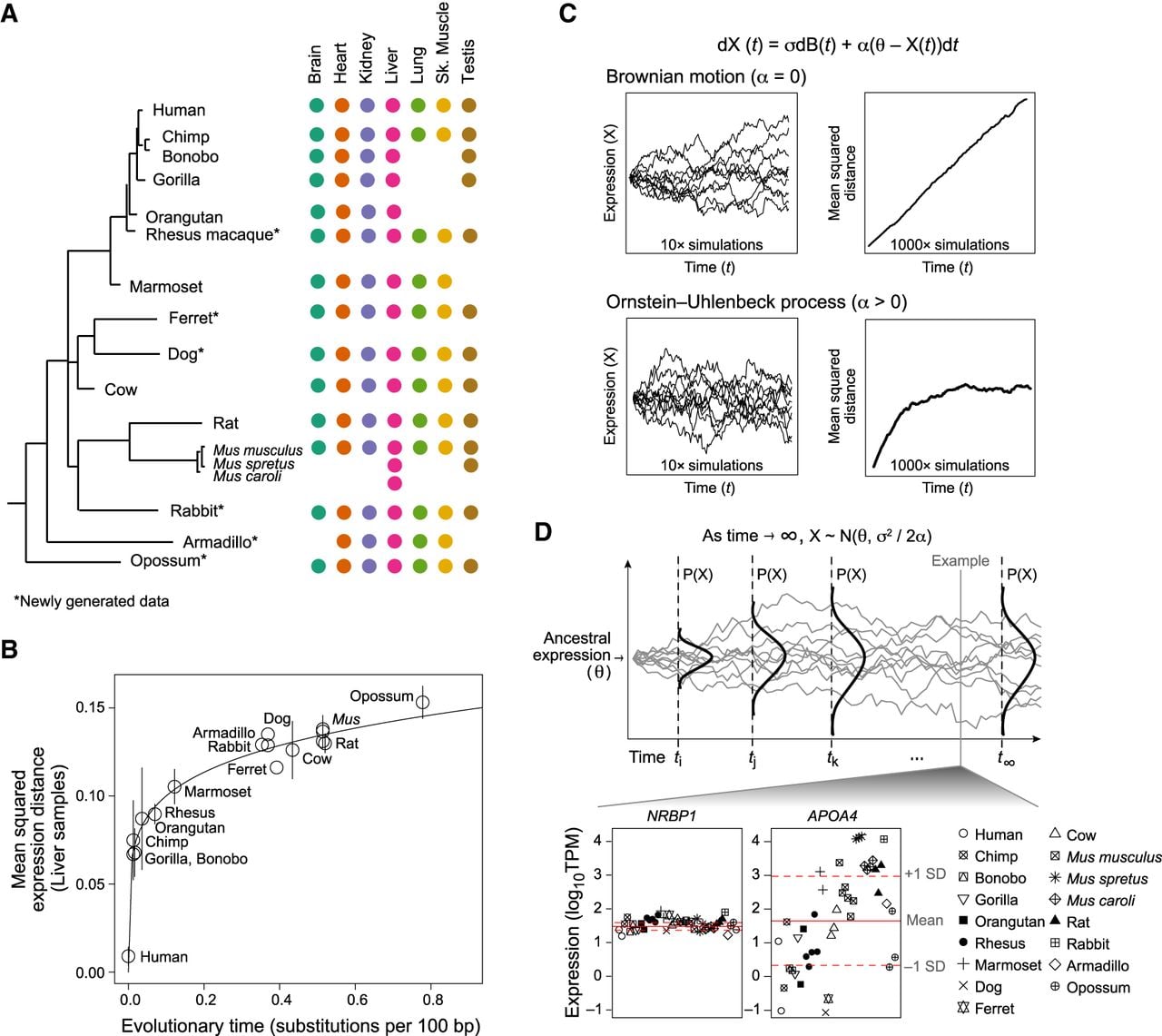
On average, pairwise expression differences between species (Supplemental Methods; Supplemental Fig. S4) saturate with evolutionary time in a power law relationship (Fig. 1B), consistent with evolutionary trends previously observed in Drosophila (Bedford and Hartl 2009). For example, when comparing each species’ profile to the corresponding human profile, differences initially diverge with increasing evolutionary distance, but this trend plateaus beyond the primate lineage. This relationship is observed in each of the five tissues for which we have expression data for all primates (brain, heart, kidney, liver, testis) (Supplemental Fig. S5) and is not driven by batch effects across different data sources or by variation in the number of samples available for each species (Supplemental Methods; Supplemental Figs. S6, S7). We observe the same relationship when using Mus musculus as the reference species in each of the two tissues for which we have expression data for multiple Mus species (Supplemental Figs. S8, S9).
Expression evolution can be modeled as an Ornstein–Uhlenbeck process
The observed pattern of expression divergence corresponds to an Ornstein–Uhlenbeck (OU) process (Fig. 1C,D), a stochastic process initially proposed as a model for evolution of general continuous phenotypes by Hansen (1997) and has more recently been suggested as an appropriate model specifically for the evolution of gene expression levels in Drosophila (Bedford and Hartl 2009).
In the context of expression levels, the OU process is a modification of a random walk, describing the change in expression (dXt) across time (dt) by dXt = σdBt + α(θ – Xt) dt, where dBt denotes a Brownian motion process. The model elegantly quantifies the contribution of both drift and selective pressure for any given gene: (1) Drift is modeled by Brownian motion with a rate σ (Fig. 1C, top), while (2) the strength of selective pressure driving expression back to an optimal expression level θ is parameterized by α (Fig. 1C, bottom). The OU process incorporates time information and fully accounts for phylogenetic relationships, thus allowing us to fit individual evolutionary expression trajectories. At longer time scales, the interplay between the rate of drift (σ) and the strength of selection (α) reaches equilibrium and, as time increases to infinity, constrains expression Xt to a stable, normal distribution, with a mean θ, and variance σ2/2α (Fig. 1D).
Thus far, OU models have primarily been used for theoretical inferences about fitness gains and selective effects of evolving expression levels (Bedford and Hartl 2009; Kalinka et al. 2010; Nourmohammad et al. 2017). There have also been limited applications of the OU model for detecting selection on expression across smaller mammalian phylogenies and incomplete gene annotations (Brawand et al. 2011; Rohlfs and Nielsen 2015). However, the complete power of using the OU model to characterize the evolutionary history of a gene's expression for biological insight has yet to be fully explored.
We thus next developed applications of the OU model to yield biologically interpretable results to evolutionary questions about gene expression levels, gene function, and disease gene discovery. First, for each tissue, we estimate from our data the asymptotic distribution of evolutionarily optimal expression for genes under stabilizing selection. We demonstrate that this distribution's OU variance (which we term “evolutionary variance”) accurately characterizes how constrained a gene's expression level is in each tissue. Second, we compare the observed expression levels in patient data to the optimal expression distributions estimated from the evolutionary model, in order to detect potentially deleterious expression levels and nominate causal disease genes. Third, we use an extension of the OU model (Butler and King 2004) that accounts for the existence of multiple distributions of optimal expression within a phylogeny to identify genetic pathways that may be related to lineage-specific adaptations. We describe each of these applications in turn.
The expression of most genes evolves under stabilizing selection within the mammalian lineage
To test whether a gene's expression is under stabilizing selection, we used a likelihood ratio test to compare the fit with no selection (α = 0; Brownian motion only) (Fig. 2A, top) to one with stabilizing selection (α > 0, OU process) (Fig. 2A, bottom). (Although we visually display patterns of expression evolution with respect to a single reference species [e.g., Fig. 1B], both models account for evolutionary distances across the entire phylogenetic tree, not just the relative distance to one reference species.) Because the expression level estimates of lowly expressed genes are associated with high technical variation and their true biological variation across species are less likely to be accurately inferred (Supplemental Fig. S10A; Silvestro et al. 2015), throughout all analyses, we focus only on genes expressed over five transcripts per million (TPM), resulting in between 3428 and 5822 genes for analysis, depending on the tissue (Supplemental Methods; Supplemental Fig. S10B).
Quantification of neutral and constrained selection on gene expression using the OU model parameters. (A) Detection of stabilizing selection. Pairwise mean squared expression distances (y-axis) between mammals and human for liver samples across evolutionary time (x-axis) for genes whose expression evolution fits better under a Brownian motion (BM) process (top), indicating neutral evolution, and genes whose expression evolution fits better an Ornstein–Uhlenbeck (OU) process (bottom), indicating the presence of stabilizing selection: (solids lines) linear regression fit for BM genes and nonlinear regression fit for OU genes. (B) Neutral and stabilizing selection across genes and tissues. Heatmap indicating genes (rows) whose expression is predicted to be evolving under neutral evolution (blue) or stabilizing selection (red) across five different tissues (columns); (gray) genes that are expressed <5 TPM. (C,D) Evolutionary variance across tissues and processes. (C) Heatmap shows estimated evolutionary variance of expression (orange: low; purple: high) across genes (columns) in five tissues (rows); (gray) genes expressed <5 TPM. (D) Bar plot of −log10 FDR values for significantly enriched GO categories of low (light gray) and high (dark gray) variance genes within each tissue; (*) category enriched in every tissue. (E) Relationship between sequence and expression evolution. Binned scatter plot of log(evolutionary variance) of liver expression (x-axis) versus sequence conservation, as measured by the phyloP score (y-axis). Median variance and phyloP scores are indicated by vertical and horizontal dotted lines, respectively. Enriched GO categories (FDR <0.001) for genes in each quadrant of the scatter plot are listed on the right.
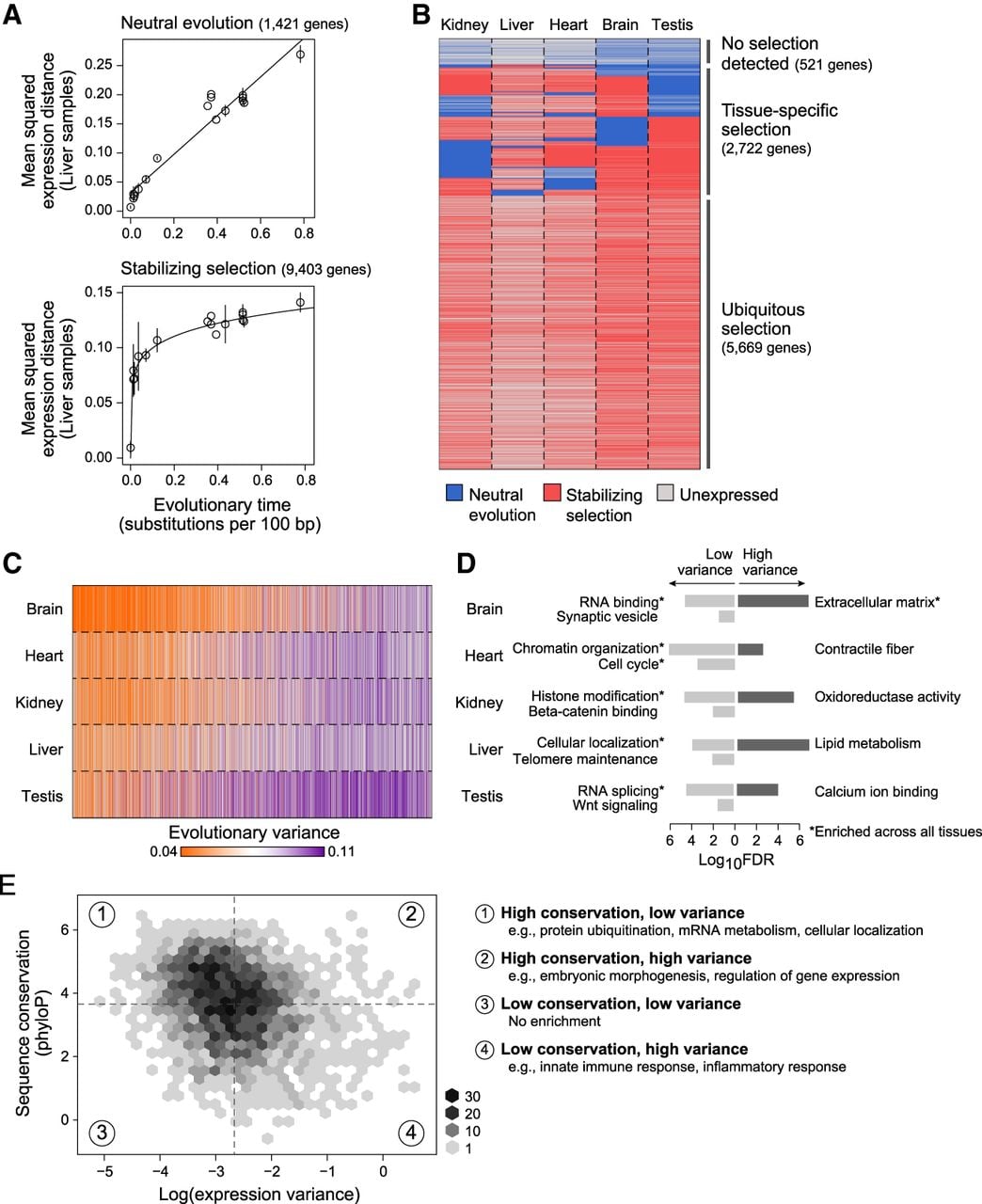
On average, the expression of 83% of genes tested (range: 77%–90%; false discovery rate [FDR] < 0.05) is better fit under a stabilizing selection model (Fig. 2A, bottom; Supplemental Fig. S11), although the expression of hundreds of genes within each tissue appeared to be neutrally evolving (Fig. 2A, top; Supplemental Fig. S11). The expression levels of 57% (5669/8912) of genes were under stabilizing selection in all tissues in which they were expressed, 39% (2722) were under stabilizing selection in only some of the tissues where they were expressed, and only 6% (521) were not under stabilizing selection in any of the tissues in our study (Fig. 2B).
We assessed our sensitivity and specificity to detect genes under expression-stabilizing selection using a jackknifing procedure, where we subsampled to consider phylogenies ranging from 3 to 16 species (Supplemental Methods). As expected, the number of genes called under stabilizing selection (i.e., rejecting the null hypothesis) increases as more species are included (Supplemental Fig. S12A), but does saturate at 14 species. Importantly, the discordance rate (relative to analysis of the full data set) is very low: <1% of genes that are found as under selection with a subsampled phylogeny are found to be neutral (i.e., accepting the null hypothesis) with the full phylogeny (Supplemental Fig. S12B).
Evolutionary distribution of gene expression levels predict gene function
The OU process was considered attractive when initially proposed for modeling expression evolution in Drosophila because of its ability to distinguish neutral from stabilizing selection. Given our finding that most mammalian genes are under stabilizing selection, we next explored the ability of the OU model to estimate the stable distribution of gene expression levels, which we reasoned is an estimate of the distribution of evolutionarily optimal expression. Thus we investigated the use of the OU model's “evolutionary variance” as a quantitative measurement of the extent of evolutionary constraint on a gene's expression in each tissue. The same jackknifing procedure as described above showed that the OU model's estimated evolutionary variance is highly robust to subsampling, as determined by the very low mean squared error (MSE < 0.005) when estimating variance from subsampled phylogenies (Supplemental Fig. S12C). In fact, when using data from less than six species, we found the evolutionary variance to be far more robust than the sample variance, which does not account for the phylogenetic relationships between the species (Supplemental Fig. S12C).
We first examined evolutionary variance patterns across tissues. To control for the number of samples in each tissue, we refitted OU evolutionary parameters on a subset of the data matched for the same number of samples across tissues (Supplemental Table S1). We found that brain had the most genes with low variance (most constraint), and testis the least, consistent with previous estimates of the rate of expression evolution for those tissues (Fig. 2C; Supplemental Fig. S13; Chan et al. 2009; Brawand et al. 2011). Across tissues, variance was reasonably correlated (mean Pearson's r = 0.70) (Supplemental Fig. S14A). For genes expressed across three or more tissues, expression level and variance were modestly inversely correlated across somatic tissues (median Pearson's r = −0.25), and the tissue of highest expression matched the tissue of lowest variance in 27.2% (1263/4645) of genes (Supplemental Fig. S14B, top); further including testis in this analysis leads to almost no correlation between expression level and variance (median Pearson's r = −0.006) (Supplemental Fig. S14B, bottom).
We next examined evolutionary variance patterns within tissues using our full data set. To avoid biases introduced by the diversity of data sources, we did not attempt to interpret absolute values of variance but rather focused on understanding the relative relationship between genes with lower and higher variance. Using a rank-based Gene Ontology (GO) enrichment test (Eden et al. 2009), we found that evolutionary variance and function were strongly associated, consistent with results from previous comparative studies that did not use phylogenetic-based methods for estimating variance (Chan et al. 2009; Yue et al. 2014): Across all tissues, genes with low variance were enriched for housekeeping functions (e.g., RNA binding and splicing, chromatin organization, cell cycle), whereas those with high variance were enriched for extracellular proteins (FDR < 0.001).
Some processes were enriched in genes with low or high variance only in specific tissues (Fig. 2D; Supplemental Table S2): Among the processes with tissue-specific conservation (low variance) were synaptic proteins in brain (FDR = 0.011) and Wnt signaling in testis (FDR = 0.014); processes with high variance included contractile fiber part in heart (FDR = 0.005), oxidoreductase activity in kidney (FDR = 6.10 × 10−6), and lipid metabolism in liver (FDR = 2.31 × 10−9). We did not find the same enriched categories when we tested for enrichments in genes ranked by expression level. Thus, we can rely on estimates of evolutionary variance as an indicator of expression constraint and gene function.
We found only a modest correlation between expression and sequence constraint (Pearson's r = −0.25) (Fig. 2E; Supplemental Methods). Genes conserved in both expression and sequence were significantly enriched for housekeeping processes (FDR < 10−4) (Supplemental Table S3), and genes divergent in both were enriched for immune and inflammatory response (FDR < 10−6). Genes conserved in sequence but divergent in expression were enriched in transcriptional regulators (FDR = 3.1 × 10−5), especially those involved in embryonic morphogenesis (FDR = 9.8 × 10−8; e.g., IRX5, HAND2, NOTCH1). Although higher evolutionary variance of expression levels may be influenced by environment, changes in cell-type composition, and genetic differences, our analysis supports the hypothesis that divergence in gene regulation without protein sequence divergence can account for species-specific phenotypes.
Using evolutionary distributions of gene expression to predict deleterious levels
In analysis of rare diseases, sequence conservation is commonly used to prioritize mutations in genes that are more essential and likely causal for rare diseases when mutated (Alföldi and Lindblad-Toh 2013; Jordan et al. 2015; Richards et al. 2015). By analogy, we hypothesized that expression conservation should also be predictive of gene essentiality. Indeed, the expression levels of genes that are either essential in culture (Hart et al. 2014), essential in mice (Georgi et al. 2013), or haploinsufficient in humans (Rehm et al. 2015) had significantly lower evolutionary variance (higher constraint) than their nonessential or haplosufficient counterparts across almost all tissues (Wilcoxon rank-sum test P-value <0.01) (Fig. 3A), a relationship was not driven by expression levels (Supplemental Fig. S15).
Evolutionary distribution of gene expression helps identify disease-contributing genes. (A) Essential genes have lower evolutionary variance. Box plots show the distribution of log(evolutionary variance) (y-axis) of genes essential in culture (top), essential in mice (middle), and haploinsufficient in human (bottom; dark gray), and their nonessential or haplosufficient counterparts (light gray) in each of seven tissues (x-axis): (***) P < 0.001; (**) P < 0.01. (B) Disease genes have lower evolutionary variance. Box plots show the distribution of log(evolutionary variance) (y-axis) of genes linked (dark gray) and not linked (light gray) to high-penetrance monogenic autism spectrum disorder (top), congenital heart defects (middle), and neuromuscular disease (bottom) in the relevant tissue (brain, heart, and muscle, respectively). (Left) Genes that are restricted in expression (>5 TPM in three or fewer tissues) in that tissue; (right) genes that are ubiquitously expressed; (***) P < 0.001; (*) <0.05. (C,D) Overview of using evolutionary distributions or GTEx RNA-seq distributions to identify outlier gene expression from RNA-seq of muscular dystrophy patients. (C) Two scoring approaches based on evolutionary distributions (left) or GTEx RNA-seq distributions (right). (D) Table shows number of significant outlier genes, −log10FDR score, and DMD’s significance rank for all patients with muscular dystrophy when using distributions estimated from evolutionary data (left) or GTEx RNA-seq data (right).
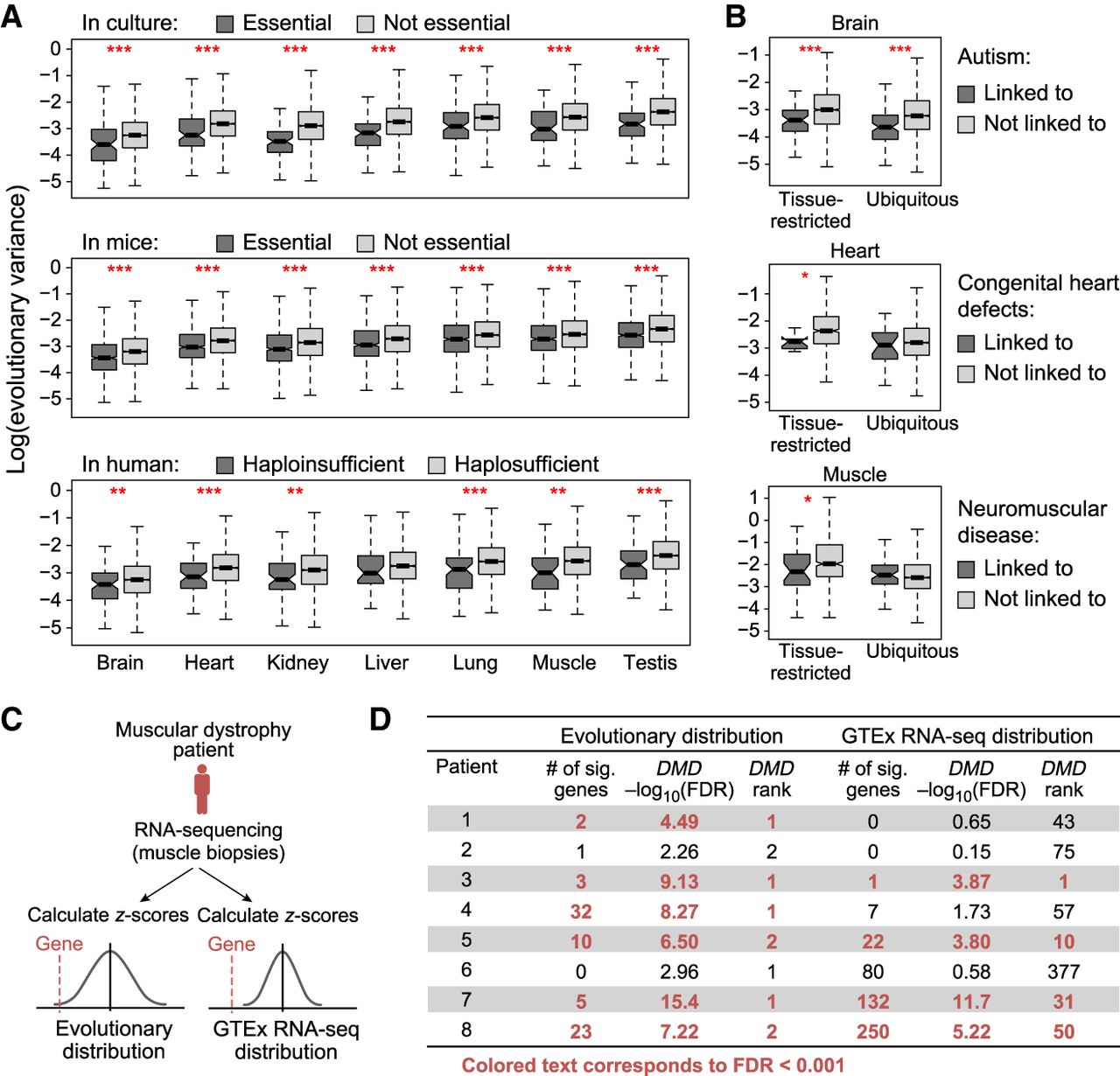
We next examined the variance of disease genes in each of three settings: rare monogenic disease genes directly linked to nonsyndromic autism spectrum disorder (ASD) (brain) (Banerjee-Basu and Packer 2010), congenital heart defects (heart) (Amberger and Hamosh 2017; Blake et al. 2017), and neuromuscular disease (skeletal muscle) (Supplemental Methods; Cummings et al. 2017). In each case, disease genes with tissue-restricted expression (>5 TPM in three or fewer tissues) (Supplemental Methods; Supplemental Fig. S16) consistently exhibited lower variance in the disease-relevant tissue than nondisease genes (P-value <0.05) (Fig. 3B; Supplemental Fig. S17). In ASD-linked genes only, we also observed significantly lower variance of ubiquitously expressed disease versus nondisease genes (Fig. 3B).
Next, we hypothesized that the parameters of each gene's optimal OU distributions can predict disease genes by highlighting outlier, likely pathogenic, gene expression levels in patient data. This is analogous to causal disease gene discovery by using conservation to identify putatively pathogenic sequence mutations in whole-exome sequencing (Choi et al. 2009; O'Roak et al. 2011). To this end, we obtained RNA-seq of muscle biopsies of 93 patients clinically diagnosed with neuromuscular disease (Methods; Supplemental Table S4). For each patient sample, we calculated a Z-score for each gene to assess how they deviate from the (optimal) evolutionary fit for that gene's expression in skeletal muscle, with correction for multiple hypothesis testing (Methods; Supplemental Fig. S18A). Compared to GTEx muscle samples from 184 healthy people (The GTEx Consortium 2013), patients had, on average, 3.2-fold more dysregulated genes overall by this measure (Wilcoxon rank-sum test P-value = 2 × 10−9) (Supplemental Fig. S18B), suggesting that the evolutionary parameters fit by the OU model can be used to detect outlier expression values that are more likely to be deleterious.
We then tested whether the OU model could be used to identify the causative gene in rare disease analysis. As a proof of principle, we focused on the subset of eight patients from the muscle disease cohort who were clinically diagnosed with either Becker or Duchenne muscular dystrophy, including confirmation of absent or decreased dystrophin protein via immunoblotting (Cummings et al. 2017). To compare our approach to a standard differential expression analysis, we ranked genes by outlier expression with Z-scores defined based either on (1) comparison to the mean and variance estimated from our evolutionary data; or (2) comparison to a mean and variance estimated from only healthy GTEx human data (Fig. 3C). By our evolutionary data, fewer genes ranked as significant outliers in each patient (median: 4, range: 0–32), and the DMD gene ranked as either the top or second most aberrantly expressed gene in six of eight patients, each showing significant underexpression (FDR < 10−3) (Fig. 3D). In comparison, scoring in reference to GTEx expression data did not yield such specific results: A median of 14.5 genes were outliers (range: 0–250), only four of eight patients were called as significantly underexpressing DMD (FDR < 10−3), and its significance in these patients ranked between 1 and 50. This difference in specificity likely reflects more accurate estimates of healthy (tolerable) variance when using evolutionary versus human data: Although estimates of mean expression are highly concordant between the two methods (Supplemental Fig. S19, left), expression variances were almost always larger when estimated using the evolutionary data set (Supplemental Fig. S19, right), reflecting the longer period of time each gene had to fully explore the space of physiologically acceptable expression levels. Thus, using human GTEx distributions resulted in many more false-positive genes that appear to be aberrantly expressed. Conversely, using the OU model's estimate of evolutionary mean and variance of optimal gene expression helps detect dysregulation of the actual disease gene and could aid novel disease gene discovery. Importantly, in contrast to methods for differential expression between patient and healthy controls, this method does not require a control population and can be conducted for an individual patient sample.
A multivariate OU model can be applied to detect lineage-specific expression changes
Finally, we explored the use of the OU model to detect directional selection in gene expression. We used an extension of the model that accounts for multiple selection regimes across a single phylogeny by modeling the distribution of expression level as a multivariate normal distribution whose mean and variance are estimated for each (predefined) subclade (Fig. 4A; Butler and King 2004; Rohlfs et al. 2014). A previous application of this extended OU model identified more than 9000 expression changes across the mammalian phylogeny (Brawand et al. 2011), but the analysis relied on a smaller phylogeny and thus focused on identifying species-specific shifts in gene expression that unfortunately could be easily confounded by environmental causes or technical effects.
Multivariate OU process enables detection of lineage-specific expression changes. (A) Multivariate OU process. Simulated trajectories of expression (y-axis) over time (x-axis) under a multivariate OU process. Trajectories in gray are sampled from the same distribution (N0) across time, while trajectories in orange start at the same ancestral distribution (N0) but evolve under a new distribution (N1) after a speciation event. (B) Three tested hypotheses of expression evolution: from left: the univariate OUall model, in which gene expression evolves under a single stabilizing regime across the phylogeny (black), and two multivariate OU models, OUprimates and OUrodents, in which gene expression evolves under the ancestral regime (black) and a new regime in the specified subclade (orange). (C) Lineage-specific expression in liver. Pairwise mean squared expression distances in liver samples (y-axis) between a reference species (labeled black point) and each of the other mammals for genes assigned to each of three tested OU models. (Black points) species evolving under ancestral distribution; (labeled orange points) species evolving under new regime after the lineage split; (solid line) nonlinear regression fit for species evolving under ancestral distribution. (D) Example processes enriched for lineage-specific expression. Heatmaps show column-normalized expression (red: high; blue: low) from genes (columns) with lineage-specific expression patterns in three enriched GO categories (FDR < 0.05): lipid transport in liver (left), immune regulation in liver (middle), and microtubule movement in testis (right).
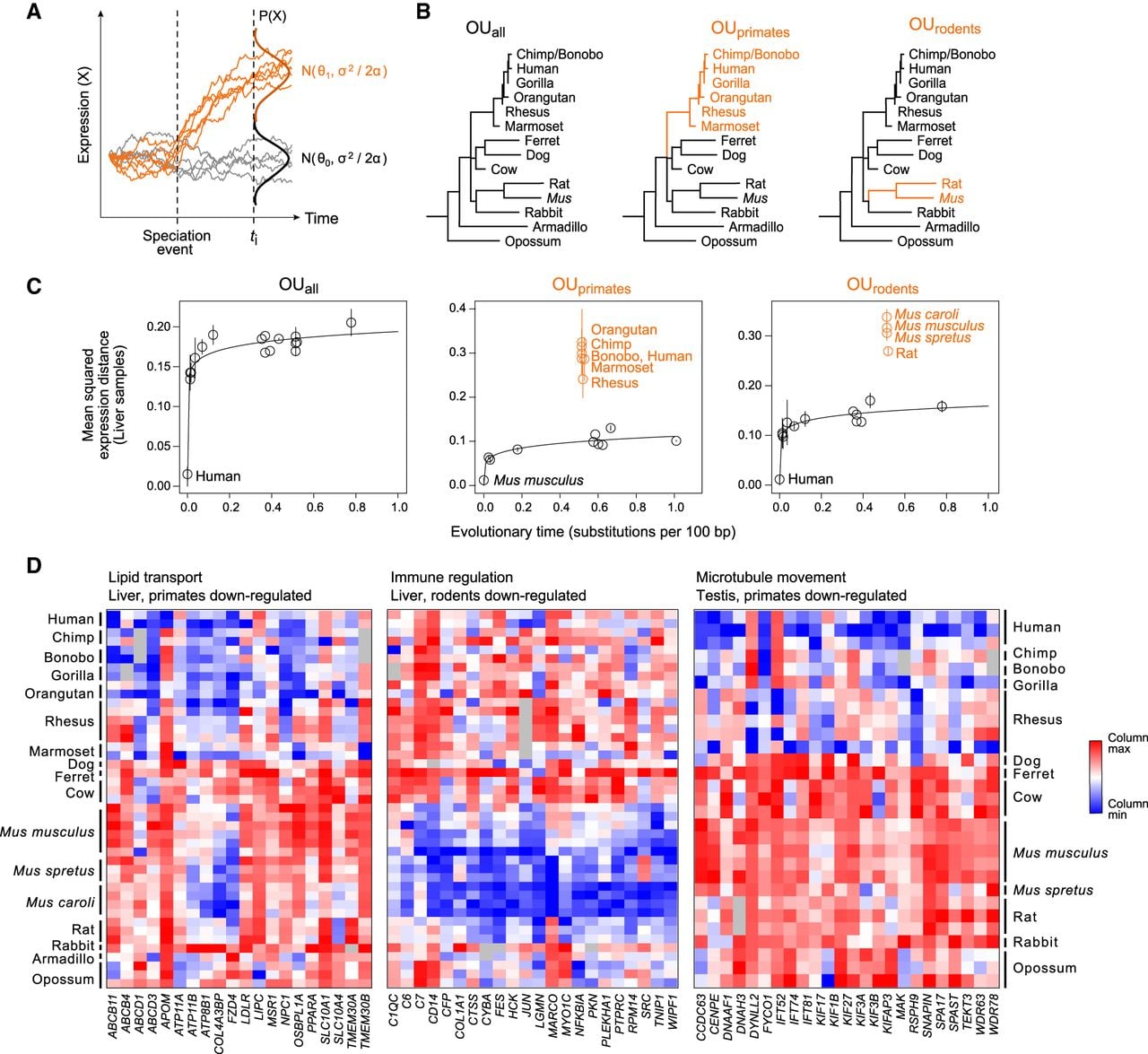
We leveraged our more comprehensive phylogenetic coverage and focused on detecting shifts in expression consistent in direction and magnitude across entire subclades of three or more species, whose samples were collected and sequenced across multiple sources to mitigate nongenetic confounders. We identified “differential gene expression” across the tree based on the approach suggested by Butler and King (2004) (Methods): For each gene, we applied the standard univariate OU model, which uses a single optimum for all species, as well as the extended OU model, which uses two optima—one for the ancestral distribution and one for the distribution within the clade of interest—and assigned the best OU model using goodness-of-fit tests. As a conservative measure, we retained only those genes that also changed at least twofold between subclades and had a mean expression level of at least 1 TPM in one of the subclades.
We first assessed the power of this approach to detect lineage-specific expression at increasing phylogenetic distances by testing for differential expression changes in liver samples shared across all primates (branch length = 0.121), rodents (branch length = 0.177), laurasians (branch length = 0.407), or lagomorphs (branch length = 0.575). We construct our data set so that we test for shared differential expression changes across three species within the clade of interest against eight comparing species and, to avoid confounders from batch effects, we choose the three species of interest such that the data for each was obtained from a different source (Supplemental Methods; Supplemental Table S1). As expected, we find that the number of differentially expressed genes detected monotonically decreases with increasing distance within the clade of interest, ranging from 470 genes in the primate clade to 327 in the lagomorph clade (Supplemental Fig. S20A). Unfortunately, our false discovery rates for this analysis, estimated by shuffling species expression data (Methods), ranged from 54% to 78%.
To improve our power to detect differential expression, we turned to our full data set and tested for differential expression within the primate clade (OUprimates, 5–7 primates versus 8–10 comparing species) and rodent clade (OUrodents, 3–5 rodents versus 10–12 comparing species) (Fig. 4B). Even with this larger phylogeny, our FDRs ranged from 18% to 52%, suggesting that future studies on differential expression should rely on larger phylogenies. The varying enrichment sizes across tissues are likely largely driven by differences in sample sizes; when using a data set matched for sample sizes across tissues, the percentage of expressed genes that are differentially expressed is fairly consistent across tissues (Supplemental Fig. S20B).
Despite the modest power of our analysis, we were able to achieve FDR < 30% in liver (primates: FDR = 0.18; rodents: FDR = 0.27) and testis (primates: FDR = 0.29; rodents: FDR = 0.29), as well as in the primate clade for lung (FDR = 0.26) and brain (FDR = 0.18) (Supplemental Fig. S21), and we further examined these sets of differentially expressed genes. For example, in liver, we identified 640 and 794 genes with lineage-specific expression changes in primates and rodents, respectively, highlighting specific metabolic processes diverging in regulation in each clade. The expression levels of lineage-specific genes deviated significantly from expectation only if there was clade-specific selection (Fig. 4C). Because of the larger set of differentially expressed genes compared to previous applications, we could identify functional enrichments among lineage-specific genes (Supplemental Table S5). We found primate-specific down-regulation of genes related to a number of lipid metabolic processes in the liver (FDR = 1.88 × 10−11). These processes include peroxisomal functions (FDR = 2.45 × 10−8), fatty acid metabolism (FDR = 1.52 × 10−8), and lipid transport (FDR = 3.36 × 10−3) (Fig. 4D), and contain known regulators of lipid metabolism such as the LDL receptor (LDLR) (Brown and Goldstein 1979), hepatic lipase (LIPC) (Guerra et al. 1997), and the transcription factor PPARA (Kersten 2014). Thus, the expression of multiple pathways may have diverged at the ancestral primate branch, consistent with observations that human lipidemia is not well-modeled by mice without further genetic modification (von Scheidt et al. 2017). In other examples, genes involved in regulation of immune response were down-regulated across rodent livers (FDR = 6.97 × 10−4), and microtubule-based movement genes (FDR = 2.82 × 10−3) and spermatogenesis (FDR = 2.82 × 10−2) were down-regulated across primates in testis (Fig. 4D), reflecting the known rapid evolution of immune-related (Kosiol et al. 2008; Areal et al. 2011; Yue et al. 2014) and reproduction-related genes (Swanson et al. 2001; Torgerson et al. 2002), respectively.
Discussion
Here, we combined a large data set of comparative gene expression profiles across mammals with systematic analysis and showed that the divergence of gene expression of one-to-one mammalian orthologs saturates across evolutionary time, and that this can be modeled well by an OU process. We further showed how to use the OU model to query for gene function, assess candidate disease genes for deleterious levels of expression, and identify lineage-specific evolution of expression.
As with any comparative species analysis, artifacts introduced from batch effects, or errors in ortholog or transcript annotation may bias our data. However, the evolutionary patterns we observed are not solely driven from a single batch as each data source comprises of species that span the entire phylogenetic tree. Additionally, our use of only one-to-one mammalian orthologs helps to mitigate errors in orthology assignment, and we confirm that the sequence identity of our annotated transcripts diverges linearly across evolutionary time, and thus is not driving the observed expression evolution patterns.
The nonlinear pattern of expression evolution is accurately modeled by the previously proposed OU process (Hansen 1997), a model which incorporates both neutral drift and stabilizing selection. Although we find that stabilizing selection plays a dominant role in expression evolution within the mammalian lineage, we note that the appropriate model is dependent on evolutionary distance: Within the primate lineage, we do find that expression differences diverge near-linearly, corroborating original studies that proposed a neutral model of expression evolution (Enard et al. 2002; Khaitovich et al. 2004); but at larger evolutionary distances, stabilizing selection has increasingly bigger effects, as has been noted in more recent RNA-seq studies in mammals (Brawand et al. 2011) and Drosophila (Bedford and Hartl 2009; Nourmohammad et al. 2017).
Importantly, although the OU process accurately models our data, there are additional factors that may constrain a gene's expression. These factors include (1) lower bound on gene expression by definition (i.e., 0 TPM); (2) upper limits of gene expression; (3) selective pressures on a gene in one tissue that have constraining effects on the same gene's expression in other tissues; and (4) selective pressure on one gene that form trans effects on expression constraints on other genes. Although cases (1) and (2) represent a small percentage of all genes tested in our study, especially with our filter for genes expressed >5 TPM, we are unable to separate genes whose expression is constrained due to indirect selective forces, as in cases (3) and (4), from expression levels under direct selective pressure using our current data. Nevertheless, the OU model remains an important quantitative tool for describing evolutionary histories of gene expression and lays groundwork for further interrogation into the mechanisms by which expression evolves.
Although previous studies using the OU model with gene expression data have focused on theoretical aspects of expression evolution, we now show how to use the OU model to estimate the distributions of optimal gene expression levels and answer physiologically and clinically relevant questions about gene function, including detecting deleterious expression levels from individual patient data. Because our data are derived from a variety of sources that may influence the accuracy of our estimates of evolutionary distributions, we are careful to only analyze relative evolutionary variances (e.g., rank-based GO enrichment tests) or construct subsets of our data set for analysis that avoid batch effects (e.g., testing for shifts in expression across multiple species, each collected from different sources). However, when directly using evolutionary mean and variance estimates for our disease expression analysis, we find that our estimates outperform healthy human data in specifically identifying outlier expression of a disease gene. This suggests that evolutionary estimates are robust to technical variance and can ultimately be provided across a variety of tissue types to aid with scientific and clinical discovery.
We finally apply a multivariate OU model (Butler and King 2004; Rohlfs et al. 2014) to identify lineage-specific expression changes across clades of species in our data. Although we improve upon previous studies that relied on smaller data sets, even with 17 species, we are unable to reach an FDR below 18%, and we uncover fewer differential expression changes shared across more evolutionarily distant clades. This suggests that future studies investigating lineage-specific expression will require data sampled from larger phylogenies and would perform better when detecting lineage-specific expression changes shared across closely related species. We also note that a drawback of this approach is that the tested hypotheses must first be constructed (e.g., primates versus nonprimates; rodents versus nonrodents), and the best-fitting model may not be truly reflective of the underlying evolutionary history. Future investigations on directional selection on expression in natural and experimental settings will help to refine the best hypotheses to be tested under this approach.
Looking forward, we anticipate that the OU model can be further developed for other biological queries, for example, testing for stabilizing selection across pathways of genes or paralog families, estimating ancestral expression states, or being applied to other transcribed regions such as short or long noncoding RNAs. As shown by our analysis, characterizations of expression across additional tissue types and species under varied developmental and environmental contexts will provide increased power and further insight into the evolution of gene expression and the relationship between genotype and phenotype.
Methods
RNA-seq for evolutionary data set
RNA samples from dog and rabbit tissues were commercially obtained from Zyagen. RNA samples from ferret tissues were a kind gift from John Englehardt (University of Iowa). RNA samples from opossum tissues were a kind gift from Paul Samollow (Texas A&M). RNA samples from armadillo tissues were a kind gift from Jason Merkin and Christopher Burge (MIT). All tissue collection was approved by IACUC and carried out in accordance with respective institutional guidelines. RNA-seq libraries were prepared as described in Levin et al. (2010) (Supplemental Methods). Samples were sequenced on an Illumina HiSeq 2000 sequencer, to a minimum depth of 35 million reads.
Alignment and expression quantification
RSEM v1.2.12 (Li and Dewey 2011) was used with default parameters to align reads to the transcriptome of each species and to quantify TPM of each gene.
Normalization of gene expression values
Gene expression values (log10TPM) were normalized using TMM normalization (Robinson and Oshlack 2010) from the Bioconductor package edgeR (Robinson et al. 2010). Briefly, TMM normalization assumes that the majority of genes are not differentially expressed (DE) between samples and estimates a scaling factor between a pair of samples such that the trimmed mean of log expression ratios (trimmed mean of M values [TMM]) is equal to 1. It is reasonable to assume that the majority of genes between pairs of species are not DE, because even between distant mammals, such as human and opossum, Pearson's correlation of expression level in a given tissue is >0.75.
Fitting OU process parameters
BM and OU models were fit to normalized expression values using the R package ouch (Butler and King 2004) with default parameters. P-values for each gene were calculated using a likelihood ratio test comparing the OU (alternative hypothesis) to the BM (null hypothesis) model, and then corrected for multiple hypothesis testing using the Benjamini–Hochberg FDR procedure (Benjamini and Hochberg 1995).
Samples for neuromuscular disease data set
The cohort of neuromuscular disease patient RNA-seq described in this study is a superset of that described in Cummings et al. (2017) (dbGaP accession phs000655.v3.p1) and 30 additional patients. Tissues were procured under Institutional Review Board (IRB)-approved protocols at the National Institute of Neurological Disorders and Stroke (Protocol #12-N-0095), Newcastle University (CF01.2011), Boston Children's Hospital (03-12-205R), University College London (08ND17), UCLA (15-001919), and St. Jude Children's Research Hospital (10/CHW/45). Patients consented to these protocols in clinic visits prior to biopsy. Patient muscle biopsies were collected as described in Cummings et al. (2017) and sequenced using the same protocol as in the GTEx project (Supplemental Methods; The GTEx Consortium 2013).
Alignment and expression quantification of human muscle data
GTEx BAM files were downloaded from dbGaP under accession ID phs000424.v6.p1 and realigned after conversion to FASTQ files with Picard SamToFastq. Both patient and GTEx reads were aligned using STAR 2-Pass v.2.4.2a (Dobin et al. 2013) using hg19 as the genome reference. Expression was quantified with RNA-SeQC v1.1.8 (DeLuca et al. 2012) using GENCODE v19 annotations.
Detecting outlier expression in patient samples
Genes expression values (log10TPM) were first normalized by TMM normalization (Robinson and Oshlack 2010) to the human skeletal muscle expression values from the evolutionary data set. For each gene in each patient sample, a Z-score was calculated using the asymptotic mean and variance estimated from the evolutionary data. Z-scores were only calculated for genes that were assessed to fit better under the OU rather than the BM model and whose asymptotic mean was estimated to be 5 TPM or higher. Z-scores were converted to P-values and then corrected for multiple hypothesis testing using the Benjamini–Hochberg FDR procedure. We used an FDR threshold of 0.01 to initially define significance. Of those, we removed another 330 genes that scored as a significant outlier in >25% of the GTEx samples. As a comparator, Z-scores were also calculated using the sample mean and variance estimated from healthy human GTEx samples. To ensure comparability between the two methods, we only calculated Z-scores for genes that were not filtered out at any steps during the evolutionary method above.
Detecting lineage-specific expression programs
In all lineage-specific differential expression analyses, P-values were calculated using a likelihood ratio test comparing each of the OU models to the BM model and then adjusted for multiple hypothesis testing using the Benjamini–Hochberg FDR procedure. For each gene, Akaike and Bayesian Information Criterion (AIC and BIC) scores were calculated on all models that were significant against the null to determine the best-fitting model. Both scores agreed for the best-fitting model in all cases. To estimate the FDR, we performed the same procedure in each tissue using shuffled expression data, in which the expression data from one species is randomly reassigned to a different species. (In a related method [Brawand et al. 2011; Rohlfs and Nielsen 2015], Q-values are derived by directly testing the alternative hypothesis θsubclade ≠ θancestral against the null hypothesis θsubclade = θancestral and adjusted for multiple hypothesis testing. However, we found that this stringent approach resulted in an even larger burden of multiple testing.) We performed GO enrichment analysis on each set of up- and down-regulated genes separately, using a background set of genes with mean expression of at least 1 TPM across all species in the appropriate tissue.
Data access
RNA-seq raw data from this study (from rabbit, dog, ferret, and opossum) have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE106077. Processed expression data and evolutionary expression distributions for all one-to-one mammalian orthologs in each tissue context are available at https://portals.broadinstitute.org/evee/.
Acknowledgments
We thank Daniel MacArthur for help with patient data; Leslie Gaffney for artwork and advice on figures; and Hopi E. Hoekstra, Daniel L. Hartl, Dawn Thompson, Yarden Katz, Akshay Krishnamurthy, Quinlan L. Sievers, Brian J. Haas, Atray Dixit, Rebecca H. Herbst, Jessica Alföldi, and Carl G. de Boer for helpful discussions and feedback. This work is supported by the BBSRC (grant no. BB/CSP1720/1), the Klarman Cell Observatory, HHMI, and the Broad Institute. K.L.-T. is a recipient of a Distinguished professor award from the Swedish Research Council.
Author contributions: J.C., K.L.-T., F.d.P., and A.R. conceived of the study. J.C. participated in its design and coordination, carried out all analyses, and wrote the manuscript. R.S. performed RNA QC and sequencing library construction. J.J. performed project management. B.B.C. contributed to analyses detecting outlier expression in neuromuscular disease patients. N.R. provided computational support. K.L.-T., W.H., F.d.P., and A.R. participated in the design and coordination of the study and in writing the manuscript.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.237636.118.
References
- ↵Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, Billis K, Carvalho-Silva D, Cummins C, Clapham P, 2017. Ensembl 2017. Nucleic Acids Res 45: D635–D642. 10.1093/nar/gkw1104
- ↵Alföldi J, Lindblad-Toh K. 2013. Comparative genomics as a tool to understand evolution and disease. Genome Res 23: 1063–1068. 10.1101/gr.157503.113
- ↵Amberger JS, Hamosh A. 2017. Searching Online Mendelian Inheritance in Man (OMIM): a knowledgebase of human genes and genetic phenotypes. Curr Protoc Bioinformatics 58: 1.2.1–1.2.12. 10.1002/cpbi.27
- ↵Areal H, Abrantes J, Esteves PJ. 2011. Signatures of positive selection in Toll-like receptor (TLR) genes in mammals. BMC Evol Biol 11: 368. 10.1186/1471-2148-11-368
- ↵Banerjee-Basu S, Packer A. 2010. SFARI Gene: an evolving database for the autism research community. Dis Model Mech 3: 133–135. 10.1242/dmm.005439
- ↵Bedford T, Hartl DL. 2009. Optimization of gene expression by natural selection. Proc Natl Acad Sci 106: 1133–1138. 10.1073/pnas.0812009106
- ↵Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Methodol 57: 289–300. 10.2307/2346101
- ↵Blake JA, Eppig JT, Kadin JA, Richardson JE, Smith CL, Bult CJ, the Mouse Genome Database Group. 2017. Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic Acids Res 45: D723–D729. 10.1093/nar/gkw1040
- ↵Blekhman R, Oshlack A, Chabot AE, Smyth GK, Gilad Y. 2008. Gene regulation in primates evolves under tissue-specific selection pressures. PLoS Genet 4: e1000271. 10.1371/journal.pgen.1000271
- ↵Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, 2011. The evolution of gene expression levels in mammalian organs. Nature 478: 343–348. 10.1038/nature10532
- ↵Brown MS, Goldstein JL. 1979. Receptor-mediated endocytosis: insights from the lipoprotein receptor system. Proc Natl Acad Sci 76: 3330–3337. 10.1073/pnas.76.7.3330
- ↵Butler MA, King AA. 2004. Phylogenetic comparative analysis: a modeling approach for adaptive evolution. Am Nat 164: 683–695. 10.1086/426002
- ↵Chan ET, Quon GT, Chua G, Babak T, Trochesset M, Zirngibl RA, Aubin J, Ratcliffe MJ, Wilde A, Brudno M, 2009. Conservation of core gene expression in vertebrate tissues. J Biol 8: 33. 10.1186/jbiol130
- ↵Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloğlu A, Ozen S, Sanjad S, 2009. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci 106: 19096–19101. 10.1073/pnas.0910672106
- ↵Cortez D, Marin R, Toledo-Flores D, Froidevaux L, Liechti A, Waters PD, Grützner F, Kaessmann H. 2014. Origins and functional evolution of Y chromosomes across mammals. Nature 508: 488–493. 10.1038/nature13151
- ↵Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Reghan Foley A, Bolduc V, Waddell LB, Sandaradura SA, O'Grady GL, 2017. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med 9: eaal5209. 10.1126/scitranslmed.aal5209
- ↵DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, Reich M, Winckler W, Getz G. 2012. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28: 1530–1532. 10.1093/bioinformatics/bts196
- ↵Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635
- ↵Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48. 10.1186/1471-2105-10-48
- ↵Enard W, Khaitovich P, Klose J, Zöllner S, Heissig F, Giavalisco P, Nieselt-Struwe K, Muchmore E, Varki A, Ravid R, 2002. Intra- and interspecific variation in primate gene expression patterns. Science 296: 340–343. 10.1126/science.1068996
- ↵Ferea TL, Botstein D, Brown PO, Rosenzweig RF. 1999. Systematic changes in gene expression patterns following adaptive evolution in yeast. Proc Natl Acad Sci 96: 9721–9726. 10.1073/pnas.96.17.9721
- ↵Fraser HB, Moses AM, Schadt EE. 2010. Evidence for widespread adaptive evolution of gene expression in budding yeast. Proc Natl Acad Sci 107: 2977–2982. 10.1073/pnas.0912245107
- ↵Georgi B, Voight BF, Bućan M. 2013. From mouse to human: evolutionary genomics analysis of human orthologs of essential genes. PLoS Genet 9: e1003484. 10.1371/journal.pgen.1003484
- ↵Gilad Y, Oshlack A, Rifkin SA. 2006. Natural selection on gene expression. Trends Genet 22: 456–461. 10.1016/j.tig.2006.06.002
- ↵The GTEx Consortium. 2013. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45: 580–585. 10.1038/ng.2653
- ↵Guerra R, Wang J, Grundy SM, Cohen JC. 1997. A hepatic lipase (LIPC) allele associated with high plasma concentrations of high density lipoprotein cholesterol. Proc Natl Acad Sci 94: 4532–4537. 10.1073/pnas.94.9.4532
- ↵Hansen TF. 1997. Stabilizing selection and the comparative analysis of adaptation. Evolution 51: 1341–1351. 10.1111/j.1558-5646.1997.tb01457.x
- ↵Harr B, Turner LM. 2010. Genome-wide analysis of alternative splicing evolution among Mus subspecies. Mol Ecol 19(Suppl 1): 228–239. 10.1111/j.1365-294X.2009.04490.x
- ↵Harris H. 1966. Enzyme polymorphisms in man. Proc R Soc Lond B Biol Sci 164: 298–310. 10.1098/rspb.1966.0032
- ↵Hart T, Brown KR, Sircoulomb F, Rottapel R, Moffat J. 2014. Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Mol Syst Biol 10: 733. 10.15252/msb.20145216
- ↵Jordan DM, Frangakis SG, Golzio C, Cassa CA, Kurtzberg J, Task Force for Neonatal Genomics, Davis EE, Sunyaev SR, Katsanis N. 2015. Identification of cis-suppression of human disease mutations by comparative genomics. Nature 524: 225–229. 10.1038/nature14497
- ↵Jukes TH, King JL. 1971. Deleterious mutations and neutral substitutions. Nature 231: 114–115. 10.1038/231114a0
- ↵Kalinka AT, Varga KM, Gerrard DT, Preibisch S, Corcoran DL, Jarrells J, Ohler U, Bergman CM, Tomancak P. 2010. Gene expression divergence recapitulates the developmental hourglass model. Nature 468: 811–814. 10.1038/nature09634
- ↵Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423: 241–254. 10.1038/nature01644
- ↵Kersten S. 2014. Integrated physiology and systems biology of PPARα. Mol Metab 3: 354–371. 10.1016/j.molmet.2014.02.002
- ↵Khaitovich P, Weiss G, Lachmann M, Hellmann I, Enard W, Muetzel B, Wirkner U, Ansorge W, Pääbo S. 2004. A neutral model of transcriptome evolution. PLoS Biol 2: E132. 10.1371/journal.pbio.0020132
- ↵Khaitovich P, Pääbo S, Weiss G. 2005. Toward a neutral evolutionary model of gene expression. Genetics 170: 929–939. 10.1534/genetics.104.037135
- ↵Kimura M. 1968. Evolutionary rate at the molecular level. Nature 217: 624–626. 10.1038/217624a0
- ↵King MC, Wilson AC. 1975. Evolution at two levels in humans and chimpanzees. Science 188: 107–116. 10.1126/science.1090005
- ↵Kosiol C, Vinar T, da Fonseca RR, Hubisz MJ, Bustamante CD, Nielsen R, Siepel A. 2008. Patterns of positive selection in six Mammalian genomes. PLoS Genet 4: e1000144. 10.1371/journal.pgen.1000144
- ↵Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A. 2010. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods 7: 709–715. 10.1038/nmeth.1491
- ↵Lewontin RC, Hubby JL. 1966. A molecular approach to the study of genic heterozygosity in natural populations. II. Amount of variation and degree of heterozygosity in natural populations of Drosophila pseudoobscura. Genetics 54: 595–609.
- ↵Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323. 10.1186/1471-2105-12-323
- ↵Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, 2011. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478: 476–482. 10.1038/nature10530
- ↵Merkin J, Russell C, Chen P, Burge CB. 2012. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 338: 1593–1599. 10.1126/science.1228186
- ↵Nourmohammad A, Rambeau J, Held T, Kovacova V, Berg J, Lässig M. 2017. Adaptive evolution of gene expression in Drosophila. Cell Rep 20: 1385–1395. 10.1016/j.celrep.2017.07.033
- ↵O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, 2011. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 43: 585–589. 10.1038/ng.835
- ↵Perry GH, Melsted P, Marioni JC, Wang Y, Bainer R, Pickrell JK, Michelini K, Zehr S, Yoder AD, Stephens M, 2012. Comparative RNA sequencing reveals substantial genetic variation in endangered primates. Genome Res 22: 602–610. 10.1101/gr.130468.111
- ↵Pierce VA, Crawford DL. 1997. Phylogenetic analysis of glycolytic enzyme expression. Science 276: 256–259. 10.1126/science.276.5310.256
- ↵Pipes L, Li S, Bozinoski M, Palermo R, Peng X, Blood P, Kelly S, Weiss JM, Thierry-Mieg J, Thierry-Mieg D, 2013. The non-human primate reference transcriptome resource (NHPRTR) for comparative functional genomics. Nucleic Acids Res 41: D906–D914. 10.1093/nar/gks1268
- ↵Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch R, 2006. Forces shaping the fastest evolving regions in the human genome. PLoS Genet 2: e168. 10.1371/journal.pgen.0020168
- ↵Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, 2015. ClinGen—the clinical genome resource. N Engl J Med 372: 2235–2242. 10.1056/NEJMsr1406261
- ↵Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. 10.1038/gim.2015.30
- ↵Robinson MD, Oshlack A. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11: R25. 10.1186/gb-2010-11-3-r25
- ↵Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. 10.1093/bioinformatics/btp616
- ↵Rohlfs RV, Nielsen R. 2015. Phylogenetic ANOVA: the expression variance and evolution model for quantitative trait evolution. Syst Biol 64: 695–708. 10.1093/sysbio/syv042
- ↵Rohlfs RV, Harrigan P, Nielsen R. 2014. Modeling gene expression evolution with an extended Ornstein–Uhlenbeck process accounting for within-species variation. Mol Biol Evol 31: 201–211. 10.1093/molbev/mst190
- ↵Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, Fortini ME, Li PW, Apweiler R, Fleischmann W, 2000. Comparative genomics of the eukaryotes. Science 287: 2204–2215. 10.1126/science.287.5461.2204
- ↵Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050. 10.1101/gr.3715005
- ↵Silvestro D, Kostikova A, Litsios G, Pearman PB, Salamin N. 2015. Measurement errors should always be incorporated in phylogenetic comparative analysis. Methods Ecol Evol 6: 340–346. 10.1111/2041-210X.12337
- ↵Swanson WJ, Yang Z, Wolfner MF, Aquadro CF. 2001. Positive Darwinian selection drives the evolution of several female reproductive proteins in mammals. Proc Natl Acad Sci 98: 2509–2514. 10.1073/pnas.051605998
- ↵Torgerson DG, Kulathinal RJ, Singh RS. 2002. Mammalian sperm proteins are rapidly evolving: evidence of positive selection in functionally diverse genes. Mol Biol Evol 19: 1973–1980. 10.1093/oxfordjournals.molbev.a004021
- ↵von Scheidt M, Zhao Y, Kurt Z, Pan C, Zeng L, Yang X, Schunkert H, Lusis AJ. 2017. Applications and limitations of mouse models for understanding human atherosclerosis. Cell Metab 25: 248–261. 10.1016/j.cmet.2016.11.001
- ↵Wang D, Marsh JL, Ayala FJ. 1996. Evolutionary changes in the expression pattern of a developmentally essential gene in three Drosophila species. Proc Natl Acad Sci 93: 7103–7107. 10.1073/pnas.93.14.7103
- ↵Wong ES, Thybert D, Schmitt BM, Stefflova K, Odom DT, Flicek P. 2015. Decoupling of evolutionary changes in transcription factor binding and gene expression in mammals. Genome Res 25: 167–178. 10.1101/gr.177840.114
- ↵Yanai I, Graur D, Ophir R. 2004. Incongruent expression profiles between human and mouse orthologous genes suggest widespread neutral evolution of transcription control. OMICS 8: 15–24. 10.1089/153623104773547462
- ↵Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, 2014. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515: 355–364. 10.1038/nature13992