
Abstract
The most widely used method for detecting genome-wide protein–DNA interactions is chromatin immunoprecipitation on tiling microarrays, commonly known as ChIP-chip. Here, we conducted the first objective analysis of tiling array platforms, amplification procedures, and signal detection algorithms in a simulated ChIP-chip experiment. Mixtures of human genomic DNA and “spike-ins” comprised of nearly 100 human sequences at various concentrations were hybridized to four tiling array platforms by eight independent groups. Blind to the number of spike-ins, their locations, and the range of concentrations, each group made predictions of the spike-in locations. We found that microarray platform choice is not the primary determinant of overall performance. In fact, variation in performance between labs, protocols, and algorithms within the same array platform was greater than the variation in performance between array platforms. However, each array platform had unique performance characteristics that varied with tiling resolution and the number of replicates, which have implications for cost versus detection power. Long oligonucleotide arrays were slightly more sensitive at detecting very low enrichment. On all platforms, simple sequence repeats and genome redundancy tended to result in false positives. LM-PCR and WGA, the most popular sample amplification techniques, reproduced relative enrichment levels with high fidelity. Performance among signal detection algorithms was heavily dependent on array platform. The spike-in DNA samples and the data presented here provide a stable benchmark against which future ChIP platforms, protocol improvements, and analysis methods can be evaluated.
With the availability of sequenced genomes and whole-genome tiling microarrays, many researchers have conducted experiments using ChIP-chip and related methods to study genome-wide protein–DNA interactions (Cawley et al. 2004; Hanlon and Lieb 2004; Kim et al. 2005; Carroll et al. 2006; Hudson and Snyder 2006; Kim and Ren 2006; Lee et al. 2006; Yang et al. 2006; O’Geen et al. 2007). These are powerful yet challenging techniques, which are comprised of many steps that can introduce variability in the final results. One potentially important factor is the relative performance of different types of tiling arrays. Currently the most popular platforms for performing ChIP-chip experiments are commercial oligonucleotide-based tiling arrays from Affymetrix, NimbleGen, and Agilent. A second factor known to introduce variation is the DNA amplification protocol, which is often required because the low DNA yield from a ChIP experiment prevents direct detection on microarrays. A third factor is the algorithm used for detecting regions of enrichment from the tiling array data. Several algorithms have been developed, but until this report there was no benchmark data set to systematically evaluate them. In this study, we used a spike-in experiment to systematically evaluate the effects of tiling microarrays, amplification protocols, and data analysis algorithms on ChIP-chip results. There are other potentially important factors that are not assessed here, and that from a practical standpoint are more difficult to systematically control and evaluate. These include the skill of the experimenter, the amount of starting material (chromatin, DNA, and antibody) used, the size of DNA fragments after shearing, the DNA labeling method, and the hybridization conditions.
There have been several studies evaluating the performance of gene expression microarrays and analysis algorithms (Choe et al. 2005; Irizarry et al. 2005; MAQC Consortium 2006; Patterson et al. 2006). However, tiling arrays present distinct informatics and experimental challenges because large contiguous genomic regions are covered with high probe densities. Thus the results from the expression array spike-in experiments are not necessarily directly relevant to tiling-array experiments. One recent study compared the performance of array-based (ChIP-chip) and sequence-based (ChIP-PET) technologies on a real ChIP experiment (Euskirchen et al. 2007). However, because this was an exploratory experiment, the list of absolute “true-positive” targets was and remains unknown. Since the experiment (Euskirchen et al. 2007) was performed without a key, the sensitivity and specificity of each technology had to be estimated retrospectively by qPCR validation of targets predicted from each platform.
In our experiment, eight independent research groups at locations worldwide each hybridized two different mixtures of DNA to one of four tiling-array platforms and predicted genome location and concentration of the spike-in sequences using a total of 13 different algorithms. Throughout the process, the research groups were entirely blind to the contents of the spike-in mixtures. Using the spike-in key, we analyzed several performance parameters for each platform, algorithm, and amplification method. While all commercial platforms performed well, we found that each had unique performance characteristics. We examined the implications of these results in planning human genome-wide experiments, in which trade-offs between probe density and cost are important.
Results
Creation of the simulated ChIP sample
To create our simulated ChIP spike-in mixture, we first randomly selected 100 cloned genomic DNA sequences (average length 497 bp) corresponding to predicted promoters in the human genome (Cooper et al. 2006), individually purified them, and normalized the concentrations of each preparation to 500 pg/μL (Fig. 1). To create enrichment levels that ranged from 1.25-fold to 196-fold relative to genomic DNA (Supplemental Tables 1 and 2), we added the appropriate volume of these stock solutions to a commercial human genomic DNA preparation (Methods; Supplemental Tables 1 and 2). The clones were validated by sequencing and PCR both before and after dilution (Supplemental Methods). We prepared one clone mixture to be directly labeled and hybridized to arrays at the given concentration (“undiluted,” 77 ng/μL), and a different clone mixture that was diluted such that amplification would be necessary before labeling and hybridization (“diluted,” 3 ng/μL). The diluted mixture was created because all of the array platforms require microgram quantities of DNA, and a typical ChIP experiment produces ∼50 ng of DNA, making amplification essential for most ChIP-chip experiments. Each amplification method is known to cause under- and over-representation of certain sequences (Liu et al. 2003), which we aimed to assess in this context.
Workflow for the multi-laboratory tiling array spike-in experiment.
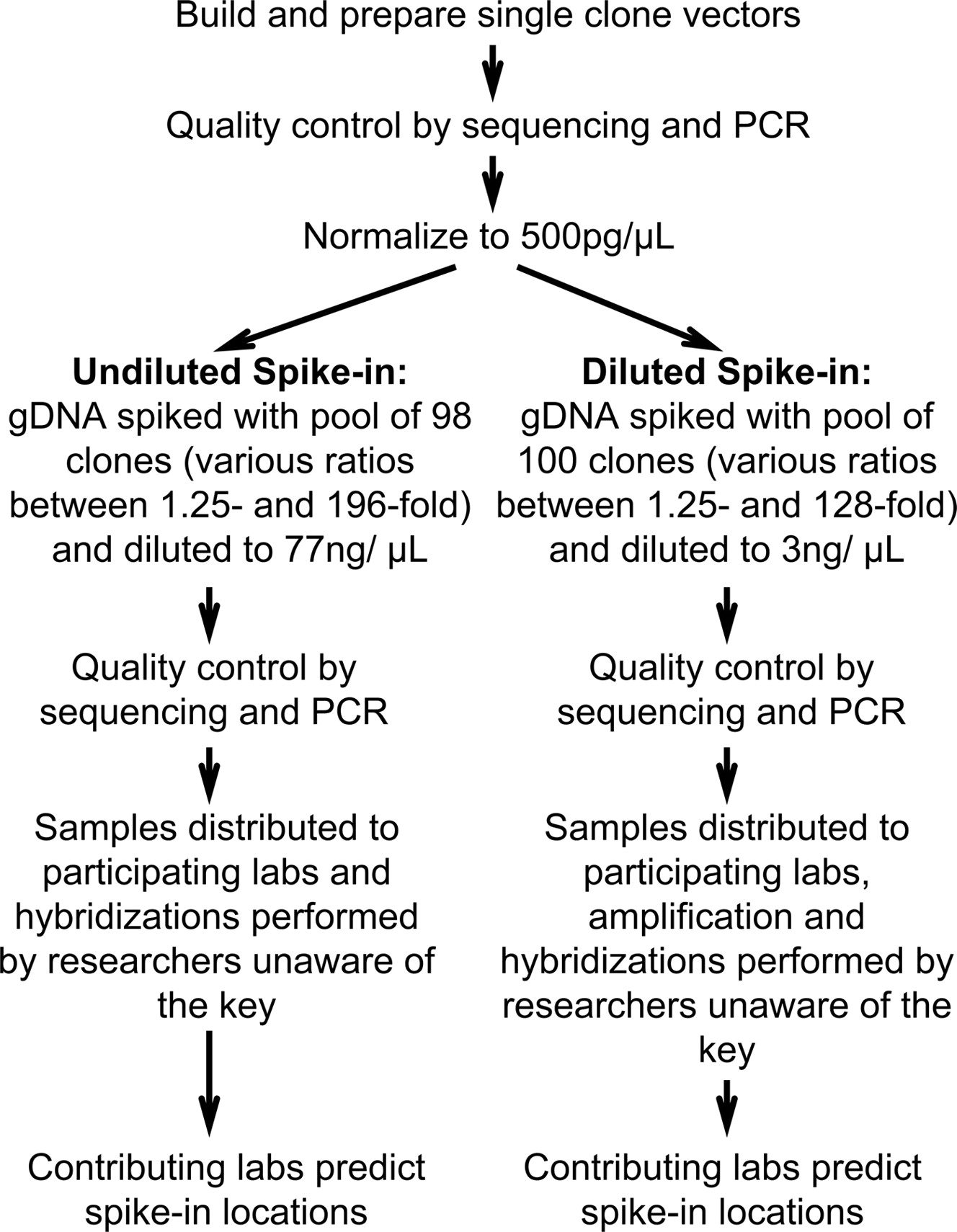
After the mixtures were prepared, the clones and their relative concentrations were again validated by sequencing and quantitative PCR (qPCR). Note that while the same spike-in clones were present in the diluted and undiluted mixtures, they were used at different enrichment levels in the two samples. In each mixture, most of the selected enrichment levels were represented by 10 distinct clones. To challenge the sensitivity of the array technologies, spike-in enrichment levels were biased toward enrichment levels less than 10-fold. We also prepared two samples containing genomic DNA at 77 ng/μL and 3 ng/μL, respectively, without any spike-ins to serve as controls. We sheared the DNA mixtures with a standard chromatin sonication procedure (Johnson et al. 2007).
Amplification, labeling, and DNA microarray hybridization of the simulated ChIP
We sent aliquots of the control DNA and the two mixtures to participating groups, who labeled, amplified (the diluted samples), and hybridized the mixtures to DNA microarrays covering the ENCODE regions (The ENCODE Project Consortium 2007) using their standard procedures (Fig. 1; Supplemental Methods). None of the individuals involved in hybridizations or predictions described below was aware of the identity of any of the clones in the spike-in mixtures, the number of spike-in clones, or the range of fold-enrichment values. For the samples requiring amplification, we tested the effect of three different amplification procedures: ligation-mediated PCR (LM-PCR), random-priming PCR (RP), and whole-genome amplification (WGA) (O’Geen et al. 2006; Supplemental Methods).
The groups labeled and hybridized the mixtures to one of three different types of tiling arrays (NimbleGen, Affymetrix, or Agilent). Each of the tiling array technologies covers the 1% of the human genome selected for study by the ENCODE Consortium (The ENCODE Project Consortium 2007). Because each array technology is unique, the total number of nucleotides and percentage of the ENCODE regions covered varies among the platforms. However, we ensured that all of the regions corresponding to the spike-in clones were well represented on all of the platforms. Affymetrix ENCODE arrays contained short 25-mer probes at a start-to-start tiling resolution of 22 bp (1.0R arrays) or 7 bp (2.0R arrays) (http://www.affymetrix.com). The probes were chosen from RepeatMasked (Jurka 2000) sequences and synthesized on the arrays in situ using photolithographic technology. Agilent ENCODE arrays consisted of isothermal 44–60-mer probes that are unique in the human genome printed at 100-bp resolution using inkjet technology (http://www.agilent.com). NimbleGen ENCODE arrays were comprised of unique 50-mers at 38-bp resolution, with the probes being synthesized in situ using maskless array synthesizer technology (http://www.nimblegen.com). We performed all hybridizations in at least duplicate, with a matched comparative hybridization using genomic DNA where appropriate. Affymetrix does not use two-channel comparative hybridization, thus spike-in and controls were hybridized on separate arrays.
This study also initially included a PCR tiling-array platform consisting of 22,180 consecutive ∼980-bp PCR products covering the ENCODE regions spotted on glass slides. However, the PCR arrays performed poorly according to our choice of evaluation metrics, apparently because of the low resolution of the PCR array platform relative to the oligonucleotide platforms. This prevented an equitable comparison of the results, and therefore the PCR array results are presented separately (Supplemental Fig. 1).
Analysis algorithms
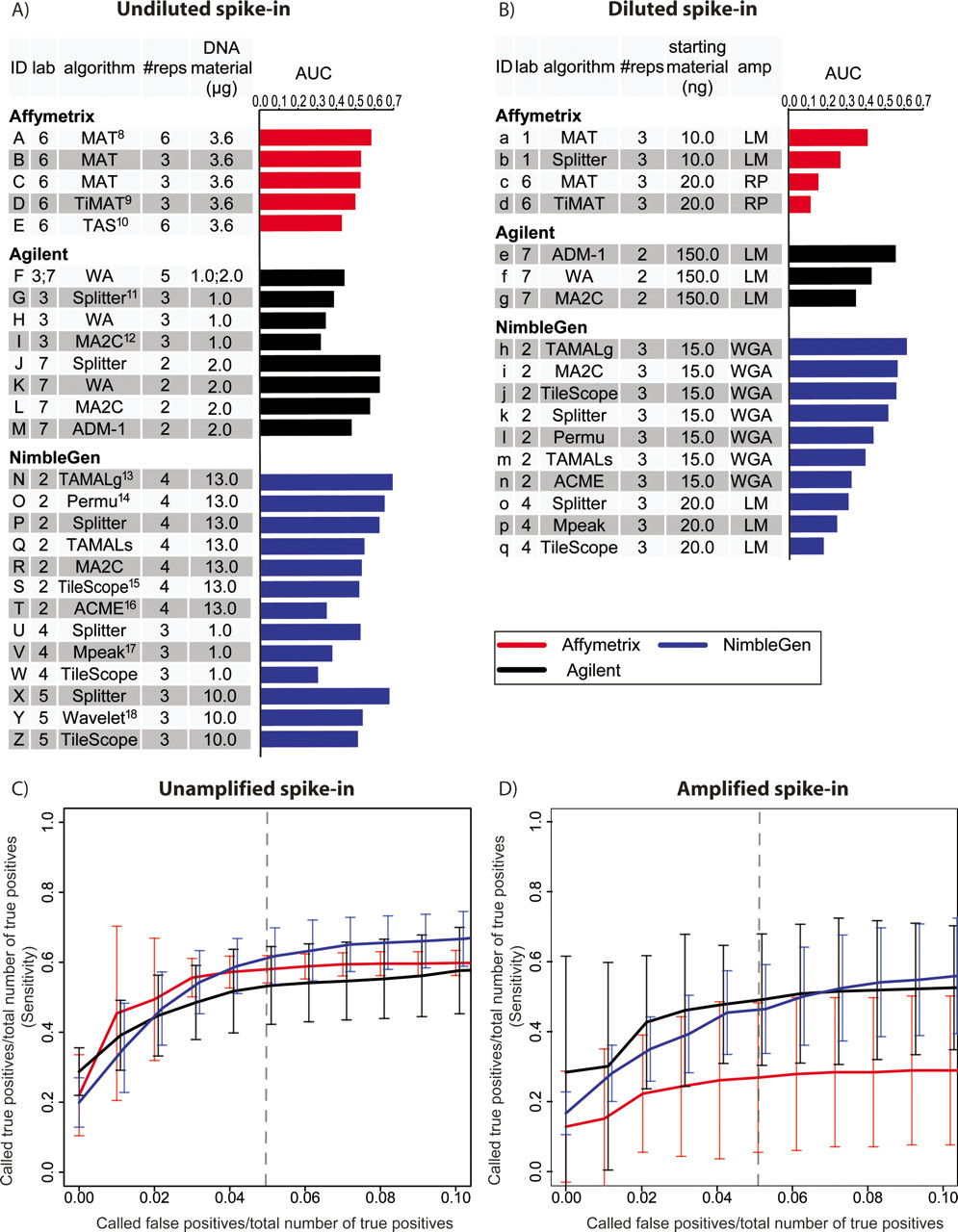
We used 13 different algorithms (Supplemental Methods) to make predictions of enriched regions from the array measurements. While most of the algorithms function only on a single platform, we used two algorithms, MA2C (Song et al. 2007) and Splitter (H. Shulha, Y. Fu, and Z. Weng; http://zlab.bu.edu/splitter), for multiple platforms. To standardize the results across algorithms, we required that each prediction consist of a rank-ordered list of predicted spike-in regions, with each region represented by a single chromosome coordinate and a quantitative value that corresponded to a predicted enrichment level. We considered a region to be predicted correctly if the single predicted coordinate was within the spike-in region. Because the total number of spike-ins was unknown to the predictors, each predictor was also asked to estimate a cutoff score above which the selected predictions were considered significant. We then used the spike-in key to assess the performance of each microarray platform, amplification method, and analysis algorithm (Fig. 2).
Summary performance statistics for spike-in predictions. (A) Undiluted and Unamplified samples. Raw data were provided by seven different labs, which are designated as follows: (1) M. Brown; (2) P. Farnham and R. Green; (3) R. Myers; (4) B. Ren; (5) M. Snyder; (6) K. Struhl and T. Gingeras; (7) S. McCuine. AUC (Area Under ROC-like Curve) values were calculated based on the ranked list of spike-in calls provided by each group. The references for the algorithms are: (8) Johnson et al. 2006; (9) D. Nix, http://sourceforge.net/projects/timat2; (10) Cawley et al. 2004; (11) H. Shulha, Y. Fu, and Z. Weng, http://zlab.bu.edu/splitter; (12) Song et al. 2007; (13) Bieda et al. 2005; (14) Lucas et al. 2007; (15) Zhang et al. 2007; (16) Scacheri et al. 2006; (17) Kim et al. 2005; (18) A. Karpikov and M. Gerstein, unpubl. (B) The same as A, for Diluted and Amplified samples. (C) ROC-like plots for Unamplified spike-in predictions. As an aid in interpretation, the dashed vertical line represents the point at which a group’s number of false-positive predictions equal 5% of the total number of true-positive spike-ins. At this point, all platforms correctly identified ∼50% of the true-positive spike-ins. Error bars represent the two-sided 95% confidence interval of the average sensitivity at each false-positive ratio (X-axis). (D) The same as C, for Amplified samples.
Assessment of sensitivity and specificity using ROC-like curves
We used an ROC (receiver operating characteristic)-like curve analysis to assess the sensitivity and specificity of the predictions from the array measurements across all spike-in concentrations (Fig. 2). All spike-in regions were considered true positives regardless of the degree of enrichment. All remaining regions represented on the array were considered true negatives. Standard ROC curves are created by plotting the sensitivity (true-positive rate; Y-axis) against 1-specificity (false-positive rate; X-axis) obtained at every rank value of predicted sites. In our simulated ChIP experiment and in many actual ChIP-chip experiments, true negatives are represented by >99% of the arrayed probes. This results in a large absolute number of false positives even at extremely low false-positive rates (false positives/true negatives). Therefore, to represent the performance of each experiment, on the X-axis we plotted the (number of true positives)/(number of spike-in clones), and on the Y-axis we plotted the (number of false positives)/(number of spike-in clones). Presented in this way, the value on either axis represents the same absolute number of true positives (Y-axis) or false positives (X-axis). Under this framework, the best possible array prediction would yield a graph that has a point in the upper left corner of the plot, which would represent a case with correct prediction of all true positives (100% sensitivity) without any false positives (100% specificity). Our benchmark for this analysis is the area under this ROC-like curve (AUC), which conceptually represents the average sensitivity over a range of specificities. We standardized the AUC values so that randomly selected sites would have an AUC of nearly zero, and a perfect performance would have an AUC of 1.
Microarray platform choice is not the primary determinant of overall performance
For all three microarray platforms, the best combination of data and analysis algorithm in the unamplified spike-in experiments generally detected ∼50% of the spike-in clones at a 5% false discovery ratio (number of false positives/total number of spike-in clones; this corresponds to about a 10% false discovery rate) (Fig. 2). Most of the missed calls were for spike-ins at very low enrichment values (see below) (Fig. 3). However, the AUC values spanned a wide range, from 0.31 (NimbleGen data from Lab 4, Tilescope algorithm) to 0.71 (NimbleGen data from Lab 2, TAMALg algorithm) (Fig. 2A). Among the platforms, the Splitter algorithm was the best on Agilent tiling arrays (AUC = 0.64), while MAT (Johnson et al. 2006) was best for Affymetrix (AUC = 0.59). For the amplified spike-in experiments, the AUC values also spanned a wide range, from 0.12 (RP amplification method, Affymetrix arrays, TiMAT [David Nix; http://sourceforge.net/projects/timat2] algorithm) to 0.57 (WGA amplification method, NimbleGen arrays, and TAMALg [Bieda et al. 2006] algorithm) (Fig. 2B). Through bootstrapping, we found the confidence interval of AUC within each array/lab/algorithm combination around ±0.07 (Supplemental Methods), thus small AUC differences may not reflect significant performance differences.
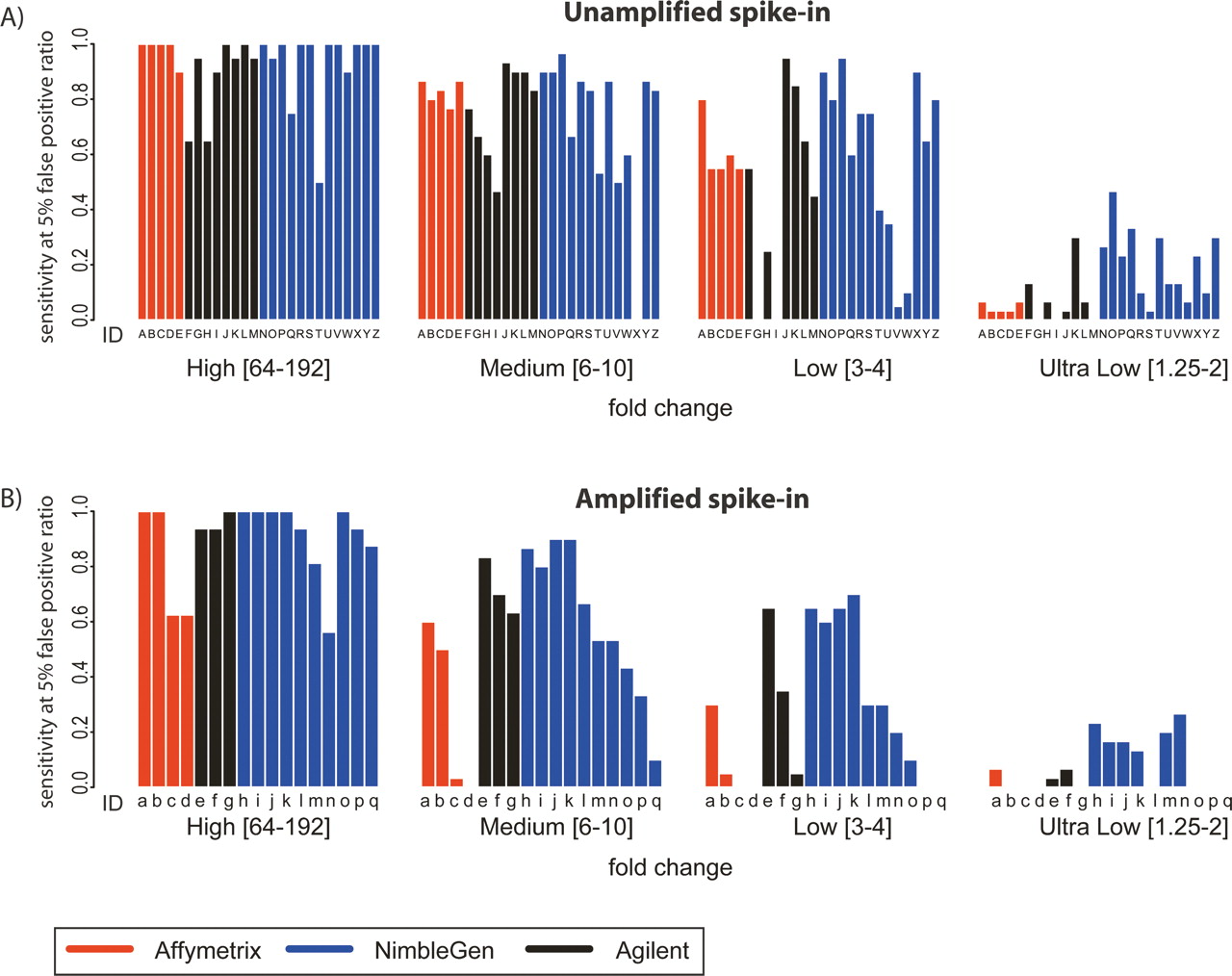
Enrichment-specific sensitivity. (A) Enrichment-specific sensitivity for Unamplified spike-in mixtures. The spike-in clones were divided into four levels of enrichment: High fold-change (64–192); Medium fold-change (6–10); Low fold-change (3–4); and Ultra Low fold-change (1.25–2). Enrichment-specific array prediction sensitivity (Y-axis) is defined as the percentage of correctly predicted enrichment-specific clones, with the total number of false positives equal to 5% of the total number of spike-in clones. Letters under each bar refer to the experiment description in Figure 2A. (B) The same as A, but for Amplified samples. Letters under each bar refer to the experiment description in Figure 2B.
The wide range of AUC values was not limited to comparisons across microarray platforms. In fact, the variance of AUC values between experiments performed within the same platform is similar to, if not greater than, the variance observed between the different platforms (Fig. 2C,D). This indicates that among commonly used experimental and analysis procedures, microarray platform choice is not the primary determinant of overall performance. Differences within a platform could arise from a variety of factors, most prominently by between-lab variability in experimental procedures and differences in the analysis algorithm used. For example, the hybridizations done in Lab 3 had a lower AUC than the hybridizations done in Lab 7 using the same Agilent microarray platform (Fig. 2A). There was at least one major difference in the experimental protocol between these labs: Lab 3 used Alexa dyes, whereas Lab 7 used Cyanine dyes for DNA labeling and detection.
All platforms were very sensitive at high enrichment levels; at extremely low enrichment levels, long oligonucleotide platforms are more sensitive
The enrichment levels produced by a typical ChIP experiment vary, from less than twofold to several thousandfold. Therefore, of particular interest is the sensitivity of arrays, amplification methods, and analysis methods across various ranges of fold enrichment. For each array, amplification method, and analysis algorithm combination, we calculated the sensitivity at high (64–192-fold), medium (sixfold to 10-fold), low (threefold to fourfold), and ultra-low (1.25–2-fold) enrichment ranges (Fig. 3). Generally, as expected, sensitivity decreases with decreasing fold enrichment. All technologies show a steep decrease in sensitivity at absolute enrichments (as opposed to measured enrichments) below threefold. Our analysis demonstrated that at a false discovery ratio of 5%, the NimbleGen platform (with four replicates) is the most sensitive platform at lower levels of enrichment (less than threefold), followed closely by Agilent (with two replicates). The differences in sensitivity among the platforms are not significant at levels of enrichment higher than threefold. These data are consistent with previous studies that showed that longer oligonucleotides are more sensitive than shorter probes (Hughes et al. 2001).
Sensitivities were lower for amplified samples than for unamplified samples regardless of the amplification method across all spike-in enrichment levels. Again, at lower-fold enrichments, lower sensitivity was observed. Holding the analysis method constant, Ligation Mediated-PCR (LM-PCR) afforded the least reduction in AUC from unamplified to amplified sample on Agilent arrays. On Affymetrix arrays, LM-PCR performed significantly better than RP amplification. The WGA method was used only on the NimbleGen platform, but also produced results with very little reduction in AUC.
The simulated ChIP-chip sample can be used to objectively assess cutoff selection
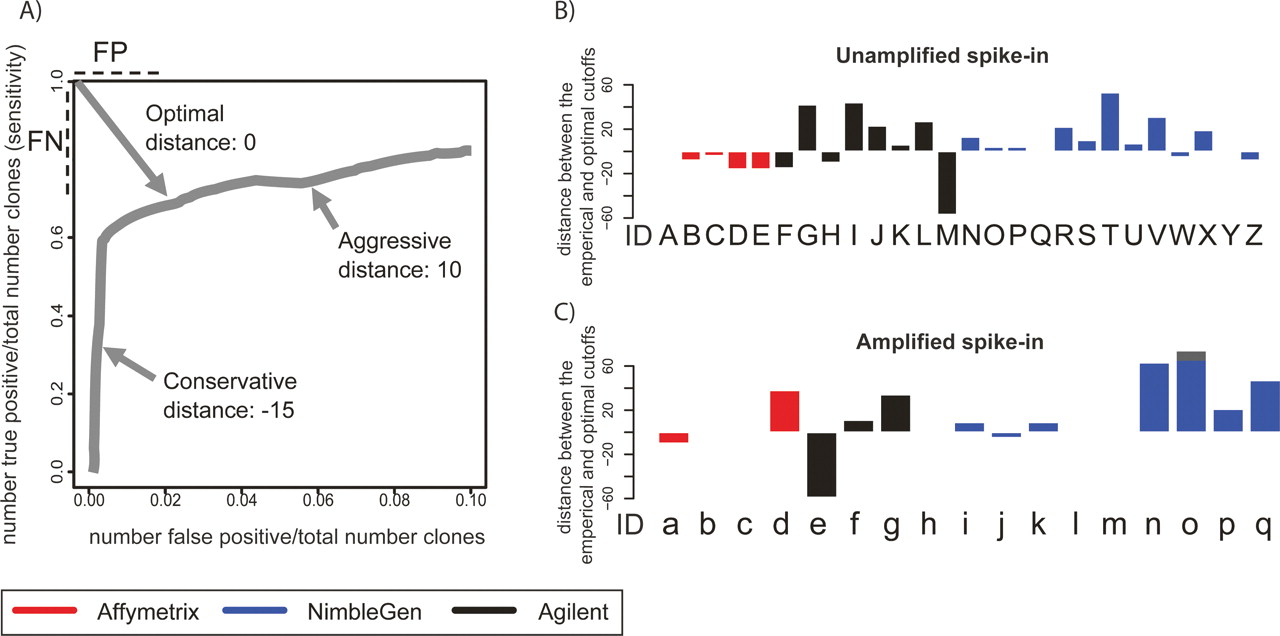
When making predictions of enriched regions based on ChIP-chip measurements, the ideal significance threshold or “cutoff” for selecting targets is generally unknown. This is because many ChIP-chip experiments are discovery efforts in which very few true binding sites are known. Therefore, it is impossible to calibrate the cutoff based on a truth model. Specificity can be improved at the cost of sensitivity, and vice versa, but in most cases a cutoff that optimally balances sensitivity and specificity produces the most useful outcome. In the context of ChIP-chip experiments, false-positive and false-negative calls are equally problematic. Because our simulated experiments have a truth model, we can calibrate the optimal threshold for each of the array experiments and peak-calling algorithms. We define the optimal threshold as the point on the ROC-like curve that is closest to the upper left corner, so long as the value on the X-axis is ≤10%. This point equally penalizes false positives and false negatives, and therefore minimizes false positives and false negatives simultaneously. The distance in rank between empirical threshold (submitted by each group) and the optimal threshold along the ROC-like curve (hereafter called the E-O distance) is a rational evaluation of the accuracy of threshold selection (Fig. 4A).
Evaluation of cutoff selection used for spike-in prediction. (A) We define the optimal threshold as the point on the ROC-like curve that is closest to the upper left corner, so long as the value on the X-axis ≤0.10. The distance in rank between empirical threshold (submitted by each group) and the optimal threshold along the ROC-like curve (hereafter E–O distance) is a rational evaluation of the accuracy of threshold selection. Aggressive and conservative thresholds will have positive and negative E–O distances, respectively. (B) The E–O distance for each set of experiments and predictions performed on the Unamplified samples. Letters under each bar refer to the experiment description in Figure 2A. (C) The same as B for the Amplified samples. Letters under each bar refer to the experiment description in Figure 2B.
Estimates of the significance threshold are often too aggressive or conservative, but do not vary with enrichment level
Overly aggressive threshold selection will produce a larger number of predicted peaks and many false positives, resulting in a positive E–O distance. Conservative threshold selection will identify fewer false positives at the cost of more false negatives than the optimal, resulting in a negative E–O distance. In the optimal situation, the empirical threshold is exactly the same as the optimal threshold, so that the E–O distance will be 0. In our simulated ChIP experiments, we found a broad range of E–O values, from −59 (very conservative, Agilent arrays, LM-PCR amplified, ADM-1 algorithm) to 74 (very aggressive, NimbleGen arrays, LM-PCR amplified, Splitter algorithm) (Fig. 4B). However, several analysis methods produced a cutoff very near the ideal threshold. In particular, MAT always produced calls with a near-optimal cutoff. We also examined the E–O distance metrics at various spike-in enrichment levels (Supplemental Fig. 2). Across all array platforms and peak prediction algorithms, E–O distances do not generally vary significantly among known spike-in enrichment levels. This suggests that it may not be necessary to calibrate prediction thresholds based on presumed enrichment levels in a ChIP experiment. Proper determination of E–O distance requires perfect knowledge of a truth model, thus spike-in experiments such as ours will remain important for labs interested in calibrating E–O distance on their particular analysis algorithm.
All platforms and most analysis methods accurately estimated actual enrichment values
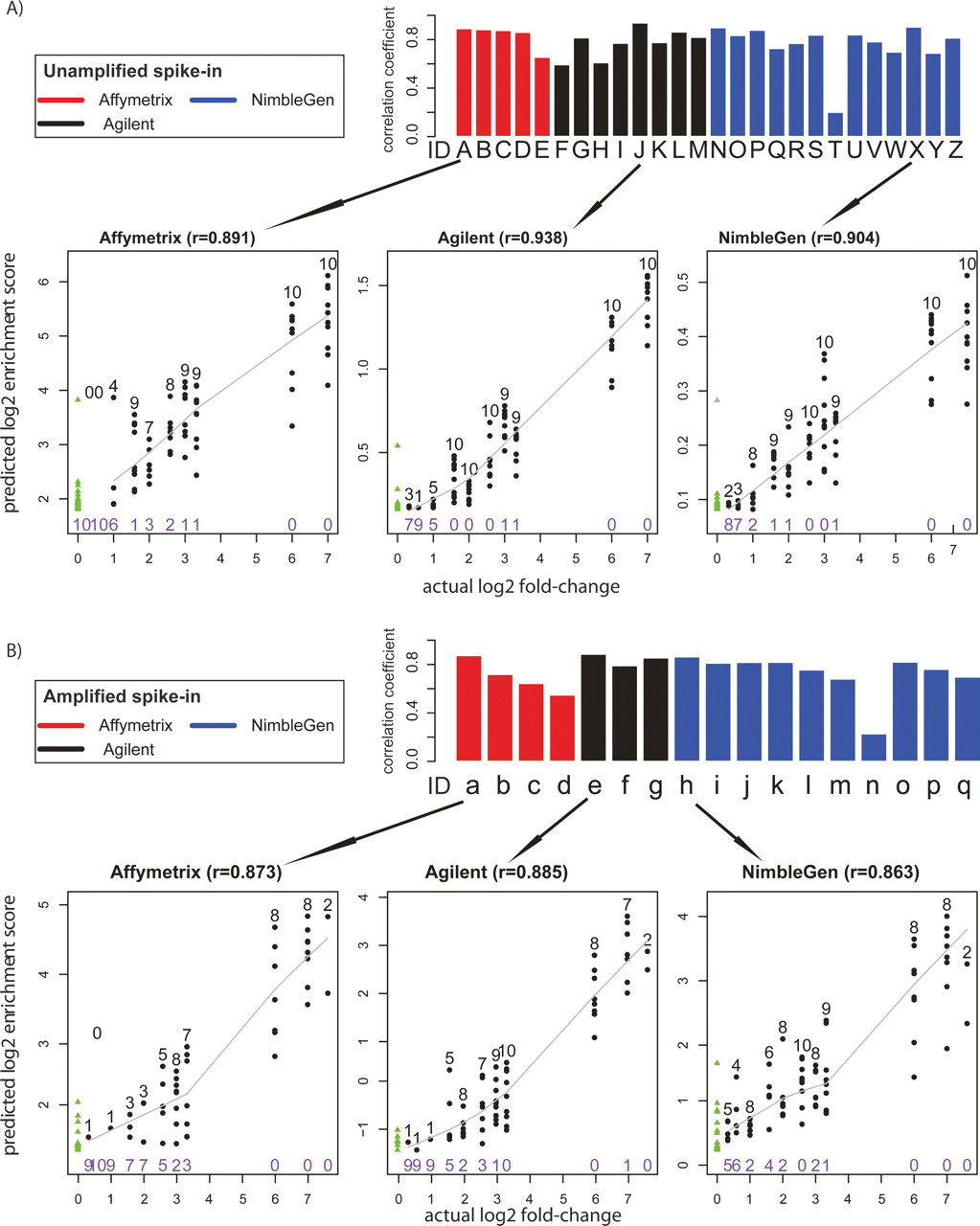
In ChIP-chip experiments, investigators are often interested in the magnitude of the relative enrichment value for any particular locus. These enrichment values may reflect an important aspect of biology such as the affinity of a transcription factor to its recognition sequences, or recruitment of multiple copies of one transcription factor to clusters of binding sites. Therefore, we evaluated the quantitative predictive power of different peak predictions from array measurements using our known quantitative truth model (Fig. 5). For each array type and peak-calling algorithm instance, we calculated Pearson’s correlation coefficient (r), between the log2 of the provided enrichment scores and the log2 of the actual spike-in fold-change of the top 100 predicted sites plus all the false-negative sites, and used this statistic as the final quantitative measurement for each prediction. Among the unamplified samples, there was a broad range of r-values across the various platforms and algorithms, ranging from 0.201 for the ACME (Scacheri et al. 2006) algorithm on the NimbleGen platform to 0.938 for Agilent arrays using the Splitter algorithm (Fig. 5A). Peak finding algorithms vary in their quantitative ability. A single data set produced by Lab 2 using NimbleGen arrays was analyzed with seven different peak detection algorithms, with one resulting in an r-value of 0.201, while all the other six methods produced r-values of greater than 0.7. The ability of each algorithm to quantitatively detect peaks in the array measurements appears to be largely unaffected by amplification (Fig. 5B). This demonstrates that each amplification method reproduces the relative enrichment levels found in the original diluted mixture with fidelity, although as shown previously, the sensitivity and specificity after amplification are usually lower.
Analysis of quantitative predictive power. (A) Unamplified samples. Bar plots represent the Pearson’s correlation coefficient r, between the log2 predicted score and the log2 actual spike-in fold-change of the top 100 predicted sites. Arrows below each bar graph point to scatterplots representative of data from each microarray platform. In the scatterplots, true positives are shown as black dots, with the number of true positives indicated above the dots in black type at each fold-change level. The number of false negatives is indicated in purple type below the points at each fold-change level. The solid line represents the LOWESS smoothed curve for all true positives. False positives are shown as green triangles, and are on the far left of the graph because of their actual log2 fold-change values of 0. (B) The same as A, but for Amplified samples.
Simple tandem repeats and segmental duplications are often associated with false calls
The ability of a tiling microarray to correctly identify a particular sequence often depends on the nucleotide content of that sequence, probe coverage in low-complexity sequences, and potential for cross-hybridization (Okoniewski and Miller 2006; Royce et al. 2007). Therefore, we used each list of predictions to examine the false positives, false negatives, and true positives with relation to GC content, repeat content, and simple tandem repeat content (Benson 1999).
The spike-in mixtures are based on predicted promoters, which are often biased toward high GC content. However, the average GC content of our spike-in clones was actually lower than the average across the entire genome (38% vs. 41%, respectively). We found that across all platforms, peak detection algorithms, and amplification methods, GC content does not vary among false positives, false negatives, true positives, and the spike-in key. Our spike-in clones harbor a significant number of RepeatMasked regions (28% of total nucleotides across all clones), which results in reduced probe coverage on most array platforms. For one algorithm, MA2C, RepeatMasked sequences accounted for a disproportionate number of false-positive predictions on both the Agilent and NimbleGen platforms, and in amplified and unamplified experiments. The other algorithms and platforms generally had fewer RepeatMasked sequences among false positives than across all spike-in clones (Supplemental Tables 3 and 4).
Simple tandem repeats (Benson 1999), which are often not masked by RepeatMasker, were frequently associated with false positives and false negatives (Supplemental Tables 3 and 4). For many algorithms and labs, false-positive predictions on NimbleGen arrays contained more than 10 times as many simple tandem repeat nucleotides as the spike-in sample key. Also, particularly in the amplified samples, false negatives on the NimbleGen platform also had significantly higher simple tandem repeat content than the spike-in sample key. Therefore, the data indicate that simple tandem repeat regions are associated with both false-positive and false-negative calls, particularly in amplified samples. It appears that a simple post-processing filter that removes peak predictions rich with simple tandem repeats could significantly reduce false positives.
Segmental duplications (Bailey et al. 2001) that are not RepeatMasked often have tiling-array coverage, but may frequently appear as false positives under normal hybridization conditions if present in sufficient copy number. We used BLAT (Kent 2002) to query the RepeatMasked spike-in clone sequences against the human genome and found that 12% of the clones in the undiluted and diluted spike-in samples had more than one significant BLAT match in the genome (Supplemental Tables 3 and 4). The same analysis on the false-positive predictions for each array and algorithm combination found that predictions on Agilent arrays consistently contain fewer regions with multiple BLAT hits genome-wide than those on other platforms. Regardless of the peak-calling algorithm or whether the samples were amplified, false positives on the NimbleGen platform had consistently more across-genome redundancy as indicated by BLAT than was present in the spike-in mixtures. In one experiment, nearly 80% of the false positives matched at least one other region in the genome (Supplemental Tables 3 and 4). The absolute number of false positives in this experiment is small, thus eliminating sequences with this simple analysis could greatly improve the overall predictions.
Cost versus detection power
As ChIP-chip efforts scale to the full genome, the considerations of sensitivity and specificity are complicated by the fact that for many laboratories, oligonucleotide densities practical for ENCODE-scale (∼30 Mb) arrays are not currently practical for genome-wide (∼3 Gb) arrays. Different platforms offer various depths of coverage of the genome, and often the coverage is flexible even within a platform type. The cost of performing such experiments varies widely (Fig. 6A). Given the variety of options, we used our simulated ChIP-chip measurements to model the prospective performance of arrays with lower probe densities (Fig. 6B).
Cost versus detection power: simulation of whole-genome experiments. (A) Summary statistics for the simulation of commercial whole-genome tiling array experiments. (B) Array performance as a function of replicate number and tiling resolution (see Methods). AUC values are indicated by color (key at bottom). Black numbers on the top indicate the percentage of probes remaining on the ENCODE array in the simulation. The red coordinates at the bottom indicate the corresponding array resolution, assuming a 1-kb region of ChIP enrichment. The currently available (August 2007) commercial whole-genome tiling array resolution is underlined. (C) Array sensitivity according to enrichment level. As in Figure 3, the spike-in clones were divided into four levels of enrichment: High (64–192 fold); Medium (6–10 fold); Low (3–4 fold); and Ultra Low (1.25–2 fold). Sensitivity at each enrichment level is defined as the percentage of correctly predicted clones, with the total number of false positives equal to 5% of the total number of spike-in clones (color key at bottom). The array platforms are indicated along the X-axis. (D). Using our deletion analysis and current (August 2007) list prices for each commercial array technology, we calculated the number of probes and dollar amount required to produce a given AUC value (left panel). The minimum number of probes required to achieve a given AUC was determined by using the information in panel B for each platform, assuming a 1.5-Gb nonrepetitive genome. For Affymetrix, a single-channel platform, the need to perform separate ChIP and control/input hybridizations was accounted for in calculating probe number. In the righthand panel, the minimum cost required to achieve a given AUC value is plotted.
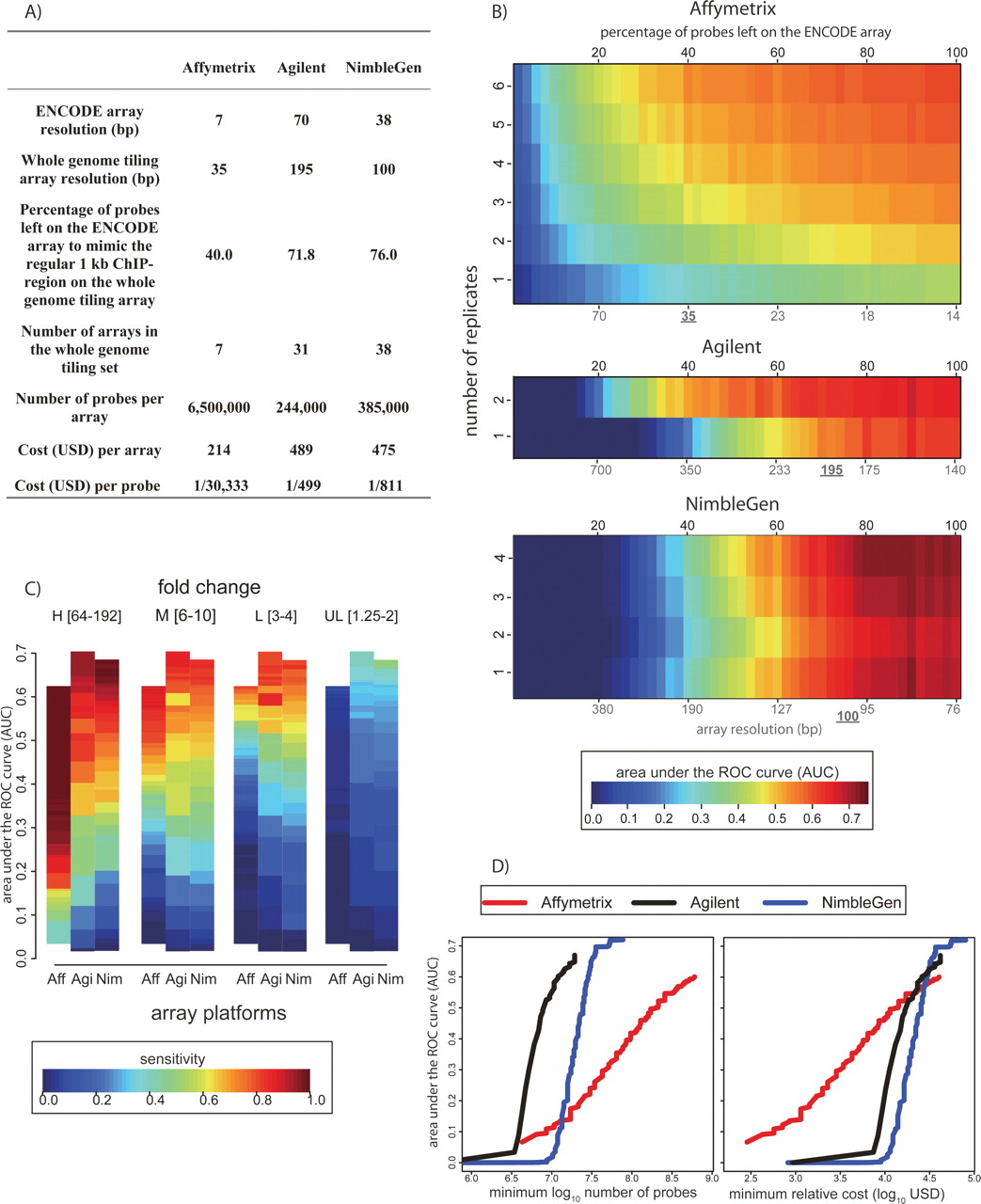
Our spike-in clones covered only ∼500 bp, but in a typical ChIP experiment ∼1 kb of DNA surrounding a site of protein–DNA interaction is enriched. To account for this in our estimation of array performance with respect to probe density, we evenly deleted probes in silico so that the absolute number of probes covering the 500-bp spike-in region would be equivalent to the number covering a 1-kb region normally enriched in a ChIP experiment. For example, an ∼1-kb region enriched in a hypothetical ChIP-chip experiment might span 10 NimbleGen probes at the 100-bp whole-genome tiling resolution, whereas an ∼500-bp spike-in clone is covered by 13 NimbleGen probes on the 38-bp resolution ENCODE array. In this scenario, to simulate whole-genome tiling array performance, we deleted NimbleGen probes (Methods) such that 10 probes would be left to cover each 500-bp region (∼50-bp resolution). For each platform, we used the same probe deletion approach, and the best and the most pragmatic current estimate for probe densities of whole-genome tiling arrays available (Fig. 6A). Since some platforms allow custom designs that make any density and number of probes theoretically possible, we extended our analysis by gradually deleting an increasing percentage of probes on the arrays so as to provide performance estimates over a wide range of potential probe densities (Fig. 6B). Lower-resolution arrays generally have a lower AUC than their denser counterparts. Furthermore, replicates are essential to increase the AUC for experiments with lower probe densities, especially for Affymetrix, which requires at the very least three replicates to generate an AUC greater than 0.4 at the projected genome-wide tiling resolution. Researchers must calibrate their desired AUC values based on the number of arrays and probe densities that are practical (Fig. 6B).
Next, we examined sensitivity at different probe densities as a function of the true enrichment values (Fig. 6C). We again find that arrays perform significantly better at higher enrichment levels, but at lower probe densities, none of the platforms were able to detect ultra-low enrichment. Particularly for the Low (threefold to fourfold) enrichment values, higher probe densities are critical for acceptable levels of sensitivity. For example, on Affymetrix arrays at the 0.5 AUC level, one would detect 100% of the High (64–192-fold), 80% of the Medium (sixfold to 10-fold), 45% of the Low (threefold to fourfold), and almost no Ultra Low (1.25–2-fold-change) targets. Therefore, investigators may wish to characterize levels of enrichment in their ChIP samples to determine the best array platforms to use and to calibrate the optimal probe density and number of replicates to perform without incurring unnecessary expenditure.
Finally, we examined the number of probes and cost required to achieve various AUC values across the three platforms. Affymetrix offers the greatest probe density of any platform, although it also requires far more probes than Agilent and NimbleGen platforms to achieve similar AUC values (Fig. 6D). However, the much lower cost per probe afforded by Affymetrix makes the cost to achieve less than 0.5 AUC values lower overall, relative to other platforms (Fig. 6E). If AUC levels greater than 0.5 are desired, the cost of the three platforms becomes virtually identical.
Discussion
We have conducted the most comprehensive study to date of tiling microarray platforms, DNA amplification protocols, and data analysis algorithms, with respect to their effect on the results of ChIP-chip experiments.
Tiling arrays from all commercial companies tested worked well at the 5% false discovery ratio (∼10% FDR) level, especially using the optimal experimental protocol with the best analysis algorithm. NimbleGen and Agilent arrays are more sensitive at detecting regions with very low enrichment (1.25- to twofold), likely owing to longer oligonucleotide probes and probe sequence optimization. The results of Affymetrix experiments benefit more from replicates than other platforms. The variation between laboratories, protocols, and analysis methods within the same platform is similar to, if not greater than, the variation between the best results from different platforms. Clearly, even investigators using the same platform must work toward better standard operating procedures and develop quality control metrics to monitor quality of reagents and arrays.
We found that both the WGA and LM-PCR protocols produce results comparable to corresponding undiluted samples and are very effective at detecting low-enrichment regions. Different analysis algorithms are appropriate for different tiling-array platforms. MAT seems to work best on Affymetrix tiling arrays. Splitter and Agilent’s internal WA or ADM-1 algorithms are the best for Agilent tiling arrays. For NimbleGen tiling arrays, TAMALg, Splitter, and NimbleGen’s internal permutation algorithms work better for the unamplified samples, and TAMALg, MA2C, and Tilescope (Zhang et al. 2007) work better for the amplified samples.
We note that the conclusions we report are supported by many aspects of the data in aggregate, rather than being dependent on a specific property of any individual experiment. Therefore, although factors such as the inclusion or exclusion of individual investigators, the particular batches of reagents or arrays used, or sets of algorithm parameters might have slightly changed the results of individual experiments reported here, the overall conclusion of the evaluation is robust with respect to these variables. Nonetheless, as with any study, there are shortcomings here. For example, NimbleGen seems to be the relatively more successful commercial platform in this study, but it is possible that this is a result of more experiments and analyses being performed with this platform. In the same way that between two people randomly drawing numbers from the same normal distribution N(μ, σ2), a person drawing 10 numbers is more likely to get the highest number than a person drawing only five, the platform with the most replicates, laboratories, and algorithms tested has an advantage among closely matched competitors. Another note of caution concerns our analysis of whole-genome array performance. All commercial tiling-array companies have proprietary algorithms for probe selection based on the hybridization quality of oligonucleotide probes. However, the effectiveness of these algorithms diminishes when probes are tiled at very high resolution, since there are simply not enough biochemically optimal probes to choose from at such resolution. Therefore, probes on the ENCODE arrays might be less optimal than those in the whole-genome arrays (which are at a lower tiling resolution) from the same platform. As a result, our simulated probe deletion analysis might underestimate the actual whole-genome array performance, especially for Affymetrix tiling arrays. Finally, the spike-in DNA used in this study has a different fragment length distribution than a real ChIP-chip sample. Real ChIP-enriched regions often have peak-shaped profiles instead of uniform enrichment across the entire region, thus algorithms modeling peak shapes may perform better with real ChIP-chip data than the spike-in signal. Nonetheless, the spike-in strategy we used provides the most feasible benchmark for the factors we are evaluating.
In this simulated ChIP-chip experiment, we have found that commercial tiling arrays perform remarkably well even at relatively low levels of enrichment. We also found that the cost to achieve similar sensitivity between the commercial tiling-array platforms is comparable. Tiling microarrays from all commercial companies continue to get less expensive and to deliver continually higher probe densities. Simultaneously, new detection technologies such as high-throughput sequencing are emerging (Johnson et al. 2007). To date, there has been no systematic comparison of ChIP-chip and ChIP-seq, or ChIP-seq performed on different sequencing platforms. Our spike-in library and data set might be used for such a purpose, and we hope that this study and our spike-in library will encourage continued rigorous competition and comparison between all of the genomic detection platforms.
Methods
Validation of the simulated ChIP sample
The simulated ChIP sample was validated in three ways: (1) sequencing of the original clone preps before dilution, (2) sequencing of the diluted clones with PCR preamplification using universal primers, and (3) inserting specific PCR of the diluted clones, followed by agarose gel electrophoresis. Our experimental validation revealed no anomalies in the spike-in mixtures, and our analysis of the array predictions adds extra evidence that the libraries were mixed at the proper stoichiometries and that the clone identities were correct.
Simulated ChIP amplification, array hybridization, and data analysis
Detailed descriptions of each experimental procedure and analysis algorithm are described in the Supplemental material.
Probe and replicate deletion simulation
We evenly and gradually deleted probes in silico at 2% intervals, such that at each step there are 100%, 98%, 96%, . . . , 2% of probes left on the arrays. At each step, we repeated this probe deletion five times with randomly selected starting positions to form five different array designs. Shown in this study is the average area under the ROC curve of all replicate combinations on all five array designs. For example, the Affymetrix analysis was generated from 15,750 different array predictions, based on 63 possible replicate combinations derived from the six available experiments (from one to six replicates: 6 + 15 + 20 + 15 + 6 + 1 = 63), five different array designs, and 50 different probe deletion steps.
Sequence analysis of array predictions
For each group of array predictions, we binned the predicted regions into false negatives, false positives, and true positives. For false positives, 200 bp of reference human sequence was added 5′ and 3′ of the predicted location. We then calculated the percent GC, the percent RepeatMasked, and the percent simple tandem repeats across the sequences in each group based on UCSC genome annotations (http://genome.ucsc.edu). For the BLAT (Kent 2002) analysis, we used a cutoff score >30 to find similar sequences in the genome for each clone.
Acknowledgments
We thank NimbleGen, Affymetrix, and Agilent for arrays and technical support, and the NHGRI ENCODE project and all of the ENCODE PIs for funding and logistical support. We thank Marc Halfon for advice. Additional funding support for the project was provided by NIH grant 1R01 HG004069-01. Affymetrix, Agilent, and NimbleGen Systems (now Roche NimbleGen) contributed reagents and expertise for the experiments presented in this paper. These companies may stand to benefit financially from publication of the results.
References
- ↵J.A. Bailey,A.M. Yavor,H.F. Massa,B.J. Trask,E.E. Eichler,(2001) Segmental duplications: Organization and impact within the current human genome project assembly. Genome Res. 11:1005–1017.
- ↵G. Benson,(1999) Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27:573–580.
- ↵M. Bieda,X. Xu,M.A. Singer,R. Green,P.J. Farnham,(2006) Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 16:595–605.
- ↵J.S. Carroll,C.A. Meyer,J. Song,W. Li,T.R. Geistlinger,J. Eeckhoute,A.S. Brodsky,E.K. Keeton,K.C. Fertuck,G.F. Hall,(2006) Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 38:1289–1297.
- ↵S. Cawley,S. Bekiranov,H.H. Ng,P. Kapranov,E.A. Sekinger,D. Kampa,A. Piccolboni,V. Sementchenko,J. Cheng,A.J. Williams,(2004) Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116:499–509.
- ↵S.E. Choe,M. Boutros,A.M. Michelson,G.M. Church,M.S. Halfon,(2005) Preferred analysis methods for Affymetrix GeneChips revealed by a wholly defined control dataset. Genome Biol. 6:R16, 10.1186/gb-2005-6-2-r16.
- ↵S.J. Cooper,N.D. Trinklein,E.D. Anton,L. Nguyen,R.M. Myers,(2006) Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Res. 16:1–10.
- ↵The ENCODE Project Consortium (2007) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447:799–816.
- ↵G.M. Euskirchen,J.S. Rozowsky,C.L. Wei,W.H. Lee,Z.D. Zhang,S. Hartman,O. Emanuelsson,V. Stolc,S. Weissman,M.B. Gerstein,(2007) Mapping of transcription factor binding regions in mammalian cells by ChIP: Comparison of array- and sequencing-based technologies. Genome Res. 17:898–909.
- ↵S.E. Hanlon,J.D. Lieb,(2004) Progress and challenges in profiling the dynamics of chromatin and transcription factor binding with DNA microarrays. Curr. Opin. Genet. Dev. 14:697–705.
- ↵M.E. Hudson,M. Snyder,(2006) High-throughput methods of regulatory element discovery. Biotechniques 41:673–681.
- ↵T.R. Hughes,M. Mao,A.R. Jones,J. Burchard,M.J. Marton,K.W. Shannon,S.M. Lefkowitz,M. Ziman,J.M. Schelter,M.R. Meyer,(2001) Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat. Biotechnol. 19:342–347.
- ↵R.A. Irizarry,D. Warren,F. Spencer,I.F. Kim,S. Biswal,B.C. Frank,E. Gabrielson,J.G. Garcia,J. Geoghegan,G. Germino,(2005) Multiple-laboratory comparison of microarray platforms. Nat. Methods 2:345–350.
- ↵W.E. Johnson,W. Li,C.A. Meyer,R. Gottardo,J.S. Carroll,M. Brown,X.S. Liu,(2006) Model-based analysis of tiling-arrays for ChIP-chip. Proc. Natl. Acad. Sci. 103:12457–12462.
- ↵D.S. Johnson,A. Mortazavi,R.M. Myers,B. Wold,(2007) Genome-wide mapping of in vivo protein–DNA interactions. Science 316:1497–1502.
- ↵J. Jurka,(2000) Repbase update: A database and an electronic journal of repetitive elements. Trends Genet. 16:418–420.
- ↵W.J. Kent,(2002) BLAT—The BLAST-like alignment tool. Genome Res. 12:656–664.
- ↵T.H. Kim,B. Ren,(2006) Genome-wide analysis of protein–DNA interactions. Annu. Rev. Genomics Hum. Genet. 7:81–102.
- ↵T.H. Kim,L.O. Barrera,M. Zheng,C. Qu,M.A. Singer,T.A. Richmond,Y. Wu,R.D. Green,B. Ren,(2005) A high-resolution map of active promoters in the human genome. Nature 436:876–880.
- ↵T.I. Lee,R.G. Jenner,L.A. Boyer,M.G. Guenther,S.S. Levine,R.M. Kumar,B. Chevalier,S.E. Johnstone,M.F. Cole,K. Isono,(2006) Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125:301–313.
- ↵C.L. Liu,S.L. Schreiber,B.E. Bernstein,(2003) Development and validation of a T7 based linear amplification for genomic DNA. BMC Genomics 4:19, 10.1186/1471-2164-4-19.
- ↵I. Lucas,A. Palakodeti,Y. Jiang,D.J. Young,N. Jiang,A.A. Fernald,M.M. Le Beau,(2007) High-throughput mapping of origins of replication in human cells. EMBO Rep. 8:770–777.
- ↵MAQC Consortium (2006) The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 24:1151–1161.
- ↵H. O’Geen,C.M. Nicolet,K. Blahnik,R. Green,P.J. Farnham,(2006) Comparison of sample preparation methods for ChIP-chip assays. Biotechniques 41:577–580.
- ↵H. O’Geen,S.L. Squazzo,S. Iyengar,K. Blahnik,J.L. Rinn,H.Y. Chang,R. Green,P.J. Farnham,(2007) Genome-wide analysis of KAP1 binding suggests autoregulation of KRAB-ZNFs. PLoS Genet. 3:e89, 10.1371/journal.pgen.0030089.
- ↵M.J. Okoniewski,C.J. Miller,(2006) Hybridization interactions between probesets in short oligo microarrays lead to spurious correlations. BMC Bioinformatics 7:276, 10.1186/1471-2105-7-276.
- ↵T.A. Patterson,E.K. Lobenhofer,S.B. Fulmer-Smentek,P.J. Collins,T.M. Chu,W. Bao,H. Fang,E.S. Kawasaki,J. Hager,I.R. Tikhonova,(2006) Performance comparison of one-color and two-color platforms within the MicroArray Quality Control (MAQC) project. Nat. Biotechnol. 24:1140–1150.
- ↵T.E. Royce,J.S. Rozowsky,M.B. Gerstein,(2007) Assessing the need for sequence-based normalization in tiling microarray experiments. Bioinformatics 23:988–997.
- ↵P.C. Scacheri,G.E. Crawford,S. Davis,(2006) Statistics for ChIP-chip and DNase hypersensitivity experiments on NimbleGen arrays. Methods Enzymol. 411:270–282.
- ↵J.S. Song,W.E. Johnson,X. Zhu,X. Zhang,W. Li,A.K. Manrai,J.S. Liu,R. Chen,X.S. Liu,(2007) Model-based analysis of 2-color arrays (MA2C) Genome Biol. 8:R178, 10.1186/gb-2007-8-8-r178.
- ↵A. Yang,Z. Zhu,P. Kapranov,F. McKeon,G.M. Church,T.R. Gingeras,K. Struhl,(2006) Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol. Cell 24:593–602.
- ↵Z.D. Zhang,J. Rozowsky,H.Y. Lam,J. Du,M. Snyder,M. Gerstein,(2007) Tilescope: Online analysis pipeline for high-density tiling microarray data. Genome Biol. 8:R81, 10.1186/gb-2007-8-5-r81.
Footnotes
[1] [Supplemental material is available online at www.genome.org. The microarray data from this study have been submitted to Gene Expression Omnibus under accession no. GSE10114.]
[2] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.7080508