
Abstract
Comprehensive identification and cataloging of copy number variations (CNVs) is required to provide a complete view of human genetic variation. The resolution of CNV detection in previous experimental designs has been limited to tens or hundreds of kilobases. Here we present PennCNV, a hidden Markov model (HMM) based approach, for kilobase-resolution detection of CNVs from Illumina high-density SNP genotyping data. This algorithm incorporates multiple sources of information, including total signal intensity and allelic intensity ratio at each SNP marker, the distance between neighboring SNPs, the allele frequency of SNPs, and the pedigree information where available. We applied PennCNV to genotyping data generated for 112 HapMap individuals; on average, we detected ∼27 CNVs for each individual with a median size of ∼12 kb. Excluding common rearrangements in lymphoblastoid cell lines, the fraction of CNVs in offspring not detected in parents (CNV-NDPs) was 3.3%. Our results demonstrate the feasibility of whole-genome fine-mapping of CNVs via high-density SNP genotyping.
Copy number variation (CNV) refers to duplication or deletion of a segment of DNA sequence compared to a reference genome assembly. Several large-scale studies have reported the presence of copy number variation in humans, suggesting that CNVs may account for a significant proportion of human phenotypic variation, including disease susceptibility (Feuk et al. 2006; Freeman et al. 2006; Eichler et al. 2007; McCarroll and Altshuler 2007). The comprehensive identification and cataloging of CNVs would greatly benefit the genetic and functional analysis of human genome variation. Results from several in silico studies (Tuzun et al. 2005; Conrad et al. 2006; Khaja et al. 2006; McCarroll et al. 2006) demonstrate that small-scale CNVs, including those <10 kb, are common in the human genome. However, previous experimental studies, performed primarily by microarray Comparative Genomic Hybridization (array-CGH) techniques, are limited to detection of CNVs of tens or hundreds of kilobases (Iafrate et al. 2004; Ishkanian et al. 2004; Sebat et al. 2004; Fiegler et al. 2006; Mills et al. 2006; Redon et al. 2006; Carter 2007; Scherer et al. 2007; Wong et al. 2007). Owing to improved resolution and genome coverage, whole-genome SNP genotyping arrays offer an alternative and more sensitive method for CNV detection. For example, a widely used whole-genome SNP genotyping platform, the Illumina HumanHap550 BeadChip (Gunderson et al. 2005; Steemers and Gunderson 2007), assays more than half a million SNPs in parallel (median SNP distance ∼3 kb), permitting kilobase-resolution detection of CNVs.
Several technical advantages in the Illumina Infinium platform make it highly suitable for high-resolution CNV detection. The assay combines specific hybridization of genomic DNA to arrayed probes with allele-specific primer extension and signal amplification, thus achieving a high signal-to-noise ratio in genotype calling (Gunderson et al. 2005). The assay does not require PCR-based amplification, thus detected signals are less susceptible to biases caused by differential amplification of given chromosomal regions. In addition to the total fluorescent intensity signals from both sets of probes/alleles at each SNP (referred to as the “log R Ratio”), the Illumina platform also allows inference of the relative ratio of the fluorescent signals between two probes/alleles at each SNP (referred to as the “B Allele Frequency”). Furthermore, data normalization at each SNP on the Infinium platform is performed by comparison of signals from a set of reference samples (e.g., HapMap samples), leading to less signal variation between SNPs.
Conventional methods for CNV identification on the Illumina platform involve examination of intensity signals (implemented in the LOH-plus module of the BeadStudio software), which identifies copy number changes by calculating the mode of B Allele Frequency for SNPs in a sliding window along the chromosome. While simple to implement, the sliding window approach has limited and relatively coarse boundary resolution for detected CNVs. A recently described algorithm, QuantiSNP, incorporates the log R Ratio and B Allele Frequency simultaneously in a hidden Markov model (HMM) framework (Colella et al. 2007). As demonstrated by simulations and by studies on individuals with known large aberrations, QuantiSNP significantly improves the resolution of CNV detection. The development of algorithms that both accurately model the signal measures and integrate more available data (e.g., genotype frequency, family relationship) can potentially lead to further improvement of CNV detection.
Here we present an integrated HMM algorithm, called “PennCNV,” to detect CNVs with high resolution using the Illumina Infinium assay. To better reflect the distribution of the intensity data, we constructed accurate models for log R Ratio and B Allele Frequency and developed more realistic models for state transition between different copy number states. In addition, PennCNV incorporates the population allele frequency for each SNP and the distance between adjacent SNPs. Several studies have demonstrated the heritability of CNVs (Locke et al. 2006; Redon et al. 2006), suggesting that using information from related family members can improve the sensitivity for CNV detection and accuracy of boundary mapping. Therefore, we incorporated a Bayesian approach into PennCNV to use family information for a posteriori CNV validation and CNV boundary mapping. The accurate prediction of CNV boundaries permits breakpoint mapping by PCR amplification and resequencing. The application of PennCNV to a large group of individuals demonstrates the feasibility of whole-genome fine-mapping of CNVs through high-density SNP genotyping.
Results
The HMM modeling strategy
To develop a strategy for detection of CNVs using the Illumina Infinium high-density SNP genotyping platform (Peiffer et al. 2006), we used the genotyping data generated on the HumanHap550 array for 112 HapMap individuals (16 CEU father–mother–offspring trios from Utah [CEU], 12 Yoruba trios from Ibadan, Nigeria [YRI], 28 unrelated Chinese and Japanese individuals from Beijing and Tokyo, respectively [CHB+JPT]), 300 disease-free children from the Children’s Hospital of Philadelphia (CHOP cohort), and 40 trios from an ongoing disease cohort study (AGRE cohort) (Geschwind et al. 2001). Compared to many algorithms that use “loss,” “normal,” and “gain” to model CNV states, we adopted a six-state definition (Colella et al. 2007) for more precise modeling of CNV events (Table 1). The BeadStudio software from Illumina displays two summary measures for a genotype signal at each SNP: the log R Ratio (LRR), a measure of normalized total signal intensity, and the B Allele Frequency (BAF), a measure of normalized allelic intensity ratio (Supplemental Table 1). To demonstrate the patterns of LRR and BAF in regions with copy number changes, we plotted these values from an individual with a 10-Mb four-copy duplication and an adjacent 2-Mb three-copy duplication on chromosome 15q (Fig. 1). The combination of LRR and BAF can be used together to determine several different copy numbers and to differentiate copy-neutral LOH (loss of heterozygosity) regions from normal state regions, supporting the utility of six distinct copy number states in the modeling strategy.
Hidden states, copy numbers, and their descriptions
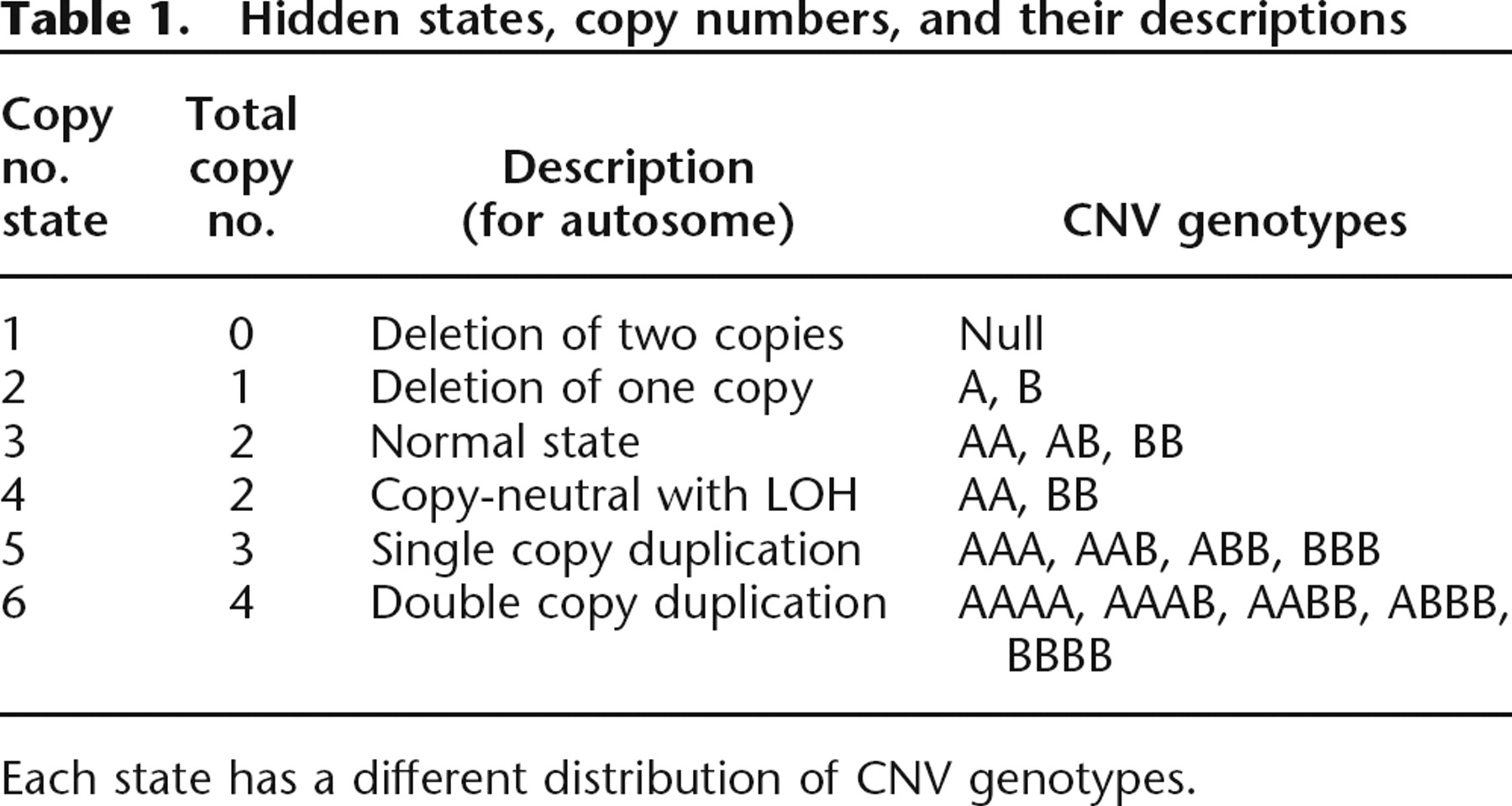
Each state has a different distribution of CNV genotypes.
An illustration of log R Ratio (LRR) and B Allele Freq (BAF) values for the chromosome 15 q-arm of an individual. A normal chromosome region has three BAF genotype clusters, as represented as AA, AB, and BB genotypes in boxes, and with LRR values centered around zero. The copy-neutral LOH region has normal LRR values, but without the AB genotype cluster. The increased copy number for a CNV region can be detected based on an increased number of peaks in the BAF distribution, as well as increased LRR values. The patterns of LRR and BAF for different CNV regions, normal regions, and copy-neutral LOH regions are distinct from each other, thus the combination of LRR and BAF can be used to generate CNV calls.
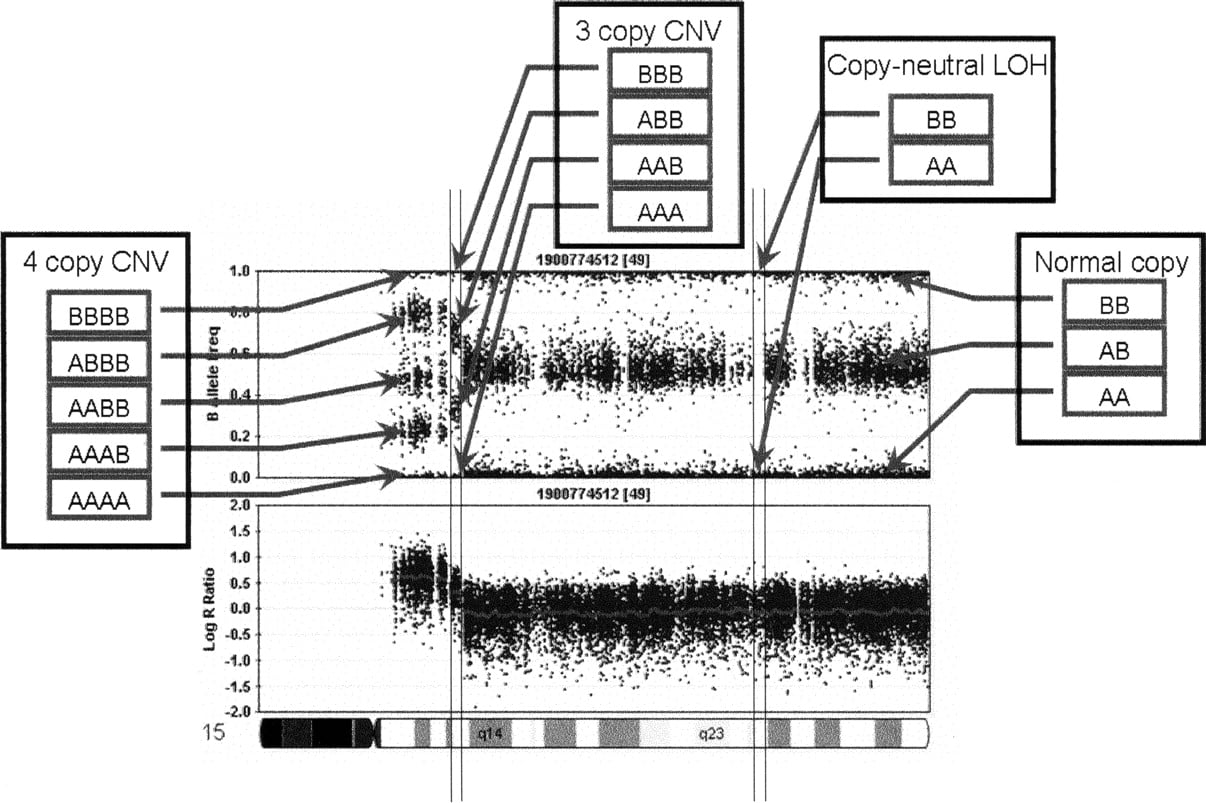
To exploit all available information for each SNP to its full potential, PennCNV incorporates several components together into a hidden Markov model (HMM), including the LRR, the BAF, the distance between neighboring SNPs, and the population frequency of the B allele (Fig. 2). Both the LRR and BAF values can be displayed and exported from BeadStudio given that there is an appropriate clustering file with canonical cluster positions for each SNP. The distance between neighboring SNPs determines the probability of having a copy number state change between them. Each SNP has two alleles referred to as the A and B alleles, thus we use the term “population frequency of B allele” to differentiate it from the BAF term that measures allelic intensity ratio. The values for population frequency of B allele for all SNPs are compiled from a large set of individuals with mixed ethnic backgrounds and of normal phenotypes; the likelihood of the copy number genotypes for each copy number state is then determined.
A flowchart outlining the procedure for CNV calling from genotyping data. The first step for LRR and BAF calculation can be alternatively performed by the BeadStudio software, given a clustering file containing canonical genotype cluster positions. The HMM integrates several sources of information to give CNV calls. When genotype data are available for family members, the pedigree information can be incorporated to model CNV events more accurately.
Since the majority of CNVs in offspring should be inherited from either parent (Locke et al. 2006), genotype data from family-based studies provide additional means for validating and fine-mapping CNV boundaries. For example, since there are several possible configurations of CNV calls in the same region in a father–mother–offspring trio (Supplemental Fig. 1), integrating the family information could help identify the most likely configuration. In our CNV detection procedure (Fig. 2), family information is used to jointly update the CNV status of all family members.
Comparative analysis of CNV detection on HapMap individuals
We applied PennCNV to genotype data derived from 112 HapMap individuals who had been genotyped by the Illumina HumanHap550 SNP genotyping platform (Table 2). In our initial analysis, the CNV calls were generated without the use of family information. We detected slightly more CNVs per individuals in YRI samples than the CEU and CHB + JPT samples (∼28 vs. ∼22), whereas the CNV size distributions are similar between populations (Supplemental Fig. 2). Deletions are approximately twofold more than duplications, but have smaller size than duplications (Supplemental Fig. 3). The detected CNVs are dispersed across the genome, with several regions showing especially high frequency of CNVs (Supplemental Fig. 4). These include an intergenic region between HTR1B and IRAK1BP1 (6q14.1), an olfactory receptor gene cluster (OR4C11-OR5L2; 11q11), and a leukocyte immunoglobulin-like receptor gene cluster (LILRB3-LILRB5; 19q13.42). These prevalent CNVs were also reported by several other CNV publications (Sebat et al. 2004; Tuzun et al. 2005; Conrad et al. 2006; McCarroll et al. 2006; Redon et al. 2006).
The CNV calling results for 112 HapMap individuals

After using family information, more CNVs with smaller sizes are detected in the CEU and YRI populations.
Multiple genome-wide studies using array-CGH have shown that chromosome rearrangements tend to occur in genomic regions exhibiting segmental duplication (Iafrate et al. 2004; Sebat et al. 2004; Sharp et al. 2005; Locke et al. 2006). However, CNVs with larger sizes are much more likely to be associated with segmental duplications than shorter CNVs (Tuzun et al. 2005; Conrad et al. 2006), thus the role of segmental duplications in generating CNVs may be overestimated in previous studies (Conrad and Hurles 2007). We retrieved segmental duplication regions from the UCSC Genome Browser (Kuhn et al. 2007) and consolidated them into a list of 8546 non-overlapping regions encompassing 154 Mb (Bailey et al. 2001). We found that 604/2633 CNVs (30 Mb/125 Mb) in HapMap individuals overlap with these regions, suggesting that fine-scale CNVs detected by high-density SNP genotyping arrays are also enriched in regions with segmental duplications.
To assess the performance of PennCNV, we next compared the CNV calls in the Illumina HumanHap550 data with those published in a recent study that examined the global variations of CNVs using HapMap individuals on two different platforms: the Whole Genome TilePath array (WGTP) and the Affymetrix 500K Early Access array (500K_EA) (Redon et al. 2006). The WGTP platform is an array-CGH technique with 26,574 large-insert clones covering 93.7% of the euchromatic regions of the human genome. The Affymetrix 500K_EA platform is another line of high-density SNP genotyping technology. Comparison of the average number of CNVs per individual and the mean length of CNVs indicates that the application of PennCNV on HumanHap550 genotyping data detects substantially smaller CNVs than the other two platforms (Table 3). The improved resolution could be due to the combination of higher data quality from the Illumina platform and the unique use of integrated information employed by our PennCNV algorithm. In addition, despite the larger size of detected CNV regions by the WGTP than the HumanHap550 platform (∼18-fold difference in base counts, 3.6-fold difference in CNV counts), the overlap of detected CNVs between the 500K_EA and HumanHap550 platforms is more than that between the 500K_EA and WGTP platforms (877 vs. 806), indicating that WGTP can only detect larger CNV regions (Redon et al. 2006).
A comparison of the number and size of CNVs in 112 HapMap individuals detected by three different technical platforms
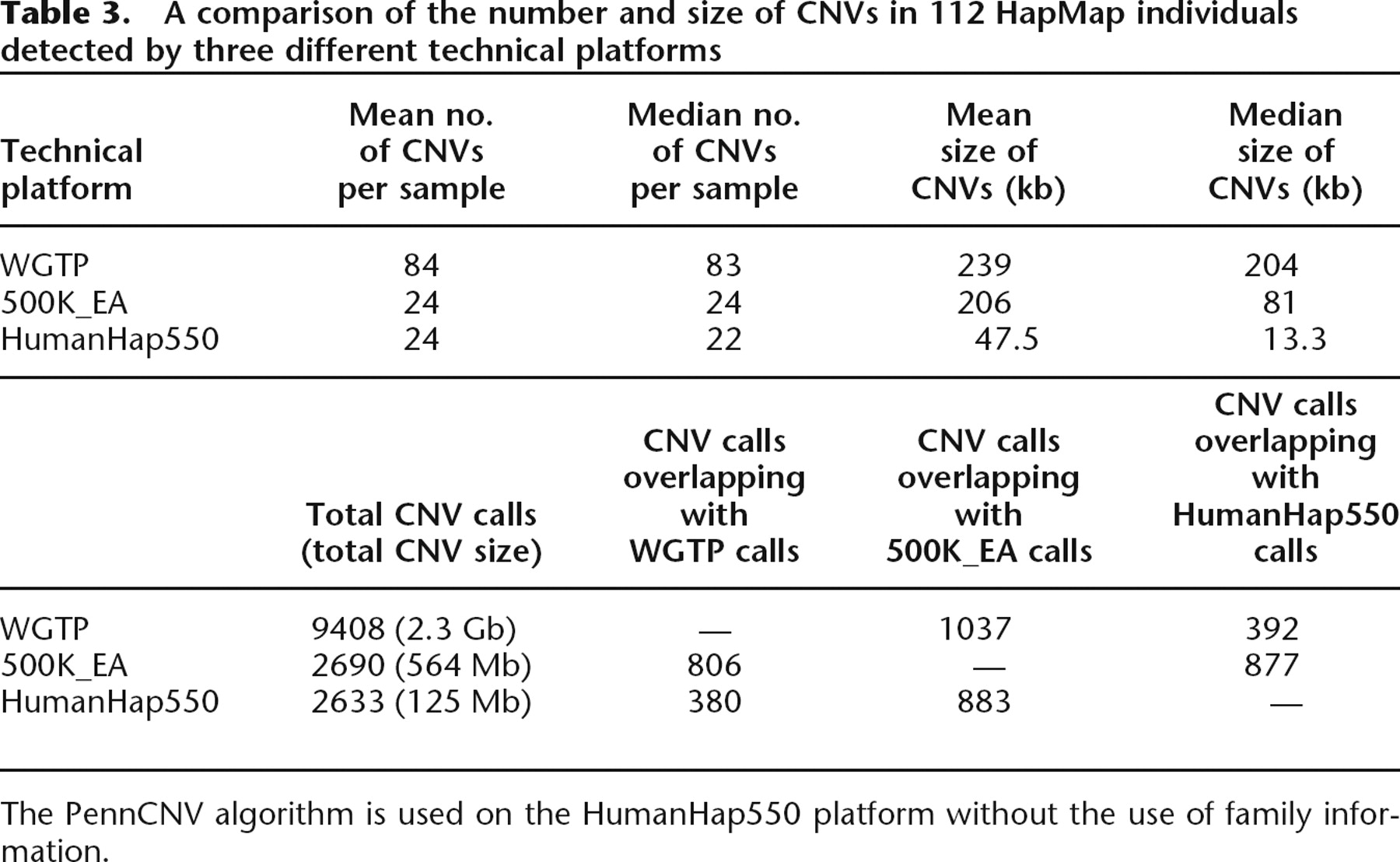
The PennCNV algorithm is used on the HumanHap550 platform without the use of family information.
The use of family information in CNV calling and validation
We believe that the vast majority of CNVs in offspring are inherited from parents (Locke et al. 2006), thus the fraction of CNVs inferred in offspring but Not Detected in Parents (CNV-NDPs) can be used as a composite measure of false-positive and false-negative rates. We therefore examined the fraction of CNV-NDPs in the HapMap CEU + YRI offspring among CNV calls generated by the three different platforms.
Firstly, with a strict criterion, we examined whether given CNVs in offspring could be detected in their parents with identical boundaries and found that 41.0% (for WGTP), 88.0% (for Affymetrix 500K_EA), and 47.4% (for Illumina HumanHap550) of the CNV calls are not inherited from parents, that is, CNV-NDPs. This criterion favors the WGTP platform because of substantially fewer probes. We next applied a relaxed evaluation criterion, by requiring that more than half of the base pairs in the offspring CNV must overlap with a parental CNV or vice versa. With this criterion, 27.1%, 20.4%, and 25.2% of offspring CNVs from the WGTP, 500K_EA, and HumanHap550 platforms are CNV-NDPs, respectively. Our comparative analysis indicates that false-positive or false-negative calls are highly prevalent in CNV detection algorithms regardless of platform or evaluation criteria and implicates the importance of using Mendelian inheritance for validation of CNV calling results and for accurate detection of CNV calls.
The PennCNV algorithm applied to data from the Illumina HumanHap550 platform allows detection of a large number of small-scale CNVs (median size: 13 kb, in comparison to 204 kb for the WGTP platform and 81 kb for the 500K_EA platform). To assess the effect of CNV length on calling accuracy, we analyzed a subset of larger CNVs, those containing >10 SNPs (median size: 69 kb), detected by the PennCNV algorithm. We found that 17.7% of offspring CNVs are CNV-NDPs with relaxed criteria, indicating that CNV-NDPs are mainly small-size CNVs. In addition, half of the CNV-NDPs actually fall within immunoglobulin regions (see below), thus ∼9% of CNV-NDPs can be explained by false-positive calls in offspring, false-negative calls in parents, or de novo CNVs.
We next examined the performance of PennCNV by incorporating family information into the calling algorithm (Table 2). After using family information, the total number of CNV calls is increased for HapMap CEU + YRI offspring (from 624 to 752) and for parents (from 1393 to 1619), indicating more sensitive CNV detection. In addition, 8.4% offspring CNVs are CNV-NDPs using the strict criterion, while 4.3% offspring CNVs are CNV-NDPs using the relaxed criterion, indicating significant reduction of CNV-NDPs after application of family information (Supplemental Fig. 5). Assuming that the vast majority of offspring CNVs are inherited from parents, we can use family-based CNV calls as a reference set to give an indirect estimate of false-positive and sensitivity measures of PennCNV in the absence of family data: 618 out of 624 offspring CNVs detected without the use of family information are also detected by family-based PennCNV, indicating a false-positive rate of 1.0% and a sensitivity of 82.2%. Similarly, using parental CNV data, we estimate that the false-positive rate is 0.2% and the sensitivity is 86%. We caution that these measures are indirect measures of algorithm performance and may be biased by the underlying assumption. Overall our analysis indicates that the use of family information significantly improves the sensitivity of CNV detection and reduces CNV-NDPs.
To examine whether our results from the HapMap individuals would apply to other study cohorts, we analyzed 40 additional trios from another ongoing study (AGRE cohort). Similar to the results on the HapMap cohort, the use of family information leads to a 24% increase of the number of CNV calls in offspring, and a 22% increase of CNV calls in parents. After using family information, the fraction of CNV-NDPs decreases from 55% to 10.1% using the strict criterion and decreases from 36% to 5.8% using the relaxed criterion. Comparing CNV calls generated with and without family information by PennCNV, we estimate that the false-positive rate is 0.8% and the sensitivity is 81.1%. Therefore, results from analysis of the AGRE cohort are in concordance with those of the HapMap individuals.
The use of family information in CNV characterization
The use of family information may also lead to better characterization of CNV-NDPs caused by somatic rearrangements or rearrangements in cell lines. The HapMap cohort and the AGRE cohort used in our study were genotyped using DNA extracted from Epstein-Barr Virus (EBV)-transformed lymphoblastoid cell lines. Recently, studies on cell-line DNA revealed high frequencies of chromosome rearrangements in several immunoglobulin regions (Simon-Sanchez et al. 2007; Wong et al. 2007). To address this issue, we compared CNVs from the HapMap cohort and the AGRE cohort with CNVs from 300 disease-free children whose DNA was isolated from whole blood. We found that three immunoglobulin regions, including IGLC1 (22q11.22, ∼800 kb), IGHA1 (14q32.33, ∼1 Mb), and the surrounding region of IGKC (2p11.2, ∼1 Mb) have elevated frequencies of CNVs in cell lines compared to whole blood (Table 4). In addition, we found that 22.6% of CNV-NDPs in HapMap offspring and 10.3% of CNV-NDPs in AGRE offspring fall within immunoglobulin regions. Therefore, the use of whole-blood samples, together with available family information, confirmed that the high frequency of chromosome rearrangements in immunoglobulin regions is a bona fide cell-line-specific phenomenon. To search for additional chromosome regions likely to be affected by non-germline copy number changes, we analyzed whether CNV-NDPs in cell line samples form frequently occurring clusters. This analysis allowed the identification of an additional locus, the T-cell alpha-chain constant region (14q11.2), which is likely to harbor CNV-NDPs. Excluding CNVs in immunoglobulin regions, the actual fraction of CNV-NDPs among all CNVs in HapMap offspring is 3.3%, indicating the high heritability of CNVs. Furthermore, examination of CNVs detected in cell lines also revealed prevalent heterosomic deletions or duplications (chromosome aberrations in subpopulations of cells). For example, regions with heterosomic deletions show decreased total signal intensity, but the allelic intensity ratio is manifested as chromosome trisomy (Supplemental Fig. 6). We found that these cell-batch-specific events usually occur throughout the entire chromosome or entire arm, especially on chromosomes X, 2, and 12 (Supplemental Fig. 7), which were also reported to form aneuploidy in other cell-line studies (Risin et al. 1992, 1993; Aardema et al. 1997; Locke et al. 2006). Thus, our analysis indicates that CNV detection algorithms based only on signal intensity can be misleading, and that genotype data on cell lines should be interpreted with caution.
Immunoglobulin-related genomic regions that show elevated frequencies of CNVs in cell line samples (HapMap cohort and AGRE cohort) compared to whole-blood samples

The increased prevalence of CNVs surrounding IGKC is less obvious, mainly because of low coverage of SNP markers in this region.
Family information can be also used to extract more biological knowledge from detected CNVs, such as inferring the parental origin of predicted de novo CNVs. To illustrate this, consider a scenario in which the father and mother genotypes at a SNP marker are AA and AB, respectively, and the PennCNV algorithm identified a de novo deletion in the offspring encompassing this SNP. If the offspring genotype call is BB (or when B Allele Frequency indicates that the actual genotype is B in the presence of the “No Call” genotype), we can infer that the de novo event happened on the paternal chromosome. Similarly, when the father, mother, and offspring genotypes are AA, BB, and AA, respectively, we can infer that the de novo event happened on the maternal chromosome. We illustrate this idea using a de novo CNV (located at 3p26, with 50 SNPs encompassing 97 kb) detected by the family-based PennCNV algorithm in the AGRE cohort (Supplemental Table 2). By manually examining the B Allele Frequency values for 50 SNPs within the CNV region in all family members (13 SNPs are informative for this analysis), we were able to unambiguously determine that the de novo event occurred on the paternal chromosome. In addition, the fact that 13/50 SNPs have Mendelian inconsistency and that all 13 SNPs support the paternal origin of the de novo event provides an additional level of validation for the predicted de novo CNV.
Identification of CNV breakpoints
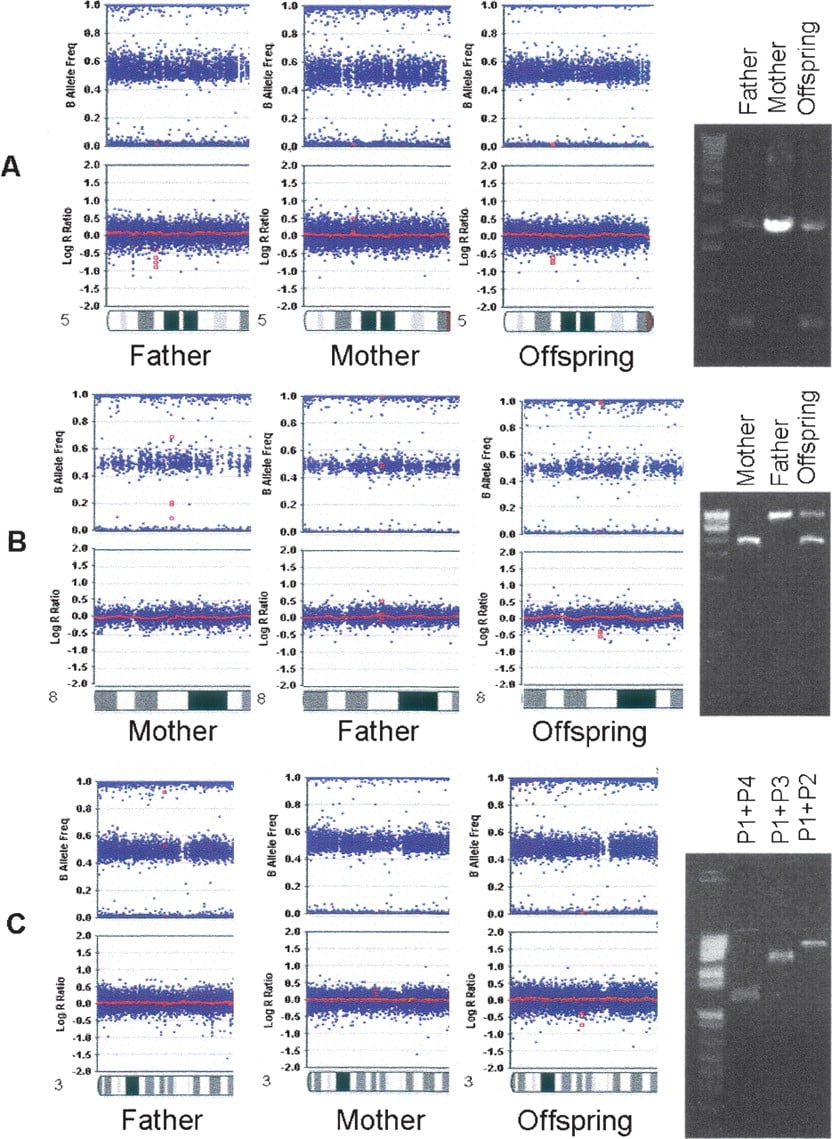
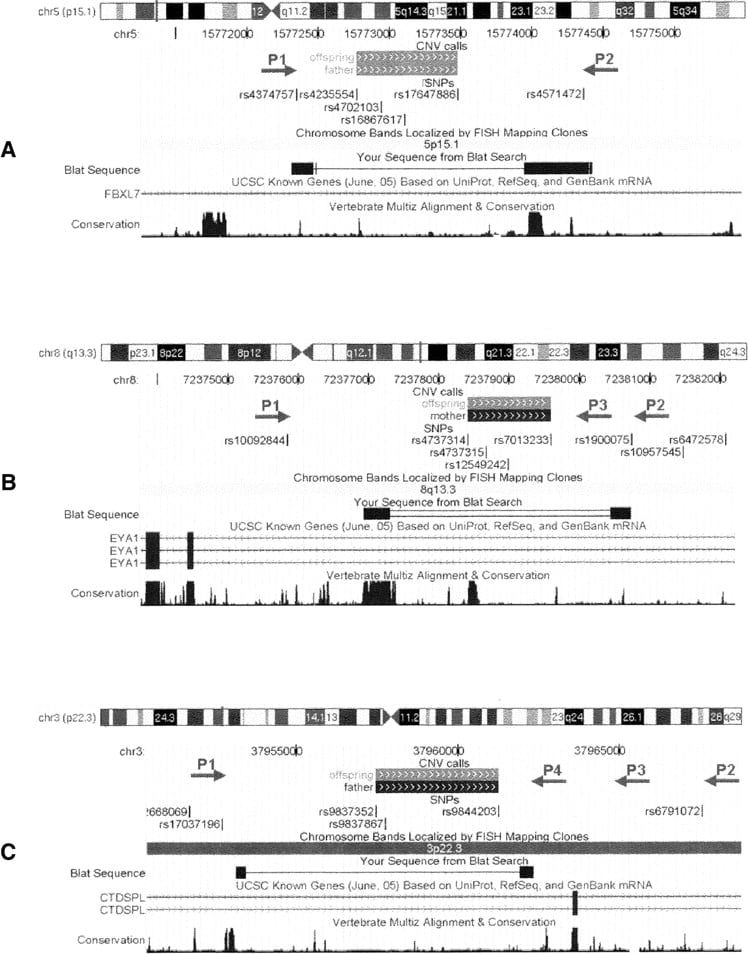
Experimental validation and precise mapping of CNV breakpoints represent an important aspect when predicting functional consequences of detected rearrangements. The high density of SNPs in the array, together with the high accuracy of CNV boundary prediction, permits selection of PCR primers for amplification of sequences around breakpoints. Figures 3 and 4 illustrate the computational prediction and experimental mapping of deletion breakpoints for three intronic CNVs (predicted size: ∼700 bp, ∼1 kb, and ∼4 kb; actual size: ∼1.4 kb, ∼3 kb, and ∼9 kb) identified within FBXL7, EYA1, and CTDSPL, respectively. These CNVs have high prevalence (>5%) in the HapMap cohort or in the AGRE cohort, and their predicted boundaries encompass or map close to conserved genomic elements (Fig. 4). We note that the CNV within FBXL7 was previously reported as 1.5 kb by Hinds et al. (2006) and as 132 kb by Redon et al. (2006), the CNV within EYA1 was previously reported as 3.2 kb by Hinds et al. (2006), and the CNV within CTDSPL was previously reported as 19 kb by Conrad et al. (2006) and 273 kb by Redon et al. (2006). Selection of PCR primers located just upstream and downstream of SNPs adjacent to the deletion in FBXL7 validated and confirmed the actual size of the CNV (Fig. 4A). PCR primer walking using one forward primer and two reverse primers outside of the deleted region in EYA1 mapped the breakpoint to an ∼700-bp genomic region between two reverse primers (Fig. 4B). PCR primer walking using one forward primer and three reverse primers outside the deleted region in CTDSPL mapped the breakpoint to an ∼1.2-kb genomic region between the forward primer and the closest reverse primer (Fig. 4C). Resequencing and BLAT alignment (Kent 2002) identified the exact breakpoints for these CNVs. In principle, similar approaches may be applied to larger deletion or duplication regions, if the outmost SNPs within a CNV are accurately predicted by a CNV-calling algorithm.
(A) A predicted ∼700-bp CNV within an intronic region of the FBXL7 gene; (B) a predicted ∼1-kb CNV within an intronic region of the EYA1 gene; and (C) a predicted ∼4-kb CNV within an intronic region of the CTDSPL gene are inherited from parent to offspring. The scatterplots for log R Ratio and B Allele Frequency are shown for the father, mother, and offspring; (red dots) the SNPs within the CNVs. The presence of CNVs and their copy numbers are validated by PCR amplification of the region encompassing breakpoints for FBXL7 and EYA1, or by PCR primer walking for CTDSPL (see Fig. 4 for more detail on primer locations).
UCSC Genome Browser (Kuhn et al. 2007) shots of the CNVs within the FBXL7 (A), EYA1 (B), and CTDSPL (C) genes, as well as the location of SNPs and PCR primers. The predicted CNV regions with (gray solid boxes) deletion of one copy or (black solid boxes) deletion of two copies on the “CNV calls” track; the actual CNV breakpoints identified by resequencing are shown in the “BLAT Search” track. For the CNV within FBXL7, a pair of PCR primers (P1 and P2) is able to generate two PCR products, thus resequencing of shorter PCR products identifies the CNV breakpoint. For the CNV within EYA1, the primer pair P1–P2, but not P1–P3, generates two PCR products, indicating that the breakpoint is between P2 and P3; thus resequencing by P2 identifies the exact breakpoint. For the CNV within CTDSPL, the primer pairs P1–P2, P1–P3, and P1–P4 all generate two PCR products, indicating that the breakpoint is between P1 and P4; thus resequencing of the shortest PCR product in Figure 3C by P1 and P4 from both ends identifies the breakpoint. These examples illustrate that the combined PCR-resequencing approach can pinpoint the exact location of predicted CNVs in the human genome.
Discussion
We developed a HMM-based algorithm for kilobase-resolution detection of CNVs using whole-genome SNP genotyping data. Comparison with previously published CNV calls generated on the same HapMap individuals indicates that our algorithm is capable of identifying fine-scale genetic structure of CNVs with a median size of ∼12 kb, which is an order of magnitude smaller than previous experimental studies but concurs with several in silico studies (Conrad et al. 2006; McCarroll et al. 2006). PCR and resequencing techniques were used to identify the exact breakpoints for several common CNVs. Our results demonstrate the feasibility of whole-genome fine-mapping of CNVs using high-density SNP genotyping technology.
The key to the performance of a CNV-calling algorithm is the ability to exploit all sources of available information to their full potential. Compared to the BeadStudio LOH-plus algorithm (Illumina) and the QuantiSNP algorithm (Colella et al. 2007), there are several advantages in the implementation of PennCNV. First, we used state-specific and distance-dependent transition probabilities in the HMM state-transition matrix, which takes into account that some state transition events (e.g., from normal state to one-copy deletion) are more likely than others (e.g., from one-copy deletion to one-copy duplication). Second, rather than treating the B Allele Frequency (BAF) with an arbitrary continuous distribution, we followed the Illumina BAF calculation procedure and appropriately modeled the “boundary truncation” event for BAF inference. Third, we indexed the CNV genotype frequency by the population frequency of the B allele estimated from a large reference population, which allows more accurate modeling of the likelihood of copy number genotypes. Fourth, for family-based genetic studies, we incorporated family information to jointly validate and re-call CNVs for related family members. To our knowledge, this is the first time that family relationship is used in CNV calling. Our results demonstrate that by incorporating family relationship a posteriori, the accuracy of CNV calls can be improved. In addition, although currently the PennCNV algorithm only generates total CNV genotypes (e.g., a CNV genotype of ABBB for a four-copy duplication), the use of family information may lead to a probabilistic model that separates total CNV genotypes into chromosome-specific CNV calls (e.g., a CNV genotype of AB in one chromosome and BB in another chromosome).
Although the PennCNV algorithm was developed specifically for data generated on the Illumina Infinium platform, it could be extended to other similar SNP genotyping platforms. There are several unique features of the Illumina data processing procedure, including the use of a group of reference samples (rather than a single reference sample) for SNP-specific signal adjustments and the use of “B Allele Frequency” for allelic intensity ratio calculation. These treatments reduce the variances of signal measures across SNPs, and make different markers more comparable to each other. In addition, these treatments also allow detection and modeling of various CNV events, such as heterosomic chromosome deletions and copy-neutral LOH. Therefore, when allele-specific signal data from a large group of reference samples are available for other genotyping platforms, it is desirable to generate similar measures as the Illumina platform, which can then be directly analyzed by PennCNV.
Our modeling procedure treats each SNP position as equally likely to be within a CNV region. However, different SNPs have different prior probability based on whether they are located within a common CNV region, thus these prior probabilities can be potentially used to improve the prediction algorithm. The prior probabilities for all SNPs can be estimated from a large set of reference samples and can then be used to construct SNP-specific state transition matrices. Alternatively, an improved algorithm can take into account the fact that some chromosomes have more CNVs than others, or that the centromeric and telomeric regions tend to have more CNVs (Nguyen et al. 2006). In this case, rather than relying on a single HMM model for all chromosomes, the use of chromosome-specific and region-specific HMM models may detect CNVs with higher sensitivity.
There are several limitations for interpreting CNV-calling data from Illumina high-density SNP genotyping arrays. These arrays were constructed using HapMap data and contain primarily tag SNPs (Steemers and Gunderson 2007). The use of linkage disequilibrium information in designing the genotyping array suggests that SNPs are not uniformly distributed; thus some small CNVs may be completely missed by the array if they are located within two neighboring SNPs far apart. In addition, the current HumanHap550 array has no SNP coverage in several heterochromatin regions, including centromeric regions. Furthermore, SNPs within common CNVs may be under-represented in the array, since they are more likely to violate Hardy-Weinberg equilibrium and may be excluded during array design. Finally, the accuracy of SNP genotyping depends on the quality of a clustering file that specifies the R and θ values for canonical genotype clusters. However, the batch-specific manufacturing process and scanning process of arrays, as well as the modification of lab protocols (including reagents) for array experiments, will lead to changes of R and θ values for many SNPs. In these cases, the clustering file is no longer accurate for some SNPs and is subject to regional variations, generating artificial signals of CNVs in some regions. (We note that array-CGH platforms and other SNP genotyping platforms are also susceptible to this problem.) For the reasons described above, we caution that despite the higher resolution of SNP genotyping arrays in detecting CNVs, different techniques can validate and complement each other to achieve the most accurate CNV calls.
In conclusion, our study demonstrates the feasibility of genome-wide CNV fine-mapping via high-density SNP genotyping technology. With the accumulation of high-density SNP genotyping data on many more individuals, we are compiling a large set of common CNVs in the human genome across populations, and we plan to fine-map the breakpoints for many of them, especially those predicted to be functionally important. This collection of common CNVs would be essential in completing the map of human genetic variation and would greatly advance our basic understanding of the dynamic human genome.
Methods
Inference of log R Ratio (LRR) and B Allele Frequency (BAF)
For each SNP, its two alleles are referred to as the A and B alleles using a set of specific naming rules (see http://www.illumina.com/downloads/TopBot_TechNote.pdf). The raw signal intensity values measured for the A and B alleles are then subject to a five-step normalization procedure using the signal intensity of all SNPs (see Illumina white paper at https://icom.illumina.com/icom/software.ilmn). This procedure produces the X and Y values for each SNP, representing the experiment-wide normalized signal intensity on the A and B alleles, respectively. Two additional measures are then calculated for each SNP, where R = X + Y refers to the total signal intensity, and θ = arctan(Y/X)/(π/2) refers to the relative allelic signal intensity ratio.
As a normalized measure of total signal intensity, the log R Ratio (LRR) value for each SNP is then calculated as LRR = log2(Robserved/Rexpected), where Rexpected is computed from linear interpolation of canonical genotype clusters (Peiffer et al. 2006). The B Allele Frequency (BAF) is a somewhat confusing term that actually refers to a normalized measure of relative signal intensity ratio of the B and A alleles:
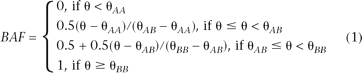
Hidden Markov model for CNV detection
The Hidden Markov Model (HMM) is a statistical technique that models a Markov process, where the probability of observing a particular state at a particular time point only depends on the states at previous time points. HMM provides a natural statistical framework for modeling dependence structures between copy numbers at nearby SNPs. To detect CNVs, we used the first-order HMM that assumes that the hidden copy number state at each SNP depends only on the copy number state of the most preceding SNP.
Let {ri, bi, zi} denote the log R ratio, B allele frequency, and copy number state at SNP i (1 ≤ i ≤ M), respectively. The likelihood of the observed data is
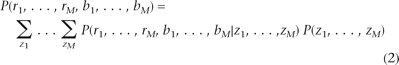
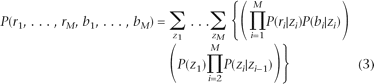
Hidden copy number states
We adopted the same definition of hidden copy number states as described in QuantiSNP (Colella et al. 2007) (Table 1). Compared to HMM implementations that only consider three states (loss, normal, and gain), we believe that the six-state definition is biologically more plausible yet still computationally feasible.
Emission probability of log R ratio
Given each hidden copy number state, the emission probability of the log R ratio is modeled as a mixture of uniform and normal distributions,
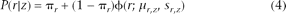
Emission probability of B allele frequency
The emission probability of BAF is slightly more complicated than the LRR. For each hidden state (except state 1), there are multiple possible genotypes with distinct patterns of B Allele Frequency (Table 1; Supplemental Table 1). Owing to the truncation procedure used in Equation 1 for BAF calculation, we treated two different scenarios separately: (1) when the BAF value is between 0 and 1, its distribution is modeled as a normal mixture; (2) when the BAF value is 0 or 1, its distribution is modeled by a mixture of point mass at 0 (denoted by M0) or 1 (denoted by M1) and truncated normal. Let K(z) denote the number of genotypes for copy number state z; then the emission probability of the B allele frequency can be written as
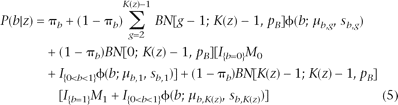
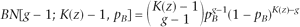
Specific treatment for chromosome X
The modeling and interpretation of LRR and BAF values for chromosome X (chrX) need special treatment. We adjust the level of LRR for all SNPs in chrX by subtracting a constant so that the average LRR value is either zero (for female) or the expected LRR value of single-copy deletion (for male). For chrX in males, the normal copy number is 1 (state 2 in HMM).
Transition probabilities of hidden states
The transition probability describes the probability of having a copy number state change between two adjacent SNPs. Intuitively, the copy number state is unlikely to change for SNPs that are nearby but is more likely to change for SNPs that are far apart. To appropriately model the dependency of transition probabilities on SNP distances, we used a modified version of a previously described state transition matrix (Marioni et al. 2006). Let zi–l and zi denote the copy number states at two adjacent SNPs i − 1 and i, respectively, and let di denote the distance between them. The transition probability is modeled as
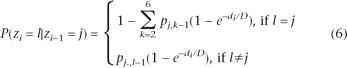
Parameter estimation and CNV calling
The initial model parameters for the HMM were estimated empirically from several large CNV regions, through manually examining the BeadStudio Genome Viewer for a set of genotyped individuals. We have found that the exponential of LRR increases approximately linearly with the copy number; therefore the expected LRR values at each given copy number state can be estimated from observed large CNV regions from a large set of training samples via simple linear interpolation. This also indicates that deletions should be easier to detect than duplications, as the deviation of LRR from zero for deletions is more than that for duplications. In addition, the duplications with four or more copies would be virtually indistinguishable, thus the maximum copy number is set as 4 in PennCNV. To optimize the HMM parameters, we relied on the Baum-Welch algorithm (Baum et al. 1970) for training the model to maximize the likelihood of the observed data for each individual, and then used the Viterbi algorithm (Viterbi 1967) to infer the most likely path (state sequences for all SNPs along each chromosome). A CNV is called from the most likely state sequence whenever a stretch of states that is different from the normal state (state 3 and state 4 for autosomes, state 2 for male chrX) is observed. In our analysis, we excluded all CNVs that contain ≤2 SNPs, because these CNVs are more likely to contain a high fraction of false positives.
A posteriori CNV validation using family information
Family information can potentially help eliminate CNVs that are incompatible with Mendelian inheritance and improve the accuracy of CNV calling and boundary prediction. To incorporate the family information a posteriori into PennCNV, we analytically derived a set of three 5 × 5 × 5 CNV inheritance matrices, for autosomes, male chromosome X, and female chromosome X, respectively (Supplemental Tables 3–5). In these matrices, state 4 (copy-neutral LOH) is combined with state 3 (normal copy number), because they are more likely to be caused by cell-line artifacts. Construction of the CNV inheritance matrices requires a priori specification of a parameter e that determines the likelihood of having a de novo CNV event, and we used 0.01 in the present study.
Let 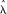 denote all HMM parameters. Given a CNV region, let bf, bm, and bo denote the vectors of B allele frequencies of the father, mother, and offspring, respectively, for all M SNPs in the region. Similarly, let rf, rm, and ro denote the vectors of log R Ratios for all SNPs, and let zf, zm, and zo denote the copy number states of the trio in the region. Using the Bayes rule, we can then calculate the posterior probability of the copy number states for the trio. When the parent(s) and the offspring have the same CNV breakpoints, or when only one individual in the trio has a CNV call, the posterior probability for the trio states can be calculated by
denote all HMM parameters. Given a CNV region, let bf, bm, and bo denote the vectors of B allele frequencies of the father, mother, and offspring, respectively, for all M SNPs in the region. Similarly, let rf, rm, and ro denote the vectors of log R Ratios for all SNPs, and let zf, zm, and zo denote the copy number states of the trio in the region. Using the Bayes rule, we can then calculate the posterior probability of the copy number states for the trio. When the parent(s) and the offspring have the same CNV breakpoints, or when only one individual in the trio has a CNV call, the posterior probability for the trio states can be calculated by
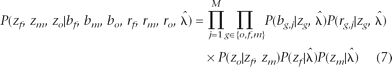
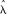 ) and P(zm|
) and P(zm|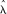 ) are the initial probabilities of copy number states in the CNV region. The most likely a posteriori trio state combination is then selected from the 125 scenarios.
) are the initial probabilities of copy number states in the CNV region. The most likely a posteriori trio state combination is then selected from the 125 scenarios.Since we initially analyze the parents and the offspring separately, it is possible that they have different CNV boundaries (Supplemental Fig. 1). In this situation, we can partition the entire combined CNV region into several smaller blocks. For example, for the scenario in the second row and the second column in Supplemental Figure 1 that contains three blocks, the posterior probability of the trio state is
All CNVs used in this study are detected using the human May 2004 genome assembly as the reference genome assembly. The PennCNV software is available from http://www.neurogenome.org/cnv/penncnv. Several support programs for processing raw genotyping data and for functionally annotating CNVs are also included. The CNV calls are publicly available for downloading from the Web site. In addition, we provide custom-made tracks for visualizing CNVs in the UCSC Genome Browser.
Acknowledgments
We wish to thank the patients and their families who donated blood samples to the Children’s Hospital of Philadelphia (CHOP), and acknowledge the technical staff at the Center for Applied Genomics at CHOP for producing the genotypes used for analyses. We also thank the Autism Genetic Resource Exchange (AGRE) Consortium6 and the participating AGRE families for the resources they provided. The Autism Genetic Resource Exchange is a program of Cure Autism Now and is supported, in part, by grant MH64547 from the National Institute of Mental Health to Daniel H. Geschwind (PI). We also thank Junhyong Kim for critical reading of the manuscript. This work is supported by a seed grant from the Penn/CHOP Center for Autism Research, by NIH grant R01 MH604687 and NARSAD distinguished Investigator Award to M.B., by the Pennsylvania Commonwealth HRFF, and by the Children’s Hospital of Philadelphia.
References
- ↵M.J. Aardema,L.L. Crosby,D.P. Gibson,G.A. Kerckaert,R.A. LeBoeuf,(1997) Aneuploidy and consistent structural chromosome changes associated with transformation of Syrian hamster embryo cells. Cancer Genet. Cytogenet. 96:140–150.
- ↵J.A. Bailey,A.M. Yavor,H.F. Massa,B.J. Trask,E.E. Eichler,(2001) Segmental duplications: Organization and impact within the current human genome project assembly. Genome Res. 11:1005–1017.
- ↵L.E. Baum,T. Petrie,G. Soules,N. Weiss,(1970) A maximization technique occurring in the statistical analysis of probabilistic functions of Markov chains. Ann Math. Statist 41:164–171.
- ↵N. Carter,(2007) Methods and strategies for analyzing copy number variation using DNA microarrays. Nat. Genet. 39:S16–S21.
- ↵S. Colella,C. Yau,J.M. Taylor,G. Mirza,H. Butler,P. Clouston,A.S. Bassett,A. Seller,C.C. Holmes,J. Ragoussis,(2007) QuantiSNP: An objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 35:2013–2025.
- ↵D.F. Conrad,M. Hurles,(2007) The population genetics of structural variation. Nat. Genet. 39:S30–S36.
- ↵D.F. Conrad,T.D. Andrews,N.P. Carter,M.E. Hurles,J.K. Pritchard,(2006) A high-resolution survey of deletion polymorphism in the human genome. Nat. Genet. 38:75–81.
- ↵E.E. Eichler,D.A. Nickerson,D. Altshuler,A.M. Bowcock,L.D. Brooks,N.P. Carter,D.M. Church,A. Felsenfeld,M. Guyer,C. Lee,(2007) Completing the map of human genetic variation. Nature 447:161–165.
- ↵L. Feuk,A.R. Carson,S.W. Scherer,(2006) Structural variation in the human genome. Nat. Rev. Genet. 7:85–97.
- ↵H. Fiegler,R. Redon,D. Andrews,C. Scott,R. Andrews,C. Carder,R. Clark,O. Dovey,P. Ellis,L. Feuk,(2006) Accurate and reliable high-throughput detection of copy number variation in the human genome. Genome Res. 16:1566–1574.
- ↵J.L. Freeman,G.H. Perry,L. Feuk,R. Redon,S.A. McCarroll,D.M. Altshuler,H. Aburatani,K.W. Jones,C. Tyler-Smith,M.E. Hurles,(2006) Copy number variation: New insights in genome diversity. Genome Res. 16:949–961.
- ↵D.H. Geschwind,J. Sowinski,C. Lord,P. Iversen,J. Shestack,P. Jones,L. Ducat,S.J. Spence,(2001) The Autism Genetic Resource Exchange: A resource for the study of autism and related neuropsychiatric conditions. Am. J. Hum. Genet. 69:463–466.
- ↵K.L. Gunderson,F.J. Steemers,G. Lee,L.G. Mendoza,M.S. Chee,(2005) A genome-wide scalable SNP genotyping assay using microarray technology. Nat. Genet. 37:549–554.
- ↵D.A. Hinds,A.P. Kloek,M. Jen,X. Chen,K.A. Frazer,(2006) Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat. Genet. 38:82–85.
- ↵A.J. Iafrate,L. Feuk,M.N. Rivera,M.L. Listewnik,P.K. Donahoe,Y. Qi,S.W. Scherer,C. Lee,(2004) Detection of large-scale variation in the human genome. Nat. Genet. 36:949–951.
- ↵A.S. Ishkanian,C.A. Malloff,S.K. Watson,R.J. DeLeeuw,B. Chi,B.P. Coe,A. Snijders,D.G. Albertson,D. Pinkel,M.A. Marra,(2004) A tiling resolution DNA microarray with complete coverage of the human genome. Nat. Genet. 36:299–303.
- ↵W.J. Kent,(2002) BLAT—The BLAST-like alignment tool. Genome Res. 12:656–664.
- ↵R. Khaja,J. Zhang,J.R. MacDonald,Y. He,A.M. Joseph-George,J. Wei,M.A. Rafiq,C. Qian,M. Shago,L. Pantano,(2006) Genome assembly comparison identifies structural variants in the human genome. Nat. Genet. 38:1413–1418.
- ↵R.M. Kuhn,D. Karolchik,A.S. Zweig,H. Trumbower,D.J. Thomas,A. Thakkapallayil,C.W. Sugnet,M. Stanke,K.E. Smith,A. Siepel,(2007) The UCSC Genome Browser Database: Update 2007. Nucleic Acids Res. 35:D668–D673.
- ↵D.P. Locke,A.J. Sharp,S.A. McCarroll,S.D. McGrath,T.L. Newman,Z. Cheng,S. Schwartz,D.G. Albertson,D. Pinkel,D.M. Altshuler,(2006) Linkage disequilibrium and heritability of copy-number polymorphisms within duplicated regions of the human genome. Am. J. Hum. Genet. 79:275–290.
- ↵J.C. Marioni,N.P. Thorne,S. Tavare,(2006) BioHMM: A heterogeneous hidden Markov model for segmenting array CGH data. Bioinformatics 22:1144–1146.
- ↵S.A. McCarroll,D. Altshuler,(2007) Copy-number variation and association studies of human disease. Nat. Genet. 39:S37–S42.
- ↵S.A. McCarroll,T.N. Hadnott,G.H. Perry,P.C. Sabeti,M.C. Zody,J.C. Barrett,S. Dallaire,S.B. Gabriel,C. Lee,M.J. Daly,(2006) Common deletion polymorphisms in the human genome. Nat. Genet. 38:86–92.
- ↵R.E. Mills,C.T. Luttig,C.E. Larkins,A. Beauchamp,C. Tsui,W.S. Pittard,S.E. Devine,(2006) An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 16:1182–1190.
- ↵D.Q. Nguyen,C. Webber,C.P. Ponting,(2006) Bias of selection on human copy-number variants. PLoS Genet. 2:e20, 10.1371/journal.pgen.0020020.
- ↵D.A. Peiffer,J.M. Le,F.J. Steemers,W. Chang,T. Jenniges,F. Garcia,K. Haden,J. Li,C.A. Shaw,J. Belmont,(2006) High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 16:1136–1148.
- ↵R. Redon,S. Ishikawa,K.R. Fitch,L. Feuk,G.H. Perry,T.D. Andrews,H. Fiegler,M.H. Shapero,A.R. Carson,W. Chen,(2006) Global variation in copy number in the human genome. Nature 444:444–454.
- ↵S. Risin,V.L. Hopwood,S. Pathak,(1992) Trisomy 12 in Epstein-Barr virus-transformed lymphoblastoid cell lines of normal individuals and patients with nonhematologic malignancies. Cancer Genet. Cytogenet. 60:164–169.
- ↵S. Risin,T. Fujimaki,C.A. Mestriner,N.M. Brown,V.L. Hopwood,I.J. Fidler,S. Pathak,(1993) Clonal expansion of cells with trisomy of chromosomes 12 and X in an EBV-transformed lymphoblastoid cell line and establishment of a tumorigenic monoclonal cell line (48,XX,+X,+12) Cytogenet. Cell Genet. 62:54–55.
- ↵S.W. Scherer,C. Lee,E. Birney,D. Altshuler,E.E. Eichler,N. Carter,M. Hurles,L. Feuk,(2007) Challenges and standards in integrating surveys of structural variation. Nat. Genet. 39:S7–S15.
- ↵J. Sebat,B. Lakshmi,J. Troge,J. Alexander,J. Young,P. Lundin,S. Maner,H. Massa,M. Walker,M. Chi,(2004) Large-scale copy number polymorphism in the human genome. Science 305:525–528.
- ↵A.J. Sharp,D.P. Locke,S.D. McGrath,Z. Cheng,J.A. Bailey,R.U. Vallente,L.M. Pertz,R.A. Clark,S. Schwartz,R. Segraves,(2005) Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 77:78–88.
- ↵J. Simon-Sanchez,S. Scholz,H.C. Fung,M. Matarin,D. Hernandez,J.R. Gibbs,A. Britton,F.W. de Vrieze,E. Peckham,K. Gwinn-Hardy,(2007) Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum. Mol. Genet. 16:1–14.
- ↵F.J. Steemers,K.L. Gunderson,(2007) Whole genome genotyping technologies on the BeadArray platform. Biotechnol. J. 2:41–49.
- ↵E. Tuzun,A.J. Sharp,J.A. Bailey,R. Kaul,V.A. Morrison,L.M. Pertz,E. Haugen,H. Hayden,D. Albertson,D. Pinkel,(2005) Fine-scale structural variation of the human genome. Nat. Genet. 37:727–732.
- ↵A.J. Viterbi,(1967) Error bounds for convolutional codes and an asymptotically optimum decoding algorithm. IEEE Trans. Inf. Theory 13:260–269.
- ↵K.K. Wong,R.J. deLeeuw,N.S. Dosanjh,L.R. Kimm,Z. Cheng,D.E. Horsman,C. MacAulay,R.T. Ng,C.J. Brown,E.E. Eichler,(2007) A comprehensive analysis of common copy-number variations in the human genome. Am. J. Hum. Genet. 80:91–104.
Notes
[1] [Supplemental material is available online at www.genome.org. The PennCNV software is available from http://www.neurogenome.org/cnv/penncnv.]
[2] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.6861907
[3] The AGRE Consortium: Dan Geschwind, UCLA, Los Angeles, CA; Maja Bucan, University of Pennsylvania, Philadelphia, PA; W. Ted Brown, N.Y.S. Institute for Basic Research in Developmental Disabilities, Staten Island, NY; Rita M. Cantor, UCLA School of Medicine, Los Angeles, CA; John N. Constantino, Washington University School of Medicine, St. Louis, MO; T. Conrad Gilliam, University of Chicago, Chicago, IL; Martha Herbert, Harvard Medical School, Boston, MA; Clara Lajonchere, Cure Autism Now/Autism Speaks, Los Angeles, CA; David H. Ledbetter, Emory University, Atlanta, GA; Christa Lese-Martin, Emory University, Atlanta, GA; Janet Miller, Cure Autism Now/Autism Speaks, Los Angeles, CA; Stanley F. Nelson, UCLA School of Medicine, Los Angeles, CA; Gerard D. Schellenberg, University of Washington, Seattle, WA; Carol A. Samango-Sprouse, George Washington University, Washington, DC; Sarah Spence, UCLA, Los Angeles, CA; Matthew State, Yale University, New Haven, CT; Rudolph E. Tanzi, Massachusetts General Hospital, Boston, MA.