
Abstract
Recent proliferation of low-cost DNA sequencing techniques will soon lead to an explosive growth in the number of sequenced genomes and will turn manual annotations into a luxury. Mass spectrometry recently emerged as a valuable technique for proteogenomic annotations that improves on the state-of-the-art in predicting genes and other features. However, previous proteogenomic approaches were limited to a single genome and did not take advantage of analyzing mass spectrometry data from multiple genomes at once. We show that such a comparative proteogenomics approach (like comparative genomics) allows one to address the problems that remained beyond the reach of the traditional “single proteome” approach in mass spectrometry. In particular, we show how comparative proteogenomics addresses the notoriously difficult problem of “one-hit-wonders” in proteomics, improves on the existing gene prediction tools in genomics, and allows identification of rare post-translational modifications. We therefore argue that complementing DNA sequencing projects by comparative proteogenomics projects can be a viable approach to improve both genomic and proteomic annotations.
Since the sequencing of the first genome, Haemophilus influenzae in 1995 (Fleischmann et al. 1995), the number of sequenced genomes has been rising sharply. Every sequencing project is followed by annotation of the genome to identify genes, pathways, etc. Comparative genomics analysis of multiple genomes has emerged as one of the key approaches for discovery of such genomic elements that greatly improves on the existing annotation tools (Batzoglou et al. 2000; Kellis et al. 2003; Xie et al. 2005). Another recent development is the application of tandem mass spectrometry (MS/MS) for genomic annotations (Jaffe et al. 2004; Kalume et al. 2005; Wang et al. 2005; Fermin et al. 2006; Gupta et al. 2007; Tanner et al. 2007). Such proteogenomic approaches further improve gene predictions and allow one to address problems that remained beyond the reach of both traditional gene prediction tools and comparative genomics.
We recently developed MS-Genome software for automated proteogenomic annotation of bacterial genomes (Gupta et al. 2007) and applied it for improving annotation of Shewanella oneidensis MR-1, a model bacterium for studies of bioremediation and metal reduction. However, the synergy between MS/MS data from different species was never explored in the past. We show that such comparative proteogenomics analysis sheds new light on the annotations of both genomes and proteomes.
Similar to Expressed Sequence Tags (EST) studies, mass spectrometry experiments generate Expressed Protein Tags (EPT) that provide valuable information about expressed proteins. However, while there are hundreds of studies on using ESTs for genome annotation, EPT studies are still in infancy (Savidor et al. 2006). This is unfortunate since EPTs may provide some advantages over ESTs and are easy to generate. In particular, unlike ESTs, EPTs are relatively uniformly distributed along the protein length and provide information about the translational starts, proteolytic events (e.g., signal peptides), and post-translational modifications (PTMs). Also, EPTs may be less affected by splicing artifacts (like trans-splicing) and sequencing errors. However, some EPTs may represent errors in peptide identifications (and are thus completely wrong), making it nontrivial to transform the existing EST approaches into the EPT domain.
While recent high-throughput MS/MS studies generated large spectral data sets for many related species, it remains unclear how to utilize these data sets across various genomes. In this study, we analyze MS/MS data sets for three Shewanella bacteria representing multiple growth conditions: Shewanella oneidensis MR-1 (∼14.5 million spectra), Shewanella frigidimarina (∼0.955 million spectra), and Shewanella putrefaciens CN-32 (∼0.768 million spectra). These data sets provide an opportunity to analyze the expressed proteomes across these bacteria (henceforth referred to as So, Sf, and Sp, respectively). In addition to predicting new genes and finding errors in existing annotations, we show that MS/MS data help to identify programmed frameshifts (as well as sequencing errors), a difficult problem in genomics. We demonstrate that comparative analysis of peptides across species is helpful in resolving the dilemma of “one-hit-wonders” in proteomics. We further discuss how comparative proteogenomic analysis enables identification of rare PTMs and proteolytic events, two difficult problems for which the high-throughput techniques are not available. Drawing parallels from gene microarray platforms, we also use mass spectrometry-based protein expression data to analyze the conserved and differentially expressed pathways across these species. Our software is available at http://proteomics.bioprojects.org/ and the proteomic data sets are available from http://ober-proteomics.pnl.gov/data.
Results
Multiple Shewanella genomes
The three Shewanella species used in this study were recently sequenced, So containing 5,131,416 base pairs (bp) being the first one (Heidelberg et al. 2002). Subsequently, Sf and Sp genomes have been sequenced (4,845,257 and 4,649,325 bp, respectively). Sf and Sp genomes, unlike So, do not have accompanying publications in the literature, although they have been cited in other studies (Yang et al. 2006). The genome sequences and annotations used in this study were obtained from the TIGR CMR database.
The protein orthology assignments across different Shewanella species were prepared using INPARANOID (Remm et al. 2001), subsequently aligned by MUSCLE (Edgar 2004) (data courtesy of LeeAnn McCue and Sean Conlan). Figure 1A shows the numbers of orthologs shared by different Shewanella species. While 2590 genes have orthologs in all three species (we call such triplets “shared genes”), for some proteins, orthologs were found in only one other species and, in many cases (for example, 1715 in So), in none. (Many Shewanella genes may be artifacts of existing gene finding tools that tend to overpredict short genes. See Clamp et al. (2007) regarding recent controversy on gene overprediction.)
Expression of orthologous genes across the three species. (A) The number of orthologs shared between different species. There are 2590 orthologous genes present in all three species (referred to as “shared genes”). (B) The number of expressed shared genes (confirmed by two or more peptides) among the three species; 1052 shared genes are expressed in all three species, 708 shared genes are expressed in none.
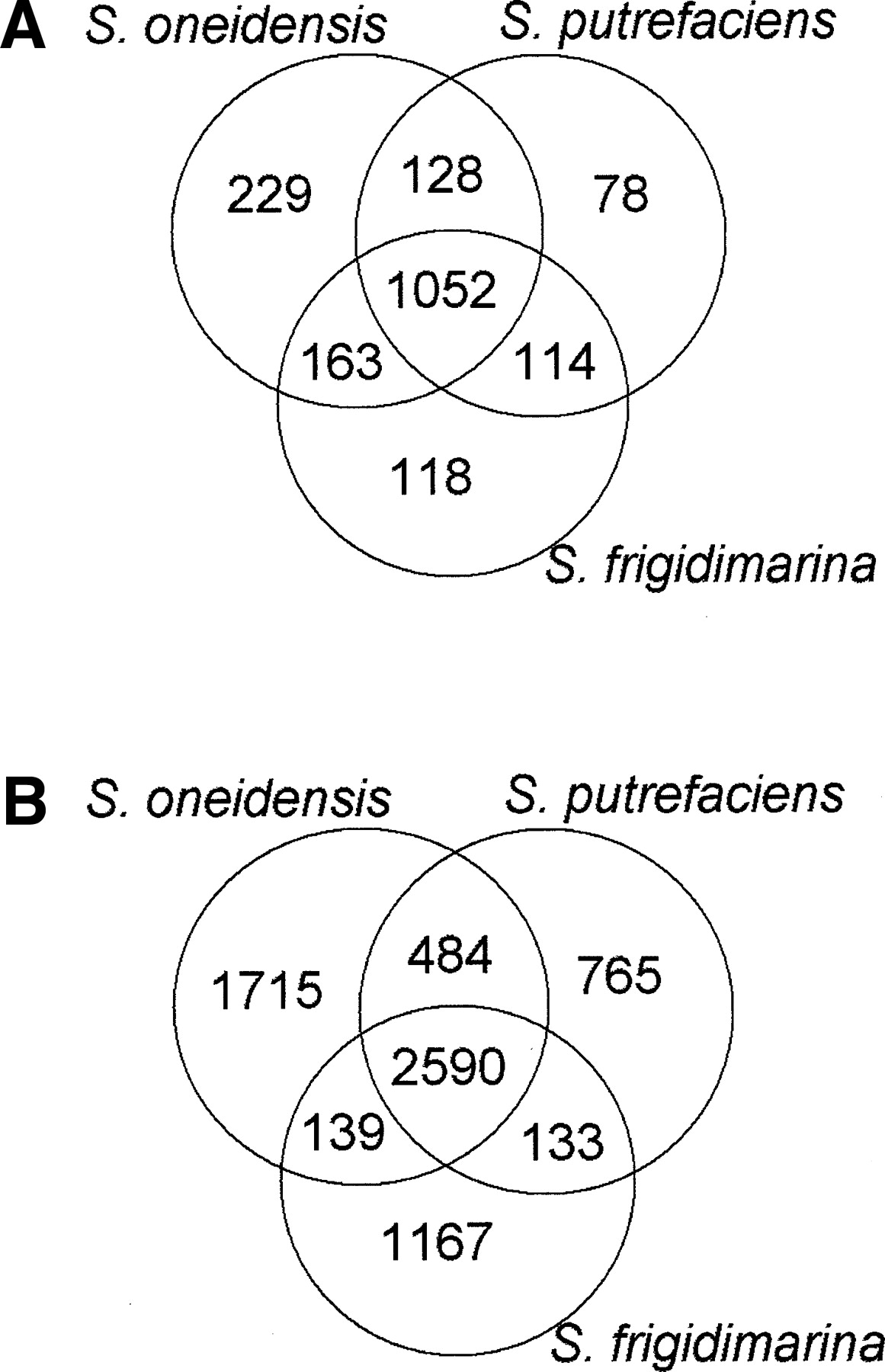
The shared genes are used for comparative analysis in this study. The protein sequence identity between So and Sp is ∼85%, while Sf is ∼70% identical to the other two species (average among all shared genes). As a result, most orthologous tryptic peptides for these species differ in at least one position.
Protein identification
Based on the peptides identified from InsPecT searches (see Methods), expression of 40%–45% proteins is confirmed in each species. Table 1 provides the number of annotated genes and our protein identifications. Interestingly, the fraction of expressed proteins among the shared genes is much higher, at ∼55%. This hints at a correlation between protein expression and sequence conservation, in agreement with the observations made in Gupta et al. (2007). In this study, we also demonstrated the use of MS-based protein identification to analyze the expression of pathways or functional categories. Having proteomic data for three species now allows us to compare the expression of pathways and identify which pathways are conserved or differentially expressed across these species. The comparative pathway analysis is described in the Supplemental material (Supplement 7).
Protein identification results
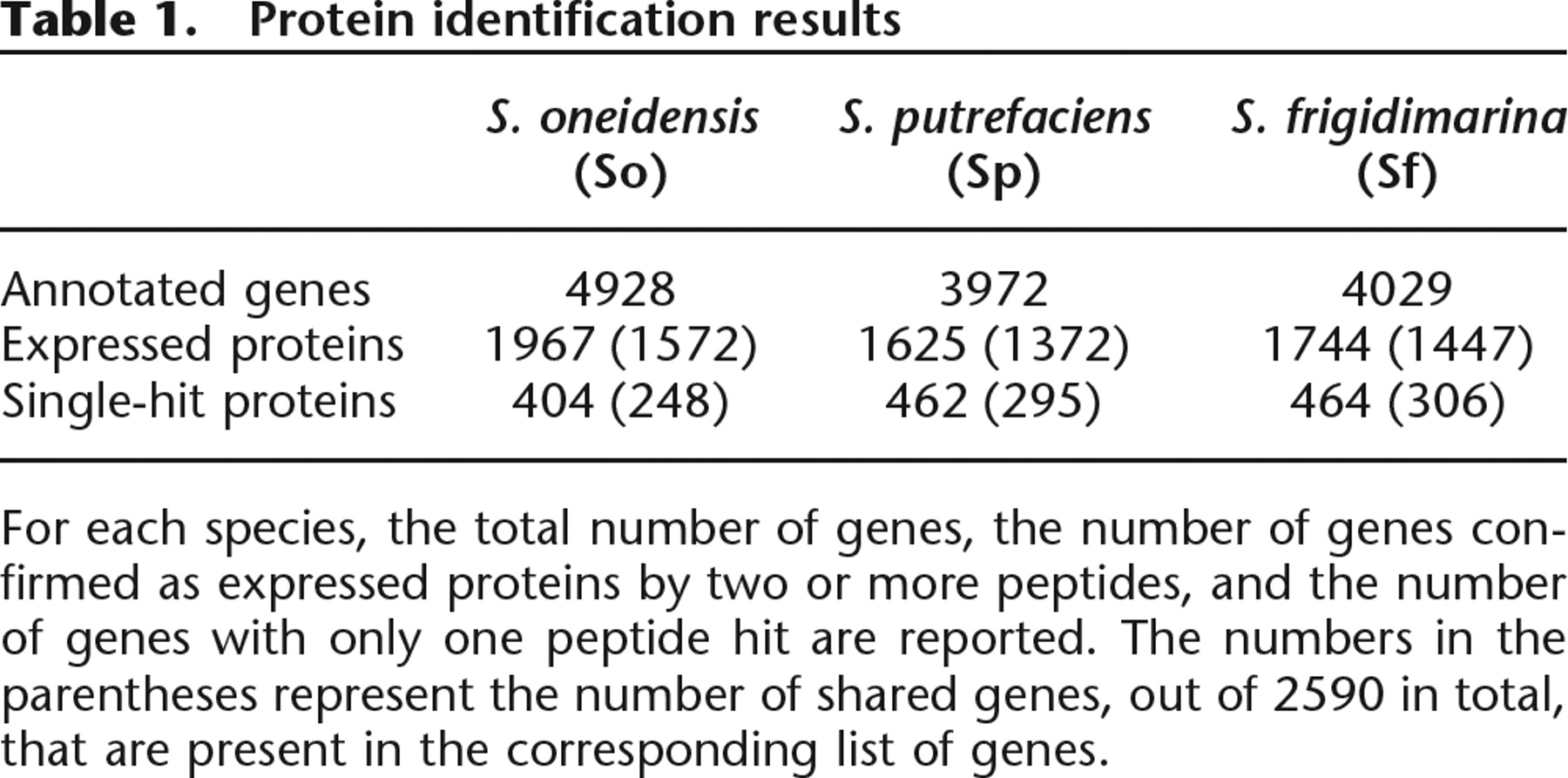
For each species, the total number of genes, the number of genes confirmed as expressed proteins by two or more peptides, and the number of genes with only one peptide hit are reported. The numbers in the parentheses represent the number of shared genes, out of 2590 in total, that are present in the corresponding list of genes.
Resolving one-hit-wonders
There are 1052 shared genes that are expressed in all three species (see Fig. 1B). However, in accordance with the Proteomics Publication Guidelines (Carr et al. 2004; Bradshaw et al. 2006), we require at least two peptides to consider a protein as expressed. Since almost every analysis of MS/MS data sets reveals a large number of proteins with a single identified peptide (one-hit-wonders), it leads to a significant reduction in the number of identified proteins (one-hit-wonders represent 21%, 28%, and 27% of all identified proteins in So, Sp, and Sf, respectively). For example, there are 404 such proteins in So that cannot be reported as reliable identifications. While many of them indeed represent expressed proteins, it is not clear how to separate them from erroneous peptide identifications (Gupta et al. 2007). Below we explore the use of comparative analysis across species to reliably select the expressed proteins among the one-hit-wonders and thus remove the term “hypothetical” from some existing gene annotations.
For each shared gene, we define an expression signature with three values that represent the number of peptide identifications in the three species. The value is 2 if the expression is confirmed by two or more peptides, 1 if only one peptide is observed, and 0 for no peptides. For example, the signature (0, 1, 2) for a shared gene represents no peptide identification in So, one peptide identification in Sp, and confirmed expression with two or more peptides in Sf. There are 27 possible distinct expression signatures that such a vector may take for a shared gene. We combine these into 10 position independent values, such that (2, 1, 1) is considered the same as (1, 1, 2) or (1, 2, 1). Table 2 shows the frequency of these 10 expression signatures among the 2590 shared genes. The argument against considering one-hit-wonders as expressed protein is that they may be unexpressed proteins with one false peptide identification. However, we note that, if the orthologous genes of a one-hit-wonder are expressed in the other two species, it adds support that the gene is a true expressed gene. Such genes are readily identified as having expression signature (1, 1, 1), (1, 1, 2), or (1, 2, 2). This approach provides extra evidence for the expression of 3 × 10 + 2 × 56 + 187 = 329 one-hit-wonders in total in the three species. [The signatures (0, 1, 1), (0, 1, 2), and (0, 2, 2) are also useful, albeit less reliable (they may represent biologically interesting cases when orthologous proteins are expressed in some species but not expressed in others).]
Expression signatures for shared genes
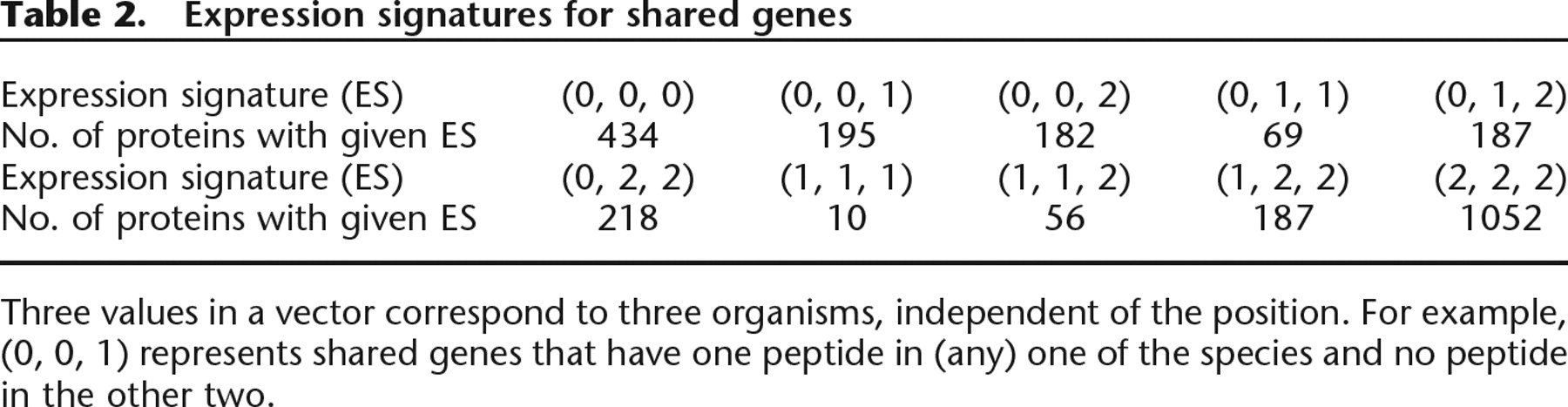
Three values in a vector correspond to three organisms, independent of the position. For example, (0, 0, 1) represents shared genes that have one peptide in (any) one of the species and no peptide in the other two.
While orthologous one-hit-wonders are strong indicators of protein expression, peptides identified at the same orthologous positions (correlated peptides) in different species provide overwhelming evidence that the proteins are expressed (see Methods for description of correlated peptides). Since the likelihood of this happening by chance is extremely small, we now dig deeper into analysis of the orthologous one-hit-wonders and demonstrate that they often have correlated peptides. Figure 2 shows the example of a shared gene (annotated as hypothetical lipoprotein) that has only one identified peptide in each organism. However, it turns out that these peptides, in spite of being slightly different from each other in their sequences, are located at the same position in the alignment of the orthologs. Thus, we argue that these proteins should be considered as expressed and re-annotated to remove the term “hypothetical” from their annotations.
Example of correlated one-hit-wonders in shared genes. Aligned amino acid sequences of the shared gene (annotated as hypothetical lipoprotein) are shown for each organism (SO_0515 in So, CN32_3345 in Sp, and Sfri_3590 in Sf). The identified peptides are shown in blue.

One reason for observing only a single peptide from a protein is the relatively few number (one in some cases) of detectable peptides in a protein (Supplement S4 in the Supplemental material describes how mutations in correlated peptides provide valuable data for studies of peptide detectability) (Tang et al. 2006; Lu et al. 2007; Mallick et al. 2007). However, if this is the case, the orthologous peptides should be observed in the closely related species. We thus check if the only peptide observed in a protein is correlated between multiple species. If the peptide identification is spurious, it is very unlikely that the peptide will be at the same position as the observed peptides in its orthologs. Interestingly, we find 46 out of 404 one-hit-wonders in So having a correlated peptide in at least one of the other two species, providing strong evidence for the expression of these proteins. Similarly, 50 and 85 one-hit-wonders in Sf and Sp, respectively, can be resolved as expressed based on correlated peptides. We note that, if the peptide identifying a one-hit-wonder is an incorrect identification, and the orthologous peptides identified in the other species are exactly the same as the one-hit-wonder peptide, they may also represent incorrect identifications of similar mass spectra (e.g., spectra from unknown contaminants). Thus, the correlated peptides are less reliable if they are identical. However, even a single change in the peptide sequences significantly changes the corresponding spectra and, therefore, the one-hit-wonder confirmations based on such distinct peptides are reliable. Noticeably, 38, 47, and 70 one-hit-wonders in So, Sf, and Sp, respectively, confirmed by correlated peptides, belong to this category.
Correcting gene predictions: Start sites
Peptides that match the genome in the non-protein-coding region upstream of a gene, within 200-bp distance, are considered candidates for early start sites. These are cases of misannotated genes that are shortened at their N terminus. Cases with stop codons between the peptide and the gene start site are discarded. To avoid spurious candidates from incorrect peptide identifications, we consider a peptide only if there is another identified peptide in the same reading frame within 200 bp (Gupta et al. 2007). The starting position of the peptide (call it position X) does not necessarily correspond to the actual start site of the gene, but only tells that the actual start should be further upstream of X.
To verify early start sites and determine their exact positions, these genes were searched against proteins in 10 other Shewanella species, and position X for each candidate was compared to the start site of the aligned homolog. These species included Shewanella loihica PV-4, S. baltica OS155, S. amazonensis SB2B, S. sp. W3-18-1, S. denitrificans OS217, S. sp. ANA-3, S. sp. MR-4, and S. sp. MR-7, besides the other two from So, Sf, and Sp (leaving the one to which the candidate gene belongs). If the start site of homolog aligned to a particular position is equal to or upstream of position X, then this new position was considered to be a putative early start site. The most frequent (supported by maximum number of homologs) of these putative starts is chosen as the new start site for the gene.
The list of early start site candidates is provided in Supplemental Table S2A. Twenty-three among 28 such candidates in So are assigned new start sites based on the comparative analysis mentioned above. Notably, 18 of these early start sites have the expected ATG, GTG, or TTG start codons, indicating that these automatically predicted start sites are indeed reliable. Two and three early start sites are identified in Sp and Sf, respectively.
As described in Methods, candidates for late start sites were generated using evidence from noncovered peptides. Such instances indicated a potential late start site either at the beginning of the noncovered peptide (call it position X) or, if N-terminal cleavage occurred, one position upstream (X − 1). The sequences of these candidate genes are aligned to the proteins in 10 other Shewanella species. Each instance where the start of a protein in the other species aligns to the potential late start site (beginning at position X or X − 1) is considered as confirmed by comparative genomics.
Supplemental Table S2B summarizes these cases in each of the three organisms. In So, five out of 33 late start candidates are confirmed, four of which start with ATG codon and one with GTG (supporting the hypothesis that these are indeed start sites). Similarly, 11 out of 16 candidates are confirmed in Sf, and four among the 11 are confirmed in Sp (all of these are also found to have ATG, GTG, or TTG start codon). The table also shows that the majority of these candidates have N-terminal methionine cleavage in the observed peptide. We find comparative proteomic evidence for one case where the late start site (10 amino acids downstream from the annotated start site) is conserved in the orthologs (ATP-dependent Clp protease, proteolytic subunit ClpP) between So (SO_1794), Sf (Sfri_2596), and Sp (CN32_1490). However, we note that this site is also found in our analysis of conserved proteolytic sites (below). While it is unclear whether this peptide corresponds to the late start site or a proteolytic event, it clearly represents a real non-tryptic peptide, as opposed to an incorrect identification.
We note that our approach assumes that a gene has only one translational start site. However, if there is a gene with alternative start sites, we will detect only the most upstream start site that has supporting peptide evidence. We also discuss an approach to detect novel short genes using comparative proteogenomic analysis in the Supplemental material (Supplement S6).
Identification of programmed frameshifts and sequencing errors
A frameshift occurs when a ribosome skips one or more nucleotides in an mRNA sequence, thereby changing the reading frame to produce a different protein sequence from the original frame. In programmed frameshifts, this phenomenon is built into the translational machinery (Farabaugh 1996). Secondary RNA structures such as pseudoknots are often responsible for the ribosomal pause and resulting frameshift (Tu et al. 1992). While many efforts went into frameshift detection (Posfai and Roberts 1992; Claverie 1993; Fichant and Quentin 1995; Brown et al. 1998; Medigue et al. 1999), accurate detection of frameshifts remains an unsolved problem. Mass spectrometry, on the other hand, provides experimental evidence for the actual translation products (proteins) and allows one to detect the frameshifts. The presence of peptides from two different reading frames within the region of a predicted gene may represent: (1) incorrect peptide identification, (2) an insertion/deletion sequencing error, (3) overlapping genes in different frames, or (4) a programmed frameshift. We demonstrate the application of comparative approaches for distinguishing between these possibilities.
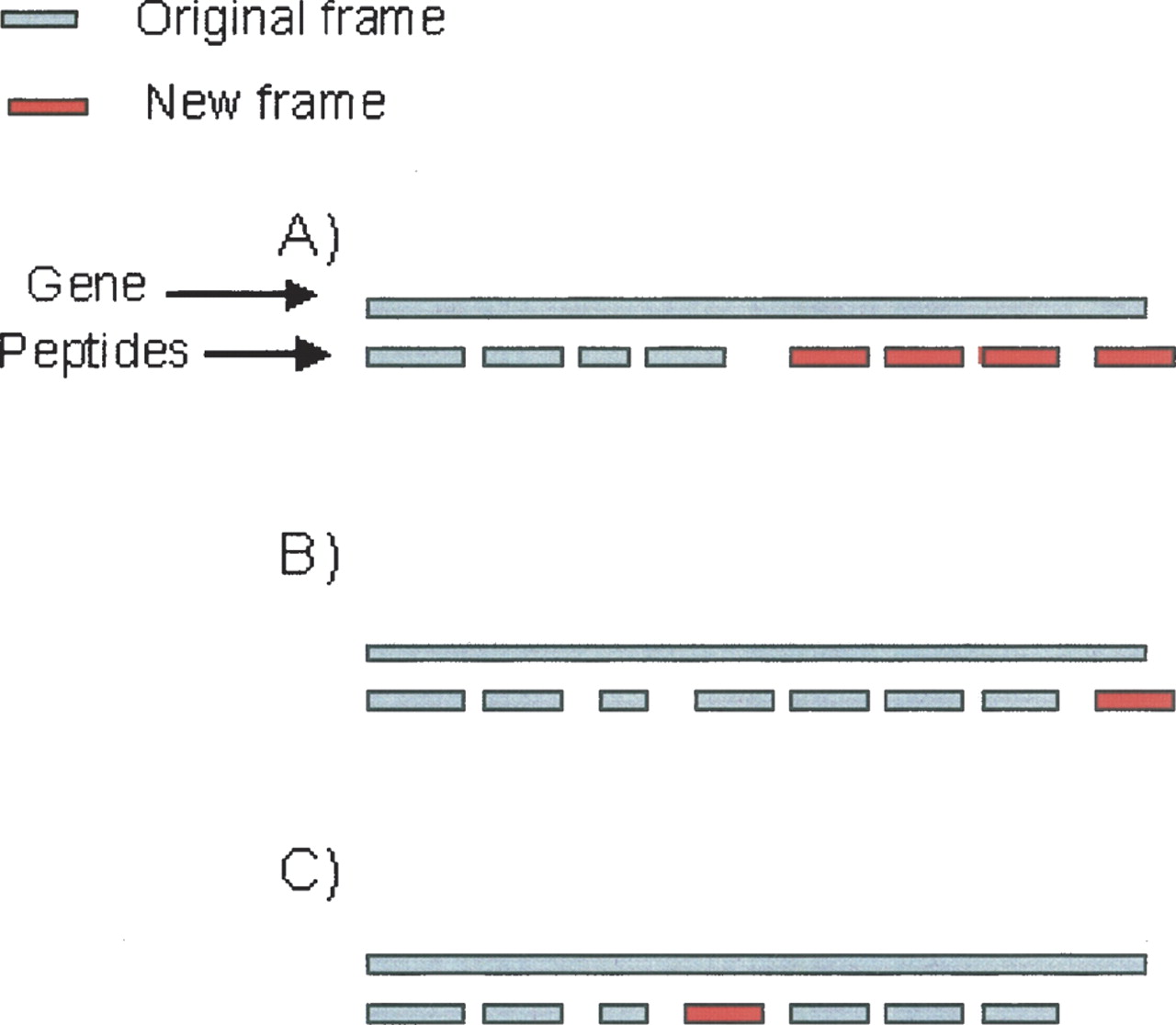
All identified peptides are mapped to the translated frames of the genome and compared with the annotated gene coordinates to determine alternate peptide reading frames in the DNA region of a single gene. As depicted in Figure 3, three types of cases are typically seen. In case A, multiple peptides are observed in two different frames (only one of them being the annotated frame of the gene) in nonoverlapping regions. In case B, only one peptide is observed in an alternative frame at one of the ends, while, in case C, one peptide is seen out of frame with in-frame peptides on both sides. We postpone the discussion of case C since in this case incorrect peptide identifications or overlapping genes are more likely explanations than a frameshift. Case A provides the most reliable evidence of a programmed frameshift since presence of multiple peptides in the same region greatly reduces the probability that these peptide identifications are spurious. The remaining case B, with only one peptide, is ambiguous and may represent either frameshifts or incorrect peptide identifications, or overlapping genes. We exploit the sequences of multiple Shewanella species to find comparative evidence for putative frameshifts in these cases.
Commonly observed configurations of peptides in alternative frame. (A) Case A: Multiple peptides are observed in two different frames (one of them being the frame of the gene) in nonoverlapping regions. (B) Case B: Only one peptide is observed out of frame at one of the ends. (C) Case C: One peptide is seen out of frame with in-frame peptides on both sides.
Protein sequence from the original frame of the gene, as well as sequence from the alternate frame implied by the identified peptides, is compared against the other Shewanella species using BLAST (Altschul et al. 1997). Good matches to the alternate-frame sequence and no matches to the gene-frame sequence provide additional evidence for a frameshift. We note that some apparent frameshifts may be caused by sequencing errors or indels in the genome sequence when a certain number (not a multiple of 3) of bases are erroneously added to or deleted from the sequence. To identify such sequencing errors, we take the nucleotide sequence of the region where frameshift occurs (region between the observed in-frame and alternate-frame peptides) and generate ClustalW (Chenna et al. 2003) multiple sequence alignment with the orthologous region in the other species. A sequencing error is visible in this alignment as an indel in the original sequence (see Fig. 4). Figure 5 shows an example of a programmed frameshift detected through this approach.
Frameshift generated by sequencing error. In top panel, the nucleotide sequence for gene SO0590 is shown in red, the amino acid sequence of the protein is shown in green, and the amino acid sequences of the three translated frames are shown in black. Peptides identified by mass spectrometry are marked in blue (surrounded by boxes). The middle panel shows the ClustalW alignment with other Shewanella species in the region where frameshift occurs. The erroneous insertion of an extra “t” stands out in the alignment. The bottom panel indicates that both peptides fall in the original frame if the extra nucleotide is removed.

An example of a programmed frameshift. The nucleotide sequence for gene SO_0991 is shown in red, the amino acid sequence of the corresponding protein is shown in green, and the amino acid sequences of the three translated frames are shown in black. This gene has been correctly annotated in TIGR, and our predicted peptides in both the original frame and the alternative frame match the protein sequence.

We identified 12 frameshift candidates in So conforming to case A (Supplemental Table S3). All these candidate frameshifts were verified with significant E-values. Nine of these instances are estimated to be sequencing errors, and three genes are putative programmed frameshifts: SO0991 (+1), SO4538 (−1), and SO4115 (−1). SO0991 (Fig. 5) is related to the peptide chain release factor 2 in Escherichia coli, that is known to undergo a programmed frameshift (Craigen et al. 1985). Fifteen frameshift candidates were identified conforming to case B but not verified by comparative evidence. No frameshift candidates could be verified in Sp or Sf. This may be attributed to the relatively small number of spectra for these two species (less than a million spectra each) as compared to 14.5 million spectra for So.
Proteolytic events
In Gupta et al. (2007), we demonstrated the use of genome scale MS/MS data set for identification of N-terminal proteolytic events such as N-terminal methionine cleavage and signal peptide cleavage. An in vivo proteolytic event can be observed as a non-tryptic peptide (assuming the proteolytic enzyme does not have the same specificity as trypsin). However, non-tryptic peptides may also be observed due to other reasons, such as degradation of tryptic peptides or incorrect peptide identifications. In Rodriguez et al. (2008), we showed that the likelihood of incorrect peptide identifications can be reduced drastically (to <0.1%) by considering only doubly confirmed cleavages and filtering out possible degradation products (Rodriguez et al. 2008).
By applying the same filtering approach as in Rodriguez et al. (2008) and removing the cuts explained by the trypsin specificity, we obtain 365, 130, and 62 putative proteolytic sites in So, Sp, and Sf, respectively. To check whether some of these sites are conserved between multiple organisms, we map them on the alignment of orthologous protein. Thirty-one proteolytic sites are found conserved between two or more organisms (see Table 3). This is a significantly larger number of conserved sites than expected by chance. For example, with proteomes of length ∼1 million amino acids (aa) each, the expected number of sites conserved by chance between Sp and Sf is less than (62/106) × (130/106) × 106 ≈ 0.01, but we observe 13. One may further challenge that these cleavages may be an artifact of in vitro peptide degradations, and that these peptides may be overrepresented in proteins containing multiple peptides. In this case, the statistical argument above must be applied to the set of these highly expressed proteins rather than to all proteins. To check this, we took proteins with 10 or more peptides (635 proteins in Sp, 671 in Sf) with total length close to 300,000 aa in each organism, and 128 and 57 putative proteolytic sites in Sp and Sf, respectively. All 13 sites conserved between Sp and Sf belongs to these highly expressed proteins. The expected number of sites conserved by chance in these proteins is (128/300000) × (57/300000) × 300,000 ≈ 0.02, still much smaller than the observed 13 sites. Thus, we argue that the conserved sites reported here cannot be results of nonspecific degradations.
List of conserved proteolytic sites
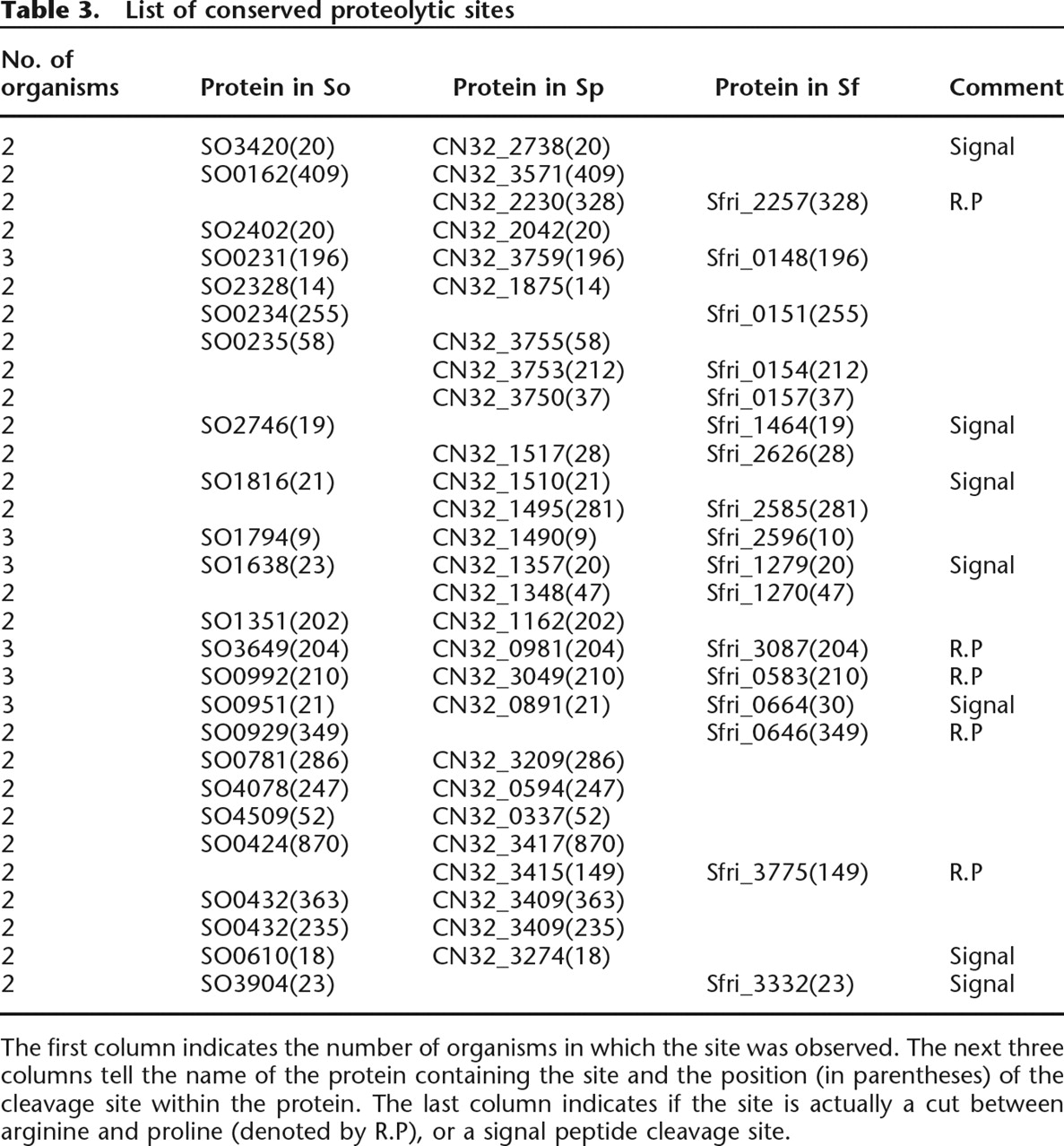
The first column indicates the number of organisms in which the site was observed. The next three columns tell the name of the protein containing the site and the position (in parentheses) of the cleavage site within the protein. The last column indicates if the site is actually a cut between arginine and proline (denoted by R.P), or a signal peptide cleavage site.
We note that many of these sites are located within peptide ladders (multiple overlapping peptides), which also raises the possibility that these cleavage sites may be a result of peptide degradation (see the example in Fig. 6). However, carefully looking into these ladders, we see that they are more likely a union of two peptide ladders, one coming from the proteolysed and the other from the unproteolysed protein product. This is supported by high spectral counts for the peptides around the cleavage site in many cases, given that one expects much lower spectral counts (usually 1) for degraded peptides as compared to the tryptic (un-degraded) peptide in a ladder. For example, the peptide LVNTGWTGGPHGIGK that supports the predicted cleavage site in Figure 6 has a spectral count of 98, even higher than the spectral counts of the covering tryptic peptides. Based on this and the statistical evidence shown above, we expect that our conserved cleavage sites represent in vivo proteolytic events. Since the knowledge of proteolytic events in bacteria is still very limited at genomic scale, we are not able to provide additional supporting information about the origin or relevance of each predicted site individually; but we make the data available for comparison with future studies. In the Supplemental material Supplement S5 provides peptides ladders for all the 31 identified sites (see Instructions.txt in the Supplemental material for details).
A cleavage site located within a peptide ladder. The first line shows a section of the protein SO_0162 (residues 399–432) with the cleavage site between Y and L marked by a downward arrow. The subsequent lines show the identified peptides along with their spectral counts in the parentheses.
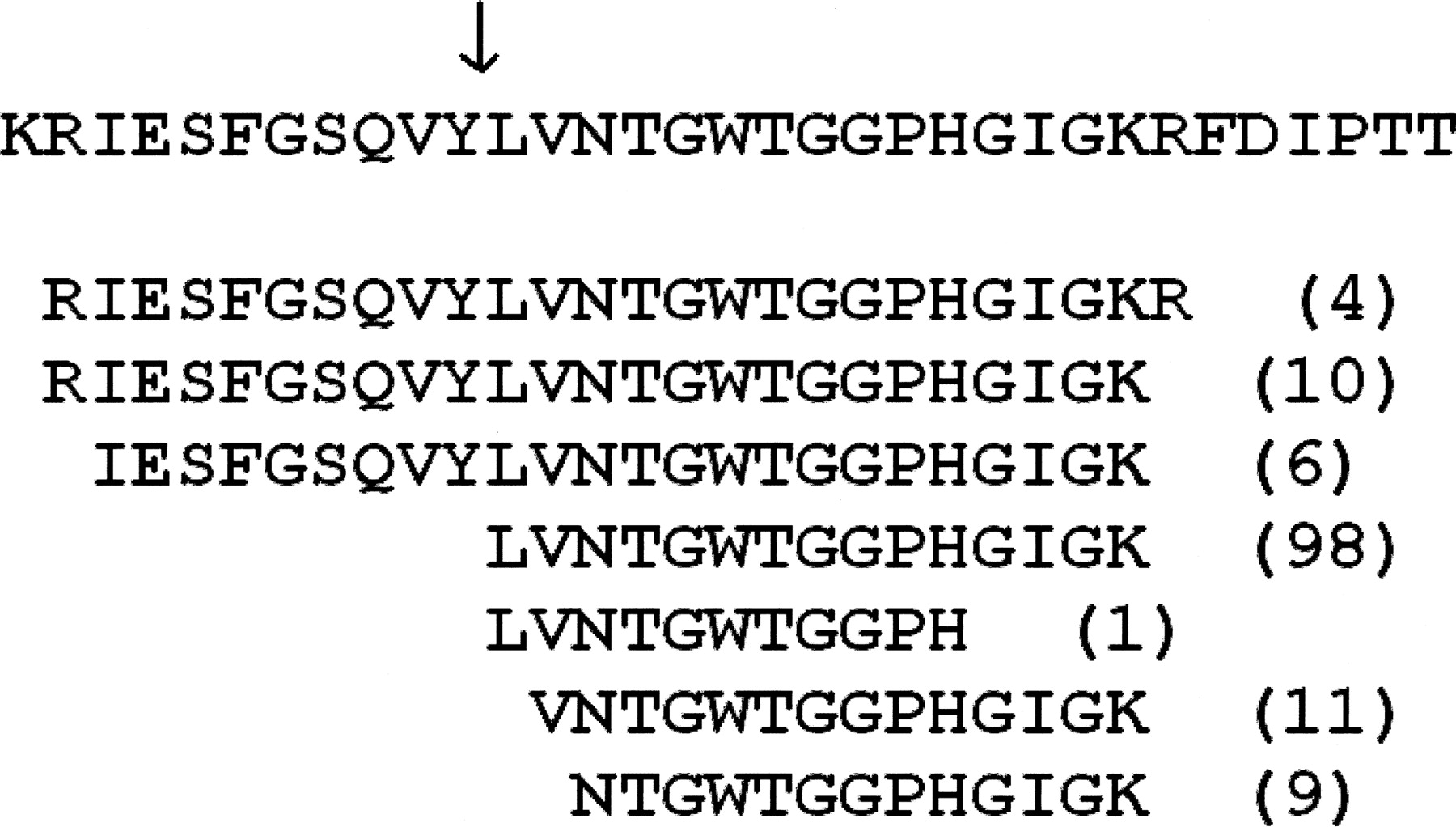
Note that here we used the traditional rules for trypsin specificity, allowing a cut after arginine or lysine but not before proline. Interestingly, five of the 31 conserved sites happen to be cuts between arginine and proline, indicating that these may be a result of trypsin digestion, further supporting the conclusion in Rodriguez et al. (2008) that the cuts after arginine and lysine followed by a proline should be considered tryptic. The other seven sites are signal peptide cleavages also predicted by SignalP (Bendtsen et al. 2004), providing additional support that our detected sites represent proteolytic events rather than statistical artifacts.
Post-translational modifications
Diphthamide is an extremely rare histidine modification that appears on a single gene (translation elongation factor 2) in the entire human genome (Moehring et al. 1980; Van Ness et al. 1980; Liu et al. 2004). Diphthamide is a target of diphtheria toxin and its position is conserved over a billion years of evolution (from yeast to human). However, systematic identification of new important and rare modifications remains a difficult, if not impossible, problem in shotgun proteomics experiments. While algorithms for blind searches for unexpected modifications have been developed (e.g., MS-Alignment) (Tsur et al. 2005), (ModifiComb) (Savitski et al. 2006), they had to rely on the “strength in numbers” principle to distinguish real modifications from computational artifacts. As a result, the biologically important modifications that appear only a few times in the genome are likely to be classified as computational artifacts. For example, each of the 25 most common modifications in So appears on at least 39 sites in the genome (Gupta et al. 2007), pushing rare modifications to the twilight zone of the statistical significance. Below we show that comparative proteogenomics allows one to identify putative rare modifications in shotgun proteomics experiments.6
In this section, we use the term post-translational modification (PTM) to denote chemical modifications of individual residues, such as phosphorylation, oxidation, methylation, etc. (Mass spectrometry experiments reveal both in vivo and in vitro modifications [chemical adducts]). Blind PTM searches with MS-Alignment (Tsur et al. 2005) or ModifiComb (Savitski et al. 2006) find all possible mass offsets (revealing potential modifications) without a priori knowledge of which modifications may be present in the sample. The first applications of these tools revealed that the world of modifications is much larger than previously thought (Nielsen et al. 2006; Wilmarth et al. 2006) and, at the same time, emphasized the still unsolved problem of finding rare modifications. Since blind searches may yield thousands of modifications (Gupta et al. 2007), the “strength in numbers” approach (Tsur et al. 2005) considers frequent modifications (e.g., offset +16 on M) as reliable and discards rare modifications as unreliable. A comparative version of this approach would be to identify modifications that are seen in multiple samples. After the post-processing of MS-Alignment results as described in Methods, we find 162 distinct modifications that are observed in all three species. While 74 of these represent chemical adducts that are expected in mass spectrometry experiments, 88 others reveal biologically interesting modifications as well as other potentially important modifications that remain unknown. The list of these modifications is provided in Supplemental Table S8A.
The strength in numbers approach, while successful, leaves many rare modifications unexplained. These modifications may represent either rare and biologically important modifications or incorrect peptide identifications. However, it is very unlikely to find a modification at the same site in orthologous genes in two different species just by chance (especially if the peptides are not identical). We find 48 such modifications that are conserved at one or more sites in the genome. For example, 48 on W are found to be conserved at three different sites. At two of these sites, the peptides covering the orthologous modification position are not identical, virtually eliminating the possibility of incorrect identifications. The list of these conserved modifications, along with the corresponding peptides is provided in Supplemental Table S8B. Most of these modifications are previously unknown, providing a refined set of candidates for experimental validations. (Experimental validation of these modifications requires chemical synthesis and remains beyond the scope of this paper.) While PTMs must be important in the metal-reducing Shewanella species, studies of modifications in Shewanella are still in infancy (Thompson et al. 2008). Although there are currently no reported experimental studies that can be used for verification of our comparative proteogenomic predictions, we hope that our analysis provides sufficient evidence to warrant some experimental verifications. Note that we cannot claim the biological significance of identified modifications; they could be either in vivo PTMs or in vitro chemical adducts, although the low-frequency modifications are less likely to be conserved if they are introduced in vitro after digestions.7
Discussion
Shewanella oneidensis MR-1 is among the most carefully annotated bacterial genomes: Gene predictions in this genome were studied in two papers (Nealson et al. 2002; Daraselia et al. 2003) and are being continuously improved by the Shewanella Federation (http://www.shewanella.org/). Significant manual effort (that took into account comparative genomics evidence) also went into the annotation of Shewanella frigidimarina and Shewanella putrefaciens CN-32. We demonstrate that comparative proteogenomics approach leads to improved annotations even for these well-studied genomes, let alone for genomes with only automated annotations available. Recent proliferation of low-cost DNA sequencing techniques will soon lead to an explosive growth in the number of sequenced genomes and will turn manual annotations into a luxury that can be afforded for only a small fraction of newly sequenced genomes. We therefore suggest that complementing DNA sequencing projects by comparative proteogenomics projects can be a viable alternative approach to improve both genomic and proteomic annotations. Below we briefly outline some other applications of comparative proteogenomics that remained beyond the scope of this paper. They refer to the biological phenomena that elude both DNA-based and MS-based “single species” analysis but become tractable with comparative proteogenomics approach.
“RNA editing” is difficult to confirm by MS-based analysis of a single genome since amino acid mutations can also be explained by DNA sequencing errors or false peptide identifications. While mass spectrometry is routinely used for confirming RNA editing events in a case-by-case fashion (Kang et al. 2005), it was never used for genome-wide discovery of RNA editing. For example, Whitelegge and colleagues used mass spectrometry to find putative RNA editing sites in plant chloroplasts but remarked that additional evidence is needed to distinguish them from DNA sequencing errors and mass spectrometry artifacts (Whitelegge et al. 2002). The comparative proteogenomics analysis of related species would be a simple way to rule out such alternative explanations and to confirm RNA editing.
“N-terminal Methionine Excision” (NME) is the process of cleaving N-terminal methionine residue that has important implications for protein half-life, food safety, and infectious diagnostics (Tobias et al. 1991; Demirev et al. 2001). The recognition rules for NME remain elusive, rendering the production of recombinant proteins of therapeutic interest risky. (This problem was originally encountered in the production of human hemoglobin [Olson et al. 1981; Ben-Bassat et al. 1987]). The key challenge for deciphering the NME code (and important exceptions from the commonly used simplistic rules) is generating large data sets with reliably annotated NME cleavages. Mass spectrometry-based NME data sets derived from single genomes are useful but intrinsically unreliable and incomplete. Comparative proteogenomics is ideally suited to generate the first reliable NME data sets and to help resolve the NME code problem.
While “signal peptides” are important for understanding protein function, they are difficult to confirm experimentally, and computational tools (e.g., SignalP) are used to fill the gap. However, since experimental data about signal peptides are limited, these tools make predictions based on a very small signal peptide database. As a result, there have been concerns regarding the quality of signal peptide predictions (Antelmann et al. 2001) since these methods may fail to identify interesting cases that are limited to a few proteins. Comparative proteogenomics opens a possibility to construct the first reliable data set of all signal peptides in a set of genomes and to study evolution of signal peptides across multiple species.
“Operon prediction” in bacterial genomes is an important but still unsolved problem. Despite the fact that many bacterial studies (e.g., prediction of regulatory motifs) critically depend on operon predictions, the accuracy of existing computational tools for operon prediction remains low (Ermolaeva et al. 2001; Price et al. 2005). Ideally, either all proteins in an operon are expressed or, alternatively, no protein in an operon is expressed. If all expressed proteins were identified (and all identified proteins were expressed), this rule would translate into the following rule: Either all proteins in an operon are identified or, alternatively, no protein in an operon is identified. However, a protein may be expressed but not identified in some genomes (false negatives) and identified but not expressed in others (false positives). Since peptide identifications errors are somewhat random, the probability that the same protein is not expressed but identified in multiple species is rather small. Also, since peptide detectability varies from species, we expect that comparative proteogenomics approach based on signatures (described in the Results section) may minimize errors and improve on existing operon predictions.
Methods
Peptide identification
Peptide identification in So was described in an earlier study (Gupta et al. 2007). The MS/MS spectra were acquired on ion-trap mass spectrometers (LCQ, ThermoFinnigan) using electrospray ionization. We used InsPecT (Tanner et al. 2005) (July 2007 version) to search the spectra of each species against a database containing the six-frame translation of the genome along with common contaminants and a decoy database of the same size. InsPecT search was run using default parameter settings (fragment ion tolerance of 0.5 Da and parent mass tolerance of 2.5 Da). The InsPecT score threshold was selected for each case to limit the number of identifications on the decoy database to at most 1% of the number of identifications on the target database, to keep the false discovery rate under control. After the filtering step, we obtained 29,160 peptides in So, 22,820 peptides in Sf, and 22,358 peptides in Sp. These include 337, 222, and 269 peptides in So, Sf, and Sp, respectively, that do not match the annotated proteins in these genomes. We demonstrate that coordinated mapping of these peptides (that are usually discarded as false identifications) represents valuable information for improving genome annotations.
Analyzing late start codons
We describe an algorithm for predicting “late” start codons, i.e., the (correct) start codons that are located downstream from the wrongly annotated start codons. While a late start codon implies a “missing” peptide in the beginning of the protein (between the wrongly annotated and correct start codons), such missing peptides can also be caused by low peptide detectability (Kuster et al. 2005) or may simply represent signal peptides. However, noncovered peptides (nontryptic peptides with no upstream coverage) (see Gupta et al. 2007 for more details) in the beginning of the protein, that cannot be explained by the signal peptide consensus sequence, point to late start codons. There are 33 cases of N-terminal most-noncovered peptides in So, within 18 residues of the start. Conspicuously, many of them either begin with ATG start codons or start immediately after a start codon (as in the case of N-terminal Methionine cleavage) (see Gupta et al. 2007). If all these peptides were artifacts, the distribution of the codons for amino acids at positions 1 (where the observed peptide begins) and −1 (corresponding to N-terminal Methionine cleavage) in these peptides would be somewhat uniform with an average 33/61 ≈ 0.5 peptides per codon. Instead, we see a nonuniform distribution at position 1 and −1 with a sharp peak at ATG (standard Methionine start codon) and overrepresentation of other start codons (TTG and GTG). We thus believe that all these cases cannot be artifacts (such as degradation products or incorrect peptide identifications).
To exclude signal peptides from consideration, we consider only noncovered peptides located within a distance of 18 aa or less from the start of the protein (signal peptides are typically longer than 18 aa). Thirty-three, 16, and 11 candidates are observed in So, Sf, and Sp, respectively. Comparative analysis of the three Shewanella species is subsequently performed to validate these candidates for late start codons.
Correlated peptides
Traditional MS/MS analysis is focused on identification of proteins and is less concerned with the question of which peptides in a protein are observed or not observed. In this study, we utilize the availability of proteomic data from related species to analyze the expression of peptides at orthologous positions. In a typical mass spectrometry experiment, some peptides with low detectability are always missed, resulting in highly nonuniform protein coverage by identified peptides (Purvine et al. 2004; Kuster et al. 2005). For example, while most ribosomal proteins in So have high coverage (>50%), a few have low coverage and one of them does not have any identified peptides. Peptide detectability may depend on several factors including protein abundance, peptide length, peptide hydrophobicity, etc., and several groups are using large data sets to develop the ability to its prediction (Tang et al. 2006; Lu et al. 2007; Mallick et al. 2007).
All identified peptides in shared genes were mapped to the alignment of the orthologs to get their coordinates with respect to the alignment. This provides a uniform reference scale to compare the positions of observed peptides between the orthologous proteins in the three species, as individual proteins may have different lengths. Peptides identified by MS/MS in two species are called correlated peptides if they are observed in the same position in the protein alignment or one of them spans another. In other words, if one peptide is located at positions (start1, end1) in the alignment, and the other peptide at (start2, end2), then peptides are considered correlated if start1 ≤ start2 ≤ end2 ≤ end1 or start2 ≤ start1 ≤ end1 ≤ end2.
Identification of post-translational modifications
MS-Alignment (Tsur et al. 2005) was used to identify PTMs in each of the three organisms in a blind mode, in the range from −200 to +250 Da. Common contaminants like keratins were included in the protein sequence databases. A decoy database of the same size as the actual protein database, containing shuffled sequences, was used to control the error rates. Any hits to the decoy database are expected to be incorrect identifications. A score cutoff is chosen such that the number of PTM sites identified in the decoy database is at most 5% of the number of identifications in the target database. This provides a controlled PTM site-specific false-discovery rate of 5%. We note that this is a more stringent criterion than a 5% error rate at the spectrum or peptide level, since several peptides in the forward database may point to the same PTM site. We further removed all spectra that were identified in the regular InsPecT search. After this post-processing of MS-Alignment results, 9917, 7649, and 6709 PTMs were obtained in So, Sf, and Sp, respectively (the complete lists along with the DTA files of spectra are available from http://proteomics.bioprojects.org/Downloads/spectra_and_peptideLists_supplement.zip). We only use tryptic-modified peptides in the subsequent analysis.
Acknowledgments
We thank Andrei Osterman for many insightful comments. This work was supported by National Institutes of Health Grant NIGMS 1-R01-RR16522 and by the Howard Hughes Medical Institute Professor Award. Part of this research at Pacific Northwest National Laboratory was supported by the Genomics:GtL Program, Office of Biological and Environmental Research, U.S. Department of Energy. Pacific Northwest National Laboratory is operated for the DOE by Batelle Memorial Institute under Contract DE-AC06-76RLO 1830.
References
- ↵S. Altschul,T.L. Madden,A.A. Schäffer,J. Zhang,Z. Zhang,W. Miller,D.J. Lipman,(1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402.
- ↵H. Antelmann,H. Tjalsma,B. Voigt,S. Ohlmeier,S. Bron,J. van Dijl,M. Hecker,(2001) A proteomic view on genome-based signal peptide predictions. Genome Res. 11:1484–1502.
- ↵S. Batzoglou,L. Pachter,J. Mesirov,B. Berger,E. Lander,(2000) Human and mouse gene structure: Comparative analysis and application to exon prediction. Genome Res. 10:950–958.
- ↵A. Ben-Bassat,K. Bauer,S. Chang,K. Myambo,A. Boosman,S. Chang,(1987) Processing of the initiation methionine from proteins: Properties of the Escherichia coli methionine aminopeptidase and its gene structure. J. Bacteriol. 169:751–757.
- ↵J. Bendtsen,H. Nielsen,G. von Heijne,S. Brunak,(2004) Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340:783–795.
- ↵R. Bradshaw,A. Burlingame,S. Carr,R. Aebersold,(2006) Reporting protein identification data: The next generation of guidelines. Mol. Cell. Proteomics 5:787–788.
- ↵N. Brown,C. Sander,P. Bork,(1998) Frame: Detection of genomic sequencing errors. Bioinformatics 14:367–371.
- ↵S. Carr,R. Aebersold,M. Baldwin,A. Burlingame,K. Clauser,A. Nesvizhskii,(2004) The need for guidelines in publication of peptide and protein identification data: Working group on publication guidelines for peptide and protein identification data. Mol. Cell. Proteomics 3:531–533.
- ↵R. Chenna,H. Sugawara,T. Koike,R. Lopez,T. Gibson,D. Higgins,J. Thompson,(2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31:3497–3500.
- ↵M. Clamp,B. Fry,M. Kamal,X. Xie,J. Cuff,M. Lin,M. Kellis,K. Lindblad-Toh,E. Lander,(2007) Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. 104:19428–19433.
- ↵J. Claverie,(1993) Detecting frame shifts by amino acid sequence comparison. J. Mol. Biol. 234:1140–1157.
- ↵W. Craigen,R. Cook,W. Tate,C. Caskey,(1985) Bacterial peptide chain release factors: Conserved primary structure and possible frameshift regulation of release factor 2. Proc. Natl. Acad. Sci. 82:3616–3620.
- ↵N. Daraselia,D. Dernovoy,Y. Tian,M. Borodovsky,R. Tatusov,T. Tatusova,(2003) Reannotation of Shewanella oneidensis genome. OMICS 7:171–176.
- ↵P. Demirev,J. Lin,F. Pineda,C. Fenselau,(2001) Bioinformatics and mass spectrometry for microorganism identification: Proteome-wide post-translational modifications and database search algorithms for characterization of intact H. pylori. Anal. Chem. 73:4566–4573.
- ↵R. Edgar,(2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797.
- ↵M. Ermolaeva,O. White,S. Salzberg,(2001) Prediction of operons in microbial genomes. Nucleic Acids Res. 29:1216–1221.
- ↵P. Farabaugh,(1996) Programmed translational frameshifting. Microbiol. Mol. Biol. Rev. 60:103–134.
- ↵D. Fermin,B. Allen,T. Blackwell,R. Menon,M. Adamski,Y. Xu,P. Ulintz,G. Omenn,D. States,(2006) Novel gene and gene model detection using a whole genome open reading frame analysis in proteomics. Genome Biol. 7:R35, 10.1186/gb-2006-7-4-r35.
- ↵G. Fichant,Y. Quentin,(1995) A frameshift error detection algorithm for DNA sequencing projects. Nucleic Acids Res. 23:2900–2908.
- ↵R. Fleischmann,M. Adams,O. White,R. Clayton,E. Kirkness,A. Kerlavage,C. Bult,J. Tomb,B. Dougherty,J. Merrick,(1995) Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269:496–498.
- ↵F. Gnad,S. Ren,J. Cox,J. Olsen,B. Macek,M. Oroshi,M. Mann,(2007) PHOSIDA (phosphorylation site database): Management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 8:R250, 10.1186/gb-2007-8-11-r250.
- ↵N. Gupta,S. Tanner,N. Jaitly,J. Adkins,M. Lipton,R. Edwards,M. Romine,A. Osterman,V. Bafna,R. Smith,(2007) Whole proteome analysis of post-translational modifications: Applications of mass-spectrometry for proteogenomic annotation. Genome Res. 17:1362–1377.
- ↵J. Heidelberg,I.T. Paulsen,K.E. Nelson,E.J. Gaidos,W.C. Nelson,T.D. Read,J.A. Eisen,R. Seshadri,N. Ward,B. Methe,(2002) Genome sequence of the dissimilatory metal ion-reducing bacterium Shewanella oneidensis. Nat. Biotechnol. 20:1118–1123.
- ↵J. Jaffe,H. Berg,G. Church,(2004) Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics 4:59–77.
- ↵D. Kalume,S. Peri,R. Reddy,J. Zhong,M. Okulate,N. Kumar,A. Pandey,(2005) Genome annotation of Anopheles gambiae using mass spectrometry-derived data. BMC Genomics 6:128, 10.1186/1471-2164-6-128.
- ↵X. Kang,K. Rogers,G. Gao,A. Falick,S. Zhou,L. Simpson,(2005) Reconstitution of uridine-deletion precleaved RNA editing with two recombinant enzymes. Proc. Natl. Acad. Sci. 102:1017–1022.
- ↵M. Kellis,N. Patterson,M. Endrizzi,B. Birren,E. Lander,(2003) Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423:241–254.
- ↵B. Kuster,M. Schirle,P. Mallick,R. Aebersold,(2005) Scoring proteomes with proteotypic peptide probes. Nat. Rev. Mol. Cell Biol. 6:577–583.
- ↵S. Liu,G. Milne,J. Kuremsky,G. Fink,S. Leppla,(2004) Identification of the proteins required for biosynthesis of diphthamide, the target of bacterial ADP-ribosylating toxins on translation elongation factor 2. Mol. Cell. Biol. 24:9487–9497.
- ↵P. Lu,C. Vogel,R. Wang,X. Yao,E. Marcotte,(2007) Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 25:117–124.
- ↵B. Macek,F. Gnad,B. Soufi,C. Kumar,J. Olsen,I. Mijakovic,M. Mann,(2008) Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell. Proteomics 7:299–307.
- ↵P. Mallick,M. Schirle,S. Chen,M. Flory,H. Lee,D. Martin,J. Ranish,B. Raught,R. Schmitt,T. Werner,(2007) Computational prediction of proteotypic peptides for quantitative proteomics. Nat. Biotechnol. 25:125–131.
- ↵C. Medigue,M. Rose,A. Viari,A. Danchin,(1999) Detecting and analyzing DNA sequencing errors: Toward a higher quality of the Bacillus subtilis genome sequence. Genome Res. 9:1116–1127.
- ↵J. Moehring,T. Moehring,D. Danley,(1980) Post-translational modification of elongation factor 2 in diphtheria-toxin-resistant mutants of CHO-K1 cells. Proc. Natl. Acad. Sci. 77:1010–1014.
- ↵K. Nealson,A. Belz,B. McKee,(2002) Breathing metals as a way of life: Geobiology in action. Antonie Van Leeuwenhoek 81:215–222.
- ↵M. Nielsen,M. Savitski,R. Zubarev,(2006) Extent of modifications in human proteome samples and their effect on dynamic range of analysis in shotgun proteomics. Mol. Cell. Proteomics 5:2384–2391.
- ↵K. Olson,J. Fenno,N. Lin,R. Harkins,C. Snider,W. Kohr,M. Ross,D. Fodge,G. Prender,N. Stebbing,(1981) Purified human growth hormone from E. coli is biologically active. Nature 293:408–411.
- ↵J. Posfai,R. Roberts,(1992) Finding errors in DNA sequences. Proc. Natl. Acad. Sci. 89:4698–4702.
- ↵M. Price,K. Huang,E. Alm,A. Arkin,(2005) A novel method for accurate operon predictions in all sequenced prokaryotes. Nucleic Acids Res. 33:880–892.
- ↵S. Purvine,A. Picone,E. Kolker,(2004) Standard mixtures for proteome studies. OMICS 8:79–92.
- ↵M. Remm,C. Storm,E. Sonnhammer,(2001) Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 314:1041–1052.
- ↵J. Rodriguez,N. Gupta,R. Smith,P. Pevzner,(2008) Does trypsin cut before proline? J. Proteome Res. 7:300–305.
- ↵A. Savidor,R. Donahoo,O. Hurtado-Gonzales,N. Verberkmoes,M. Shah,K. Lamour,W. McDonald,(2006) Expressed peptide tags: An additional layer of data for genome annotation. J. Proteome Res. 5:3048–3058.
- ↵M. Savitski,M. Nielsen,R. Zubarev,(2006) Modificomb, a new proteomic tool for mapping substoichiometric post-translational modifications, finding novel types of modifications, and fingerprinting complex protein mixtures. Mol. Cell. Proteomics 5:935–948.
- ↵H. Tang,R. Arnold,P. Alves,Z. Xun,D. Clemmer,M. Novotny,J. Reilly,P. Rejivojac,(2006) A computational approach toward label-free protein quantification using predicted peptide detectability. Bioinformatics 22:e481–e488, 10.1093/bioinformatics/btl237.
- ↵S. Tanner,H. Shu,A. Frank,L. Wang,E. Zandi,M. Mumby,P. Pevzner,V. Bafna,(2005) InsPecT: Fast and accurate identification of post-translationally modified peptides from tandem mass spectra. Anal. Chem. 77:4626–4639.
- ↵S. Tanner,Z. Shen,J. Ng,L. Florea,R. Guigo,S. Briggs,V. Bafna,(2007) Improving gene annotation using peptide mass spectrometry. Genome Res. 17:231–239.
- ↵M. Thompson,D. Thompson,R. Hettich,(2008) Systematic assessment of the benefits and caveats in mining microbial post-translational modifications from shotgun proteomic data: The response of Shewanella oneidensis to chromate exposure. J. Proteome Res. 7:648–658.
- ↵J. Tobias,T. Shrader,G. Rocap,A. Varshavsky,(1991) The N-end rule in bacteria. Science 254:1374–1377.
- ↵D. Tsur,S. Tanner,E. Zandi,V. Bafna,P. Pevzner,(2005) Identification of post-translational modifications via blind search of mass spectra. Nat. Biotechnol. 23:1562–1567.
- ↵C. Tu,T. Tzeng,J. Bruenn,(1992) Ribosomal movement impeded at a pseudoknot required for frameshifting. Proc. Natl. Acad. Sci. 89:8636–8640.
- ↵B. Van Ness,J. Howard,J. Bodley,(1980) ADP-ribosylation of elongation factor 2 by diphtheria toxin. NMR spectra and proposed structures of ribosyl-diphthamide and its hydrolysis products. J. Biol. Chem. 255:10710–10716.
- ↵R. Wang,J. Prince,E. Marcotte,(2005) Mass spectrometry of the M. smegmatis proteome: Protein expression levels correlate with function, operons, and codon bias. Genome Res. 15:1118–1126.
- ↵J. Whitelegge,H. Zhang,R. Aguilera,R. Taylor,W. Cramer,(2002) Full subunit coverage liquid chromatography electrospray ionization mass spectrometry (LCMS+) of an oligomeric membrane protein: Cytochrome b6f complex from spinach and the cyanobacterium Mastigocladus laminosus. Mol. Cell. Proteomics 1:816–827.
- ↵P. Wilmarth,S. Tanner,S. Dasari,S. Nagalla,M. Riviere,V. Bafna,P. Pevzner,L. David,(2006) Age-related changes in human crystallins determined from comparative analysis of post-translational modifications in young and aged lens: Does deamidation contribute to crystallin insolubility? J. Proteome Res. 5:2554–2566.
- ↵X. Xie,J. Lu,E. Kulbokas,T. Golub,V. Mootha,K. Lindblad-Toh,E. Lander,M. Kellis,(2005) Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434:338–345.
- ↵C. Yang,D. Rodionov,X. Li,O. Laikova,M. Gelfand,O. Zagnitko,M. Romine,A. Obraztsova,K. Nealson,A. Osterman,(2006) Comparative genomics and experimental characterization of N-acetylglucosamine utilization pathway of Shewanella oneidensis. J. Biol. Chem. 281:29872–29885.
Notes
[1] [Supplemental material is available online at www.genome.org.]
[2] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.074344.107.
[3] The first evolutionary studies of modifications were published by the Matthias Mann lab (Gnad et al. 2007; Macek et al. 2008) for the case of phosphorylations. We emphasize the difference between these recent papers focusing on a single known modification and our approach that attempts to identify multiple unknown modification types via comparative analysis.
[4] We also cannot exclude the possibility that they represent a “combined” modification, i.e., two different modifications (let us say with offsets X and Y) on neighboring residues that are misidentified as a single modification (with offset X + Y). However, many of our identifications have excellent b/y ladders, indicating that such artifacts are unlikely.