
How Does Replication-Associated Mutational Pressure Influence Amino Acid Composition of Proteins?
Abstract
We have performed detrended DNA walks on whole prokaryotic genomes, on noncoding sequences and, separately, on each position in codons of coding sequences. Our method enables us to distinguish between the mutational pressure associated with replication and the mutational pressure associated with transcription and other mechanisms that introduce asymmetry into prokaryotic chromosomes. In many prokaryotic genomes, each component of mutational pressure affects coding sequences not only in silent positions but also in positions in which changes cause amino acid substitutions in coded proteins. Asymmetry in the silent positions of codons differentiates the rate of translation of mRNA produced from leading and lagging strands. Asymmetry in the amino acid composition of proteins resulting from replication-associated mutational pressure also corresponds to leading and lagging roles of DNA strands, whereas asymmetry connected with transcription and coding function corresponds to the distance of genes from the origin or terminus of chromosome replication.
There are many mechanisms in nucleic acids metabolism that introduce asymmetry into nucleotide composition of the two DNA strands (for review, see Francino and Ochman 1997; Mrázek and Karlin 1998). DNA asymmetry can be described in terms of relations between numbers of the four different nucleotides in DNA strands, or it can be visualized in diagrams representing different kinds of DNA walks. Filipski (1990) first interpreted the asymmetry in G/C content as a result of asymmetric replication-associated mutational pressure in viruses. Lobry showed asymmetry in nucleotide composition of some prokaryotic genomes by two-dimensional DNA walks (Lobry 1996a) and by the analysis of sliding windows (Lobry 1996b). He pointed out that this asymmetry changes its polarity at the origin and terminus of chromosome replication, where DNA strands change their role from leading to lagging or vice versa. Later, Freeman et al. (1998),Mrázek and Karlin (1998), Grigoriev (1998), and McLean et al. (1998) described DNA strand asymmetry in different numerical ways. Mechanisms that might be responsible for the observed asymmetry have been also discussed many times (Freeman et al. 1998; Grigoriev 1998;Mrázek and Karlin 1998). One of the accepted hypotheses states that the potential cause of asymmetry is deamination of methylated cytosines, which leads to thymines. Some investigators believe that this type of substitution differentiates between sense and antisense strands of coding sequences, and transcription mechanism introduces the asymmetry into DNA strands (Beletskii and Bhagwat 1996; Francino et al. 1996; Francino and Ochman 1997; Freeman et al. 1998).
Intergenic noncoding sequences seem to be the most likely to accumulate substitutions. However asymmetry in the third position in codons, which could be the effect of silent substitutions, is also observed. These substitutions are not necessarily neutral; they can lead to asymmetry in the distribution of genes on the chromosome according to the rate of translation of their products. The average codon adaptation index (CAI;Sharp and Li 1987) for genes on the leading strand is different from that for genes located on the lagging strand. Such effects were observed in the Escherichia coli (Francino and Ochman 1997) and Borrelia burgdorferi (McInerney 1998) genomes. These workers observed preferences for transcription of the DNA strand in the direction of replication rather than in the inverse direction, which was reflected by higher numbers of coding sequences on leading than on lagging strands in many genomes (Brewer 1990; Blattner et al. 1997;Kunst et al. 1997). Conversely, some experiments have proved that the frequency of mutations introduced into the nontranscribed DNA strand is higher than those in the transcribed strand (Francino et al. 1996).
Replication is thought to be another cause of strand compositional asymmetry in genomes. Although it is not clear if replication of only one or both strands is discontinuous (Okazaki et al. 1968; Kornberg and Baker 1992; Wang and Chen 1992, 1994), the topology of the replication fork requires different enzymatic mechanisms for the synthesis of leading and lagging DNA strands with different error rates (Kornberg and Baker 1992; Kunkel 1992; Waga and Stillman 1994). Moreover, some experiments have shown that differences in processivity of leading and lagging DNA strands may be responsible for the unequal fidelity of replication of these two strands (Fijalkowska et al. 1998).
Usually, DNA asymmetry analyses of genomes were performed on sliding windows. We have performed detrended DNA walks for nucleotide composition analysis of coding and noncoding sequences. This enables us to distinguish between the mutational effect of replication and the effect of transcription and/or coding functions (Cebrat et al. 1999). We have performed separate analyses of the two DNA strands: the Watson (W) strand (GenBank), and the Crick (C) strand (complementary to W). The asymmetry introduced by replication-associated mechanisms into open reading frames (ORFs) lying on the leading and lagging DNA strands is of the reciprocal sign. Thus, when detrended DNA walks on ORFs situated on the W strand in the scale of the chromosome are added to DNA walks performed on ORFs from the C strand, the values of asymmetry compensate each other and disappear, leaving the effect of asymmetry introduced by other mechanisms (see Methods for details). In contrast, the asymmetry in ORFs resulting from their coding function or transcription is of the same sign independent of their location on leading or lagging strands. Thus, the addition of DNA walks cumulates asymmetry introduced by mechanisms not related to replication-associated mutational pressure. Addition on both DNA strands results in asymmetries that are the result of the same, unbalanced composition of linked genes from complementary DNA strands.
In this paper we have shown that replication, as well as other mutational pressure mechanisms, is responsible for introducing nucleotide substitutions into DNA that are not silent and change amino acid composition of coded proteins.
RESULTS
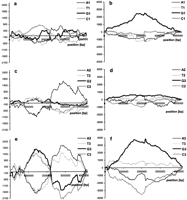
In Figure 1a detrended DNA walks on the Treponema pallidumchromosome have been shown, illustrating a nucleotide composition of ORFs of >150 codons, not counting shorter ORFs from overlapping pairs of ORFs, situated on the W strand (i.e., the coding strand lies on the W strand). In the T. pallidum genome, ∼60% of coding sequences are located on the leading DNA strand. Because the walks in Figure 1a are presented in the scale of the new sequence of the spliced ORFs, the measure of asymmetry in coding density is the value of shifts of the extrema at the middle of thex-axis. In these DNA walks numbers on the x-axis correspond to coordinates of the walker on the sequences of spliced conescutive ORFs situated on the W strand, not to their real coordinates on the chromosome. Numbers on the y-axis represent differences between the found number of the analyzed nucleotide and its expected number if ORFs were distributed evenly on the chromosome independently of W or C strands or leading/lagging DNA strands. Figure1b presents the same DNA walks for T. pallidum but in the scale of the chromosome. In these walks numbers on the x-axis represent the real coordinates of ORFs on the chromosome. These walks lose their information on coding density. The extrema have to be in the middle of the plot, where the replication terminus is situated. Figure1, c and d, shows analogous walks for the C strand. In Figure 1e the results of subtraction of the walks presented in Figure 1, b and d, (for strands W and C) are presented. Let us assume (after Beletski and Bhagwat 1996; Francino et al. 1996; Freeman et al. 1998) that transcription introduces asymmetry by preferentially high mutation rate in the nontranscribed strand. Then, whether the ORF is on a leading or a lagging strand, asymmetry is of the same sign and subtraction should eliminate it. Because the effect of asymmetry introduced by replication is of the reciprocal sign on leading and lagging strands, subtraction cumulates it. Thus, Figure 1e represents the cumulative effect of replication-associated mutational pressure on ORFs lying on both DNA strands. In contrast, the addition of walks performed on W and C strands eliminates the asymmetry introduced by replication, leaving the cumulative effect of transcription-associated mutational pressure (Fig.1f). One can also expect to see local asymmetry after addition of DNA walks if genes mapped on chromosomes in the same region, independent of the leading or lagging strand, have specific bias in nucleotide composition. In Figure 2, the results of subtraction of DNA walks (W–C) for six eubacterial genomes are shown. Note that in all plots but Figure 2f, the replication terminus is in the center of the x-axis. Figure 2f represents DNA walks on the linear genome of B. burgdorferi, where the origin of replication is in the middle of the chromosome (the center ofx-axis). In all of these genomes the leading strand is relatively richer in G than the lagging strand. This is also true for other eubacterial genomes (supplementary information available atwww.genome.org and http://smorfland. microb.uni.wroc.pl).
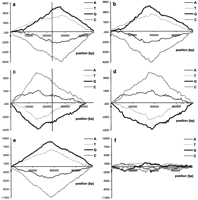
Detrended DNA walks on T. pallidum ORFs longer than 150 codons. (a) All ORFs of the W strand were spliced, and the walks were performed in the scale of the new sequence (x-axis). The vertical line indicates the middle of the analyzed sequence; numbers on the y-axis indicate the relative cumulative abundance of the nucleotide (see Methods). (b) The same walks but the scale of the x-axis shows the position of ORFs on the chromosome. (c,d) The same kinds of walks done on the C strand. (e) The result of subtraction of walks presented on b and d. (f) The results of the addition of walks presented in b and d. The terminus of replication is at the extrema of the diagrams.
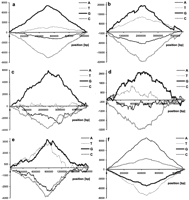
The result of subtraction of the DNA walk on strand C from the DNA walk on strand W for ORFs longer than 150 codons for the genomes ofChlamydia trachomatis (a); Mycobacterium tuberculosis (b); Escherichia coli (c);Helicobacter pylori (d); Haemophilus influenzae (e); and Borrelia burgdorferi(f). In all plots but f the terminus of replication is in the center of the x-axis. In the plot representing theB. burgdorferi genome, the origin of replication is in the center of the diagram.
The analysis of DNA walks on W and C strands done for the third positions in coding sequences and for intergenic sequences of theT. pallidum genome is shown in Figure 3. These results suggest that the asymmetry introduced by replication-associated mutational pressure into the third codon positions resembles that of intergenic sequences. Analyses of the results of addition of DNA walks on intergenic sequences have not shown any traces of asymmetry introduced by mechanisms other than replication-associated ones.
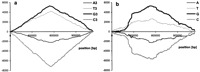
The result of subtraction of the DNA walk on the C strand from the DNA walk on the W strand for the third positions in codons (a) and the DNA walk on the W strand of intergenic sequences (b) of the T. pallidum genome. To make possible a quantitative comparison of a and b we have multiplied the values on the scale of y-axis for intergenic sequences—sequences that do not belong to any ORF longer than 70 codons—by a factor of 1.8 (the number of nucleotides in analyzed intergenic sequences was 1.8 times lower than the number of nucleotides in the third positions.
Some mutations of the third positions in codons, for example, almost all transitions, are silent, but others are not and belong to the class of missenes mutations. If we assume that most of the accumulated mutations are in the fourfold-degenerated codons, in which each mutation in the third position is silent, we should find differences in the accumulation of mutations in codons where transversions in the third positions are missense (twofold-degenerated codons). To check this, we have performed separate walks on twofold- and fourfold-degenerated codons. Both classes of codons accumulate mutations, and some of these mutations (transversions in twofold-degenerated codons) are of the missense class. The DNA walks presenting asymmetric accumulation of mutations in the twofold- and fourfold-degenerated codons in eubacterial genomes are presented atwww.genome.org and http://smorfland.microb.uni.wroc.pl.
Because a transversion even in the third positions can change the encoded amino acid, we have performed walks on amino acids coded by ORFs lying on the two DNA strands, and we have subtracted and added the resulting walks to separate the effect of replication-associated mutational pressure from the effect of transcription and/or other effects. In Figure 4 the effect of replication on amino acid composition of proteins coded by genes on leading and lagging strands of T. pallidum, Chlamydia trachomatis, andB. burgdorferi genomes is shown. Analyzing the results of the subtraction of walks, we have found amino acids that prevail on the leading or lagging strand in different genomes. In E. coli,Bacillus subtilis, T. pallidum, B. burgdorferi, and C. trachomatis Gly, Val, and Asp were coded relatively more frequently on the leading strand, whereas Ile, Thr, and His were more prevalent on the lagging strand. Nevertheless, eubacterial genomes differ significantly in prevalence of specific amino acids on leading or lagging strands (supplementary information available at www.genome.org and http://smorfland.microb.uni.wroc.pl). These results prove that the skew found previously in the prevalence of some codons in genes transcribed in the direction of replication (Fraser et al. 1998) is connected to replication-associated mutational pressure.
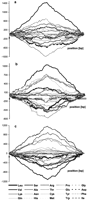
The effect of subtraction of walks on amino acids for the genomesT. pallidum (a), C. trachomatis(b), and B. burgdorferi (c). Numbers on they-axis indicate the relative cumulative abundance of amino acids.
In the T. pallidum genome no effects other than those connected with the leading/lagging role of DNA strands on protein composition have been observed. However, in large genomes (E. coli and B. subtilis) addition of DNA walks done for ORFs from W and C strands differentiates between regions proximal and distal to the origin of replication of the chromosome (Fig.5). Note that replication-associated effects divide chromosomes into two replichores—left and right—with extrema in the center of plots. Other effects that we have observed are connected with proximal/distal parts of chromosomes with extrema near the middle of replichores. The trends at the left and right ends of the plot (Fig. 5) are the same and reciprocal to the trends in the central part of the plots. The central part of the plot corresponds to the region close to the terminus of replication (from both sides), and both ends of plots correspond to regions close to the origin of replication (from both sides).
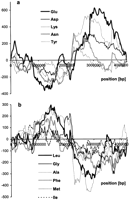
The effect of addition of walks on amino acids for two groups of amino acids coded by the B. subtilis genome.
Thus, in the region close to the replication terminus of the B. subtilis genome (Fig. 5) different trends are observed for different groups of amino acids. Generally, hydrophobic amino acids are more abundant in proteins coded by the proximal region of the chromosome, whereas hydrophilic amino acids are more abundant in proteins coded by regions close to the terminus of chromosome replication.
Information about the asymmetry in DNA nucleotide composition that reflects the asymmetry in amino acid composition of proteins can be shown in a more degenerated form by analyzing DNA walks done for the first, second, and third positions in codons (Fig. 6, results for theB. subtilis genome). The asymmetry is seen even in the second positions (Fig. 6c), which are crucial for the properties of the amino acids coded. Still, the effect of the replication-associated mutational pressure on the second positions is weaker (Fig. 6d) than that of transcription and/or other coding functions (Fig. 6c). Especially in the second positions, asymmetry is seen in the A/T ratio in proximal versus distal parts of the chromosome. Asymmetry in the third positions is seen for both leading versus lagging strands (Fig. 6f) and proximal versus distal parts of chromosomes (Fig. 6e).
Addition (a,c,e) and subtraction (b,d,f) of DNA walks performed on nucleotides in different positions in codons of ORFs longer than 150 codons of the B. subtilis genome. (a,b) First positions; (c,d) second positions; (e,f) third positions.
DISCUSSION
The effect of replication-associated mutational pressure on nucleotide compositional bias of eubacterial chromosomes can be separated from the effects introduced by other mechanisms. Usually, the transcription itself and transcription-coupled repair are blamed for introducing mutations into coding sequences. In such cases, these substitutions should be similar, independent of the location of the transcribed strand of the gene on the leading or lagging DNA strand. If it is purely an effect of transcription, it should be also independent of the distance from the origin or terminus of replication, unless there is some other correlation between the rate or frequency of transcription and the location of genes on chromosome (in respect of proximal/distal location). In such a case, the observed trends can be created by various nucleotide substitutions resulting from different transcription rate in proximal and distal regions of the chromosome.
We have observed some classes of substitution that seem to be characteristic of ORFs lying in the same region of the chromosome but in opposite directions. For example, after the addition of walks, there is an evident surplus of adenine over thymine in the second positions of codons in the distal region of chromosome in the B. subtilis genome. The bias observed in the three positions of codons is affected very weakly by replication-associated mutational pressure (see subtraction of walks in Fig. 6) and possesses some specific features:
- 1.
- There is no correlation between this asymmetry and types of substitutions in any other positions of codons or intergenic sequences.
- 2.
- It is not introduced by replication-associated mutational pressure (it has the same sign for ORFs of leading and lagging strands).
- 3.
- The A/T relations in the second positions reflect the hydrophilic/hydrophobic amino acid composition in coded proteins.
Thus, it seems reasonable to accept the hypothesis that this effect is caused by nonrandom selection of recombinants with preferential location of genes coding for hydrophobic (transmembrane) proteins near the origin of replication. One can argue that this asymmetry can be generated by insertion of phage genomes or a local grouping of genes with the same compositional bias. This might explain irregularities near the origin of the B. subtilis genome, where >20 ribosomal proteins are coded. Nevertheless, almost half of the B. subtilis genome (halves of replichores from both sides of the terminus) are relatively richer in adenine in the second positions in codons.
On the other hand, the nonrandom topology of microbial genomes can be a mechanism of gene control by discrimination. In fast-dividing cells, the copy number of proximal genes can be up to eight times higher than that of distal genes (Cooper and Helmstetter 1968). This reflects the topology of replication when the cell cycle is shorter than the time needed for replication of the whole chromosome. Nevertheless, it is possible that the composition of the third positions in codons, influencing the rate of translation, is superimposed on other levels of gene control. We have observed different codon usage in proximal and distal regions in relatively large genomes of B. subtilis and E. coli (Fig. 6e), but we have not found such differences in the smaller genomes of Treponema or Borrelia (as observed previously byKarlin et al. 1998). Thus, the relations in abundance of products of proximal versus distal genes can change under different growing conditions.
Whereas the effect seen after the elimination of replication-associated mutational pressure seems to depend on recombinational events, the role of replication-associated pressure itself in amino acid composition of proteins is not so obvious. Amino acid composition of proteins depends on the location of coding genes on leading or lagging DNA strands. In many genomes the effect of position on the leading or lagging strand is seen not only in the third codon position but also in the first position (Fig 6b). It is difficult to assume that all of the accumulated substitutions are neutral. Thus, it seems that the selection for appropriate positions of genes and/or codon usage controlling rate of translation are also responsible for the observed leading/lagging strand nucleotide composition asymmetry. The topology of the B. burgdorferi genome supports this view. There is an extremely strong effect of position on the leading versus the lagging strand in that genome. Codon usage on both strands is quite distinct (McInerney 1998), and coding sequences of the two DNA strands are so different that they form two nonoverlapping sets of genes when their contents are analyzed (data not shown). However, genes are not randomly distributed into these two sets—all genes coding for ribosomal proteins are located on the leading strand—which supports the hypothesis that recombination is also responsible for the specific distribution of the genes generating the nucleotide bias in a genome. This could explain the relative conservation of the general genetic map topology of related bacterial genera, for example, Escherichiaand Salmonella (Wilkins 1988).
Because amino acid composition of proteins strongly depends on the positions of their genes on the chromosome, any phylogenetic analysis, as well as algorithms recognizing coding sequences by content sensors, should respect the location of ORFs on the chromosome. Furthermore, for some genomes it is important to know only the leading/lagging positions (i.e., T. pallidum, B. burdorferi, but for other genomes additional information about proximal/distal location might be required (i.e., E. coli, B. subtilis).
Data for Analysis and Methods
The results presented in this paper were obtained by analysis of prokaryotic genomic sequences downloaded from the following:ftp://ftp.pasteur.fr (2/18/98): B. subtilis (Kunst et al. 1997); http://utmmg.med.uth.tmc.edu (2/16/98): T. pallidum(Fraser et al. 1998); http://www.genetics.wisc.edu (11/14/97): E. coli (Blattner et al. 1997); http://www.ncbi.nlm.nih.gov(10/13/97): Haemophilus influenzae (Fleischmann et al. 1995);http://www.ncbi.nlm.nih.gov (10/16/98): Helicobacter pylori(Tomb et al. 1997); http://www.ncbi.nlm.nih.gov (10/30/98):Mycobacterium tuberculosis (Cole et al. 1998);http://www.ncbi.nlm.nih.gov (11/13/98): C. trachomatis(Stephens et al. 1998); and http://www.ncbi.nlm.nih.gov (3/3/98):B. burgdorferi (Fraser et al. 1997). The data have not been updated after the date of retrieval.
To show DNA compositional bias, different DNA walks and their transformations were done. Detailed descriptions of DNA walks, their possible interpretation, and nomenclature are according to Cebrat and Dudek (1998).
To demonstrate local trends independent of coding functions, we performed detrended DNA walks, in which we eliminated strong trends resulting from base composition of coding ORFs (Cebrat et al. 1997;Cebrat and Dudek 1998) because they mask the asymmetry of strands introduced by mutational pressure.
To eliminate these coding trends we counted the following value for a given ORF:J = [N] − (F × L), where J = the value of the walker jump for the ORF,N = the number of nucleotides (A, T, G, or C) in the analyzed positions of the ORF, F = the frequency of the given nucleotide at the examined positions in the whole set of analyzed ORFs, and L = the length of the given ORF in codons. When intergenic sequences were analyzed, F was the frequency of the nucleotide in the whole set of intergenic sequences and L was the length of the visited sequence in nucleotides. We applied an analogous procedure to the distribution analysis of amino acids on the chromosome. In this case, in the above equation, we inserted the number of analyzed amino acid residues instead of N and the frequency of the given amino acid in the set of the analyzed ORFs instead ofF.
When walks for the two strands were added, the walker visited nonoverlapping ORFs of both strands as they appeared on the chromosome, scanned them in the proper reading frame, and moved according to the result of scanning. When the walks for the C strand were subtracted from the walks for the W strand, the value of the walker jump for each ORF in the C strand was multiplied by −1. Note that detrended walks done in the scale of the chromosome lose their information on asymmetry in the total length of ORFs on leading and lagging strands. That is why addition of these walks done for ORFs of W and C strands eliminates the effect of replication associated with mutational pressure and does not depend on differences in coding density of leading versus lagging strands.
Availability of the Results
The detailed results of coding sequence asymmetry for all eubacterial and archaebacterial genomes completed so far are available on the World Wide Web at the following addresses:www.genome.org and http://smorfland.microb.uni.wroc.pl.
Acknowledgments
This work was supported by The State Committee for Scientific Research (6PO4A 030 14).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL cebrat{at}angband.microb.uni.wroc.pl; FAX 48-71-3252-151.
-
- Received July 10, 1998.
- Accepted March 17, 1999.
- Cold Spring Harbor Laboratory Press








