
An Effective Approach for Analyzing “Prefinished” Genomic Sequence Data
- Peter M. Kuehl1,2,3,
- Jane M. Weisemann3,
- Jeffrey W. Touchman2,
- Eric D. Green2, and
- Mark S. Boguski3,4
- 1University of Maryland, Department of Molecular and Cellular Biology, Baltimore, Maryland 21201; 2Genome Technology Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892; 3National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland 20894 USA
Abstract
Ongoing efforts to sequence the human genome are already generating large amounts of data, with substantial increases anticipated over the next few years. In most cases, a shotgun sequencing strategy is being used, which rapidly yields most of the primary sequence in incompletely assembled sequence contigs (“prefinished” sequence) and more slowly produces the final, completely assembled sequence (“finished” sequence). Thus, in general, prefinished sequence is produced in excess of finished sequence, and this trend is certain to continue and even accelerate over the next few years. Even at a prefinished stage, genomic sequence represents a rich source of important biological information that is of great interest to many investigators. However, analyzing such data is a challenging and daunting task, both because of its sheer volume and because it can change on a day-by-day basis. To facilitate the discovery and characterization of genes and other important elements within prefinished sequence, we have developed an analytical strategy and system that uses readily available software tools in new combinations. Implementation of this strategy for the analysis of prefinished sequence data from human chromosome 7 has demonstrated that this is a convenient, inexpensive, and extensible solution to the problem of analyzing the large amounts of preliminary data being produced by large-scale sequencing efforts. Our approach is accessible to any investigator who wishes to assimilate additional information about particular sequence data en route to developing richer annotations of a finished sequence.
[Our software system is available via an extensive web supplement to this article at http://www.ncbi.nlm.nih.gov/Kuehl/prefinished.]
The systematic sequencing of the human genome has begun as part of the ongoing Human Genome Project (Olson 1995; Boguski et al. 1996). The dominant strategy being used in this effort is clone-by-clone shotgun sequencing (Wilson and Mardis 1997). Typically, this process first involves deriving large numbers of sequence reads from subclones derived from a bacterial artificial chromosome (BAC) or P1 artificial chromosome (PAC) to provide a high-redundancy sampling of the starting clone. Preliminary sequence contigs are then assembled in an automated fashion by use of software tools that are becoming increasing powerful (Ewing et al. 1998; Ewing and Green 1998; Gordon et al. 1998). In the second stage of this process, additional sequence reads are obtained in a highly directed fashion, so as to close the remaining sequence gaps, to ensure the presence of high-quality data throughout the assembled sequence, to prevent predictable artifacts from producing errors in the sequence, and to produce a final finished product. Assembled sequence at the various intermediate stages (i.e., not yet a finished product) is often referred to as “prefinished” sequence. Because the first stage in shotgun sequencing is more straightforward, there is often a considerable time lag between the generation of prefinished sequence data for a clone, which typically corresponds to nearly all of its sequence (albeit not always properly assembled or contiguous), and the ultimate production of a finished sequence. It is important to note that the groups participating in human genome sequencing within the international Human Genome Project have agreed to make prefinished sequence data freely available to other investigators (Statement on the Rapid Release of Genomic DNA Sequence 1998), either through the public databases (e.g., GenBank) and/or on their individual web sites (Pruitt 1997). Ultimately, all finished sequence will be submitted to GenBank, EMBL, or DDBJ (Ouellette and Boguski 1997).
In addition to the publicly funded project, private efforts to generate large amounts of human genome sequence over the next few years are planned (Venter et al. 1998). In this case, a shotgun sequencing strategy will be applied to the whole human genome en masse, an approach whose efficacy has been critically debated in the past (Green 1997; Weber and Myers 1997). Regardless of the nature of its ultimate product(s), such an initiative should, at a minimum, produce large amounts of prefinished (i.e., partially assembled) human genome sequence.
The combined public and private sequencing efforts are thus certain to generate large amounts of prefinished sequence data that will be of great interest to investigators wishing to identify genes, polymorphisms, and other important sequence elements. Two inherent features of prefinished sequence data that can make analysis challenging are (1) its sheer size (even for individual BAC or PAC clones) and (2) its dynamic nature (i.e., prefinished sequence can change on a regular basis as additional data is generated, as new assemblies are made, and as problems are resolved). These challenges prompted us to develop a simple but thorough approach for analyzing and annotating prefinished genomic sequence, such as that being generated at the sequencing centers. We were also motivated by the fact that there is little or no commercial software that can be readily used for managing, analyzing, and transmitting large amounts of sequence data, and most research groups lack the resources and infrastructure to develop their own systems. As a result, investigators typically resort to the use of ad hoc methods for this purpose, such as manual storage of the frequently changing data or annotations in spreadsheets or laboratory notebooks. The latter approaches make long-term maintenance of the data difficult and hinder the convenient sharing of results with remote collaborators or other investigators. Here we describe that three publicly available tools—PowerBLAST (Zhang and Madden 1997), Musk (Chao et al. 1995), and Sequin (Kans and Ouellette 1998)—can be used in combination for the systematic analysis of prefinished sequence data. We illustrate the utility of these tools by analyzing sequences generated from human chromosome 7.
RESULTS AND DISCUSSION
We sought to develop and implement a systematic approach for analyzing genomic sequence data, even when it is at a prefinished stage (i.e., prior to complete assembly and verification). The approach we established for this task (Fig. 1 and Table1) was developed around three publicly available software tools: PowerBLAST (ftp://ncbi.nlm.nih.gov/pub/sim2/PowerBlast) (Zhang and Madden 1997), Musk (Chao et al. 1995), and Sequin (Kans and Ouellette 1998). PowerBLAST is a graphical network client for sequence similarity searching. Musk is a graphical viewer for visualizing ASN.1 objects (Ostell and Kans 1998), such as PowerBLAST output. Sequin is a software tool for annotating sequence and submitting it to GenBank (Benson et al. 1998), but for this application we have used it as a local database and visualization system for managing and exchanging sequence data. Importantly, PowerBLAST, Musk, and Sequin run on computers that are universally available in typical molecular biology laboratories. These programs are freely available and already designed for use with large sequence files. In addition, they have features that make them uniquely suited for use with genomic sequence data from ongoing shotgun sequencing projects. These features include
- 1.
- Scalability for use with sequences ranging in size from several kilobase pairs to hundreds of kilobase pairs.
- 2.
- The ability of PowerBLAST to output data in a format (ASN.1) that can be directly read by Sequin.
- 3.
- The efficiency with which multiple analyses of the sequence [e.g., repeat masking followed by comparison to dbEST, dbSTS, and the nonredundant nucleotide (nr) database (Zhang and Madden 1997)] can be performed in a single action within PowerBLAST.
- 4.
- The ability to track all the analyses performed on a clone insert within a single database (Sequin) record.
- 5.
- The interactive graphical interface that facilitates the interpretation of the resulting patterns of sequence matches.
- 6.
- The ease with which such analyses can be propagated as sequences progress from prefinished to finished stages.
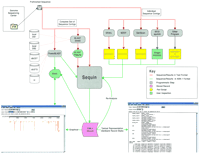
Overview of approach for analyzing prefinished genomic sequence data. Prefinished sequences for individual clones are posted on a public access ftp site. By use of either a web browser or ftp client, this data can be downloaded and analyzed by two main classes of programs: (1) those that compare the sequence with existing databases (e.g., BLAST, PowerBLAST; left) and (2) those that predict or identify features intrinsic to the sequence (e.g., GRAIL, MZEF, GenScan, SEG; right). The first class of programs can be used with public (e.g., dbEST, dbSTS, nr) as well as locally created databases (DB), such as specialized collections of cDNA sequences. The results generated with both classes of programs are collected as annotations within a common Sequin record. Several filter programs (indicated in yellow) must be used to take the textual output from some analysis programs and convert it into ASN.1 format. Such filters are relatively easy to implement and make this system usable with most (if not all) sequence analysis programs. This conversion allows all of the analyses to be stored in a computable format that can be readily updated as more complete versions of the sequence become available (see Fig. 3). The spatial relationships among all annotations can be edited and viewed by either Sequin or Musk. See Table 1 for additional details about the various programs and resources.
Computational Tools Used for Sequence Analysis
As a demonstration of this approach, Figure2 depicts an initial analysis performed on a ∼45-kb prefinished sequence contig assembled from PAC DJ1099C19, a clone derived from human chromosome 7 and sequenced by the Washington University Genome Sequencing Center (http://genome.wustl.edu/gsc/Search/ftp.shtml). Figure 2A is a global view of the PowerBLAST analysis, as displayed by Musk (Zhang and Madden 1997). Nucleotide matches from BLASTN queries against dbEST, dbSTS, and GenBank are contained within a single boxed region labeled BLASTN. Although, in this instance, no significant protein matches were found by BLASTX (Altschul et al. 1994), any protein matches would normally be indicated within a separate boxed region distinct from the nucleotide matches. Such an overview of database matches provides an appreciation of the larger spatial relationships among the sequence matches. Figure2A contains an artificially generated cluster of ESTs demonstrating how multiple ESTs can be compressed into a single feature, thereby reducing the complexity of the display (note that many and varied additional examples are included in the web supplement). The color of each depicted match varies with the number of spatially separate alignments between the matching sequence and the query sequence. This is illustrated more clearly in Figure 2B, a higher resolution view of the last ∼6 kb of the sequence contig. Note the occurrence of single EST sequences matching multiple sites in the query sequence, suggesting division of those ESTs into multiple exons. In addition, more than one EST often aligns under a single region in the query sequence, indicating a cluster of ESTs. Often, but not always, such sets of ESTs would already correspond to a single UniGene cluster (Schuler et al. 1996).
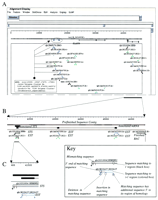
Three representative Musk views of the ASN.1 output generated for a single sequence contig by PowerBLAST from the lowest (A) to highest (C) resolution. Sequence matches to records in various databases are shown in a boxed region directly below the schematically depicted sequence contig. In addition, sequence annotations of the query sequence (e.g., Annotated mRNA) are shown as black lines and boxes immediately below the depicted query sequence. Regions in which the query sequence has been masked for repetitive elements are shown as crosshatched. (A) Global view of entire contig. Multiple sequence matches are shown within the boxed region labeled BLASTN. In addition, several user-added annotations (colored lines and boxes) are shown immediately below the contig sequence. In particular, a UniGene cluster has been annotated as an mRNA (blue). The ESTs corresponding to this hypothetical UniGene cluster are linked with dotted lines. (B) A higher resolution view of the last ∼6 kb of the sequence contig. Various sequence matches are shown, with additional text added to indicate the different types of matching sequences (e.g., EST, STS, finished BAC). Sequences matching to more than one region of the query sequence are colored to reflect the number of such matches. One EST (N22627) matches three regions (green), whereas another EST (AA347250) matches two regions (blue). Also indicated are the putative intron–exon boundaries for an mRNA containing several of the matching EST sequences, an STS (G31804), and the 5′ end of a finished BAC clone (AC000064). (C) A 200-bp region within the ∼6-kb segment shown in B. At this high level of detail, the degree of identity between the query sequence and matching sequences can be more clearly seen. Various such features are graphically indicated, including mismatches, insertions, and deletions (see the inset box for a key).
The higher-resolution views of the data within Musk (Fig. 2) reveal important additional features of the sequence matches. For example, the GenBank accession number of each matching sequence is provided; each of these is in turn electronically linked to the corresponding GenBank record, allowing quick access to additional information about the matching sequences. Mismatches between the query sequence and a matched sequence are indicated by an orange line in the boxed region. Insertions and deletions are marked as gapped regions and vertical black lines, respectively. This coloring allows a rapid, rough assessment of the degree of identity between the query and aligned database sequences. Finally, additional information about the nature of the matching sequences is indicated in a color-coded fashion as described above. The latter feature is especially useful for characterizing genes of interest and defining their intron-exon boundaries.
A key aspect of our approach for sequence analysis/annotation is its ability to manage the inevitable maturation of sequence contigs, especially when used for the repeated analysis of sequences at various prefinished stages. Figure 3 illustrates how the PowerBLAST/Musk/Sequin combination can deal with such a progression. At an earlier stage of analysis, PAC DJ1099C19 was represented by multiple sequence contigs, with the analysis and annotation performed for two such contigs shown in Figure 3A. As sequencing progressed with this clone, contigs merged to create a single, larger sequence contig. All of the earlier annotations could be simply carried over and used for the larger contig without having to recompute and reanalyze the stable features.
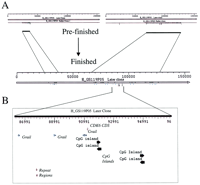
Evolution of sequence analysis with the progression of prefinished sequence data. During the sequencing of genomic clones, prefinished sequence contigs eventually merge together. The approach described here allows the retention of information derived from the earlier analysis of the smaller sequence contigs. (A) Two small prefinished sequence contigs and their associated annotated feature are shown relative to their eventual positions within the finished sequence. During the subsequent analysis of the finished sequence (or even the sequence obtained by the merger of the two small contigs), all of the features deduced at an earlier stage can be directly incorporated into an updated Sequin record. (B) A more detailed view of a portion of the final sequence is shown, which reveals the presence of sequence features annotated at an earlier stage of analysis (e.g., CpG islands, CDK6 CDS).
In summary, we have devised and implemented a straightforward approach for analyzing genomic sequence data, either at a prefinished or finished stage. Such a strategy is useful both for organizing additional studies of regions of particular interest (e.g., searching for disease genes, such as during a positional cloning project) and as a framework for more detailed or specialized sequence annotation (e.g., third party annotation). In either context, the annotation of genomic sequence is an evolving process that requires both initial analysis followed by more thorough experimental investigations and refreshed comparisons with the growing sequence databases. The structured, computer-readable (ASN.1) product of our approach could also serve as an ideal electronic supplement to publications that focus on annotation of sequence data beyond that supplied in the original GenBank sequence record (Wheelan and Boguski 1998). In any event, our strategy represents a particularly practical approach for mining the wealth of information that will become increasingly available over the next few years as a working draft of the human genome is assembled (Collins et al. 1998).
A users guide to the software system reported here is provided on an extensive web supplement to this article athttp://www.ncbi.nlm.nih.gov/Kuehl/pre-finished.
METHODS
Prefinished sequences were retrieved by ftp from the Washington University Genome Sequencing Center (http://genome.wustl.edu/gsc/Search/ftp.shtml). Intermittently, the status of sequence data was checked, with changes identified by a new date for the corresponding file. Following ftp downloading, all sequence files were stored locally and subjected to the analyses described below.
PowerBLAST analyses were performed by use of the following parameters: BLASTN (M = 1, N = −3, S = 40, S2 = 40) and BLASTX (S = 90, S2 = 90, FILTER = SEG). The results from PowerBLAST were then output as ASN.1 objects for ease of importation into Sequin (Kans and Ouellette 1998). Output from PowerBLAST was examined by the Musk viewer [included as part of the PowerBLAST package (ftp://ncbi.nlm.nih.gov/pub/sim2/PowerBlast)]. For additional details about the PowerBLAST/Musk programs, seehttp://www.genome.org/cgi/content/full/7/6/649.
A separate ASN.1 file was generated for the PowerBLAST results from each sequence contig of a BAC/PAC clone. Each ASN.1 file was then examined for sequence matches with >95% identity across 50 nucleotides. Furthermore, sequence matches were identified as ESTs, STSs, or known genes. ESTs were compared against the UniGene database (http://www.ncbi.nlm.nih.gov/UniGene). When multiple ESTs were represented in a UniGene cluster, the ESTs were annotated as single features for both the 5′ and 3′ ends, thereby reducing the complexity of the Musk views. STSs were annotated as STS features. Finally, nucleotide matches were checked for consistency with protein match (BLASTX) information within the region. In addition to searching the public sequence databases, each sequence contig was compared with a series of local databases with BLASTN. As before, matches of >95% identity across 50 nucleotides were annotated in the Sequin record of that sequence contig.
In a separate step, sequences likely to contain polymorphisms were identified by two approaches. First, individual sequence contigs were compared with a local database of simple nucleotide repeats with BLASTN. Second, the SEG program was used to identify regions of low compositional complexity; such regions often contain simple sequence repeats or pseudorepeats. Regions containing five or more copies of a repeat element were annotated.
As part of our sequence analysis process, several gene prediction programs [GRAIL (Xu et al. 1994), GenScan (Burge and Karlin 1997), and MZEF (Zhang 1997)] were used to identify putative genes. Typically, these programs were used once a sequence contig reached ∼50 kb in size. To reduce the manual effort involved in the visual examination of these results, a series of Perl scripts (available on request) were written to convert the output from each gene prediction program to ASN.1 sequence records that were compatible with Sequin.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL boguski{at}ncbi.nlm.nih.gov; FAX (301) 435-7794.
-
- Received August 31, 1998.
- Accepted December 10, 1998.
- Cold Spring Harbor Laboratory Press








