
A Sequence-Ready BAC Clone Contig of a 2.2-Mb Segment of Human Chromosome 1q24
Abstract
Human chromosomal region 1q24 encodes two cloned disease genes and lies within large genetic inclusion intervals for several disease genes that have yet to be identified. We have constructed a single bacterial artificial chromosome (BAC) clone contig that spans over 2 Mb of 1q24 and consists of 78 clones connected by 100 STSs. The average density of mapped STSs is one of the highest described for a multimegabase region of the human genome. The contig was efficiently constructed by generating STSs from clone ends, followed by library walking. Distance information was added by determining the insert sizes of all clones, and expressed sequence tags (ESTs) and genes were incorporated to create a partial transcript map of the region, providing candidate genes for local disease loci. The gene order and content of the region provide insight into ancient duplication events that have occurred on proximal 1q. The stage is now set for further elucidation of this interesting region through large-scale sequencing.
[The sequence data described in this paper have been submitted to GenBank under accession nos. G42259–G42312 and G42330–G42335.]
Construction of large-scale contigs of bacterial clones has been an important problem in genomics and genetics for over a decade. Large-scale bacterial clone contigs of mammalian DNA were first constructed to characterize selected genomic regions such as the mouse major histocompatibility locus (Steinmetz et al. 1982), to characterize entire human chromosomes (Tynan et al. 1992), or as part of positional cloning projects aimed at identifying human disease genes (Zuo et al. 1993). The success of the positional cloning paradigm has seen a wider application of the methodology to organisms as distant from human as plants. More recently, the problem of generating large-scale contigs as substrates for sequencing the human genome has framed much of the discussion of appropriate strategies for contig construction (McPherson 1997). The current application of genomic methods to a broad range of organisms, combined with the inevitable growth in DNA sequencing that will follow the successful completion of the human genomic sequence, make it likely that construction of large-scale contigs of bacterial clones will continue to be important in the future.
The q24 segment of human chromosome 1 has been the focus of a great deal of activity because of its gene content. Two human disease genes, one involved in inherited open-angle glaucoma (Stone et al. 1997) and another mutated in familial trimethylaminuria (Dolphin et al. 1997;Treacy et al. 1998), have already been identified in the region. The FAS ligand (TNFSF6), a molecule crucial to apoptotic signal transduction in the immune system, is also encoded by a gene in the region, and increased copy number of chromosome 1q has been associated with progression of a wide variety of pediatric and adult tumors (Douglass et al. 1985; Knuutila et al. 1998). Ancient genomic duplication events have resulted in gene families with members located at proximal chromosome 1q and 1p (Moseley and Seldin 1989; Maresco et al. 1996), and, in some cases, chromosomes 6q, 9q, or 19p (Katsanis, et al. 1996; Kasahara et al. 1997). Sequencing of 1q24 will not only uncover genes responsible for phenotypes that map to the region, but may give clues to the gene content of other interesting regions and yield insight into the nature of ancient duplication events. Although three independent yeast artificial chromosome (YAC) contigs of this region have been described (Clépet et al. 1996; Sunden et al. 1996; Belmouden et al. 1997), a bacterial clone contig has not been available. As an initial step toward sequencing of 1q24, we have constructed a single BAC clone contig that spans >2 Mb of this important region of the human genome. We used a strategy of simultaneous walking from multiple entry points. Our results indicate that this approach is a viable alternative to contig construction methods that require the generation of a high density of STSs at the beginning of the process.
RESULTS
We began our contig construction effort with information and reagents that are typical of those available for the great majority of the human genome. A YAC/STS content map of the region was constructed (D. Vollrath, unpubl.), and 19 roughly ordered STSs (an average of ∼1 STS per 100 kb) were screened against a fivefold redundant, commercially available BAC library (see Methods). A collection of 59 clones was obtained, grouped into 9 contigs. The initial screening set of STSs consisted of all markers publicly available in the region at the time and the STSs were expected to be randomly distributed across the region. The markers included genetically mapped microsatellite repeats, genomic STSs generated by the Whitehead Institute Center for Genome Research or the Stanford Human Genome Center, and markers taken from the literature that we mapped to the YAC contig (Dolphin et al. 1991, 1992; Shephard et al. 1993; Seltmann et al. 1994; McCombie et al. 1996).
To close gaps, additional STSs were generated from BAC clone insert ends by DOP-vector PCR (Wu et al. 1996). The insert size of each clone was determined by pulsed field gel electrophoresis (PFGE) followingNotI digestion of miniprep DNA. Clones smaller than ∼75 kb were usually excluded from end isolation. A total of 40 STSs were created during the first round of end STS generation. Individual STSs were tested against their cognate BAC clone, other clones in the same contig, and clones at the edges of adjacent contigs. (In some cases, the orientation of contigs was known from YAC/STS content mapping.) Previously undetected overlap was revealed in a number of cases, fusing the original 9 contigs into 4 contigs of 31, 4 , 12, and 12 clones, centromere distal to proximal, respectively.
An initial walking step was taken by screening the BAC library with six end STSs (2481, 2492, 2527, 2585, 2751, and 2753) located at the borders of the three gaps or at the telomeric end of the largest contig. Eleven new clones were identified and these were verified and precisely positioned in the contig by testing with nearby STSs. Concurrently, STSs derived from radiation-hybrid-mapped ESTs were placed on the YAC map (see below). An EST (stsG8166) that appeared to fall in or near a gap was tested against the BAC clones and found to be absent. This STS was therefore used to screen the BAC library and five clones were identified, one of which (359N20) was among the eleven obtained in the first walking step. The largest contig was thereby extended into one of the gaps.
Insert sizes were determined for the newly identified clones, ends of selected clones were isolated and sequenced, and sixteen new end STSs were created and located on the contig. These new end STSs were then tested against nearby BAC clones, resulting in detection of clone overlap and closure of all three gaps to produce a single large contig (Fig. 1). In total, STSs were generated from both ends of 14 clones and from a single end of 32 clones, resulting in 60 end STSs (Table 1).

BAC clone contig and locations of STSs across a 2.2-Mb region of human chromosome 1q24. (Left) 1q telomere; (right) centromere. The Figure is drawn to scale; the length of individual BAC clones (solid rectangles) reflects measured insert sizes. The locations of STSs are listed across the top with shaded lines extending through the contig as guides. Groups of STSs that are not ordered with respect to one another are indicated by a solid line under the STS names. STSs for which location was not strictly constrained were spaced at roughly equidistant intervals between constrained markers. Some STSs contain two names separated by a (/) to indicate that two independent primer sets were tested on the contig and subsequently determined to be from the same location. We have added a prefix C to the addresses of three clones obtained from the CEPH BAC library (C283F8, C207A5, C258G3). STS names that are a four digit number beginning with 2 are BAC end STSs derived from contig clones. (See Table 1 for the correspondence between STS number and clone insert end.) YAC end STSs L733F12, R902C7, L928G11, R912H6, L766E4, L885B7, R821E8, R834F6, and R719G2, are from Belmouden et al. (1997), and 650G9L and 933H12L are from Clépet et al. (1996). D1S3666 is from Sunden et al. (1996). C1E13 and C1E117 are from Seltmann et al. (1994). See Table 2 for STSs from genes and ESTs. Primer pairs for FMO1-4 were obtained from the literature (Dolphin et al. 1991, 1992; Shephard et al. 1993; McCombie et al. 1996). The remaining STSs are from publicly available physical maps from the Whitehead Institute Center for Genome Research, NCBI, or the Stanford Human Genome Center.
BAC End STSs
In an effort to confirm overlap and increase clone coverage in sparse regions of the contig, a final round of BAC screening was carried out with several STSs. An additional clone was isolated from the CITB library and three BACs were obtained from a library constructed at Centre d’Etude du Polymorphisme Humain (CEPH). In the course of constructing the contig, all clone inserts were sized by PFGE after digestion with NotI. No NotI sites were found within clone inserts, consistent with a restriction map of the region based on digestion of YAC clones (Belmouden et al. 1997). In summary, two rounds of end STS generation and one walking step were required to achieve closure and a single contig of 78 clones.
To integrate our map with previously published physical and genetic maps of the region, additional STSs were placed on the contig by testing the collection of BAC clones with PCR assays from YAC ends (Clépet et al. 1996; Belmouden et al. 1997) or a polymorphic microsatellite marker (Sunden et al. 1996). We also obtained a partial transcript map of the region by locating ESTs from a low resolution radiation hybrid map of the human genome (Schuler et al. 1996) or from the literature (Stone et al. 1997). ESTs that had been broadly localized to the region were tested against DNA from somatic cell hybrids containing portions of 1q, and ESTs that passed this test were further localized on the YAC contig. Thirteen ESTs were excluded from the region by this approach, and 10 ESTs were placed on the BAC contig. The latter, when combined with other genes or ESTs already present on the BAC contig, yielded a total of 20 genes or ESTs localized in the region (Table 2), including one end STS (2562) that matched a previously unmapped EST cluster.
Genes and ESTs
To assess clone integrity and confirm overlap, a HindIII fingerprint was obtained for all of the clones in the contig. Clone digests were loaded on agarose gels according to their position in the contig to facilitate comparison of fingerprints among related clones (Fig. 2). Following electrophoresis and staining of the gel with a sensitive intercalating dye, fingerprints were manually inspected and no inconsistencies were observed. As further confirmation, fingerprinting gels were blotted to nylon membranes and clones from different sections of the contig were probed with one of six end STS probes (2499, 2596, 2606, 2608, 2663, and 2751). In all cases, an STS hybridization probe detected the same-sizedHindIII fragment in only those clones identified previously by PCR as containing the STS (data not shown).
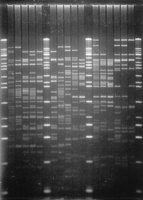
HindIII fingerprint of contig clones. DNAs isolated from 16 clones corresponding to a section of the contig were digested withHindIII and resolved by agarose gel electrophoresis. Counting from left to right, lanes 1, 7,13, and 20 contain size standards, a mixture ofHindIII-digested λ DNA and 1-kb ladder (Life Technologies, Gaithersburg, MD). In these lanes, the smallest visible band is 0.5 kb in size and the largest is 23.1 kb.
DNA not used in fingerprinting was used in a final PCR confirmation step. Groups of related clones were tested with STSs from the relevant region of the contig. An effort was made to test at least three STSs, and usually more, beyond each end of a clone to produce a halo of negative data points. In this way, we could be confident that potential interstitial deletions or errors in STS order would be detected. The confirmation step eliminated a small number of apparent interstitial deletions that resulted from initial false-negative data, but three inconsistencies remained. All three inconsistencies required the placement of an STS outside of the end STS generated from a particular clone. In all three cases, the inconsistencies were found to be due to the fact that two STS had been created from the same sequence because two BACs ended at the same HindIII site (see Discussion).
DISCUSSION
We have constructed a redundant, verified BAC contig of a region of 1q24 by library screening and walking from clone ends. The size of the contig is >2 Mb, on the basis of two independent methods. First, the distance between L733F12 and WI-10286 is ∼2 Mb, as determined by restriction mapping of YAC clones (Belmouden et al. 1997), and our BAC contig extends beyond these markers. Second, we combined STS content information and knowledge of the correspondence between end STSs and the BAC clones from which they were derived, with BAC insert size information, obtained for every clone in the contig, to create an internally consistent representation of STS and clone locations (Fig.1). The fact that one-half of all STSs in the contig were generated from BAC ends and that at least one end STS was created from 46 of 78 clones in the contig, places significant constraints on the location of STSs and clones within the contig. This analysis yields an estimated contig size of 2.2 Mb and a distance of 2.1 Mb between L733F12 and WI-10286, in pleasing congruence with the earlier estimate. The BAC/STS map that we have created thus has an average resolution of 1 STS per 22 kb, making it one of the few multimegabase regions of the genome with this level of STS resolution. Most of the STSs are ordered with respect to one another, resulting in an average of one ordered marker per 24 kb. These STSs may be useful for searching for single nucleotide polymorphisms even after the entire sequence of the region has been obtained (Kwok et al. 1996).
The contig is sequence ready on the basis of several criteria. First, the contig is redundant; on average, each marker is present in 4.5 clones. (One marker, D1S1619, is present only once in the contig, but the size of the single coverage segment is estimated to be <10 kb.) Second, the clones that make up the contig appear to be faithful replicas of genomic DNA. Our approach of walking from clone ends is especially sensitive to chimerism, and no evidence of this type of artifact was found for any of the clones. Similarly, no evidence of interstitial deletion was found for any of the clones, as assessed by STS content and HindIII fingerprints. Finally, there is no evidence of clone rearrangement on the basis of comparison ofHindIII fingerprints among overlapping clones. However, although we used methods capable of detecting HindIII fragments 500 bp or larger, we cannot account for smaller fragments. Also, the number of HindIII fragments present in the largest clones (>175 kb) made it difficult to unambiguously assign every band for a small number of clones. Finally, the degree of clone overlap can be estimated and a minimal tiling path of clones can be defined. By incorporating clone insert size information into our representation of the contig, a credible estimate of clone overlap is available. There is some uncertainty in these estimates due to uncertainties in insert sizes (± ∼5%) and uncertainty in the precise location of some clone ends or STSs. The fact that the estimated overall size of the contig fits well with a previous estimate, indicates that errors in overlap are not likely to be large.
Construction of the contig required ∼1 year of effort by a skilled technician. We learned several lessons in the course of the work that will benefit future contig construction projects. After one round of STS generation and detection of overlap, walking in both directions from internal contigs appeared to be an efficient strategy for closure. However, for the smallest of the two internal contigs, our walking efforts were stymied because we were only able to generate two end STSs from the four clones in the contig (180K5, 208D18, 365B4, and 122N7) due to repeat sequences at insert ends. Initial screening of a more highly redundant library would provide more ends and mitigate this problem.
During the walking process, clone orientation within contigs was not known, so about one-half of all end STSs generated were internally located in an existing contig. Although such STSs were not useful for walking, they ultimately contributed greatly to the contig by constraining the location of insert ends and increasing overall STS density. We also learned that generating STSs from the largest BACs increased the efficiency of walking. Sizing BAC inserts prior to end STS generation was not a great burden and insert sizes provided useful distance information after the contig was completed.
We observed at least three instances in which pairs of BACs ended at the same HindIII site. The Sp6 ends of clones 27D3 and 186A4 (STSs 2505 and 2507, respectively) were the same, as were the Sp6 ends of clones 260O11 and 378J17 (STSs 2542 and 2540, respectively). Clones that shared ends were clearly independent as demonstrated by differences in STS content and well addresses. [Two other BACs (224I8 and 233O4) also shared identical end sequences, but we could not be certain that the clones were independent because they have the same STS content and similar insert sizes.] In another case, two clones, 169I3 and 37L6, were found to overlap by only the 6 bp of a HindIII site. PCR was carried out with one primer from STS 2564 (T7 end of 169I3) and a single primer from 2529 (T7 end of 37L6). A 500-bp fragment was generated and its sequence revealed a singleHindIII site near the middle (data not shown). The two BAC end sequences corresponded perfectly to the two halves of the 500-bp fragment. The two STSs lie within the first intron of the myocilin (MYOC) gene (D. Vollrath and V. Jaramillo-Babb, unpubl.), which contributed to our discovery of the 6-bp overlap. Additional, undiscovered occurrences of these types may account for some of the apparent clustering in the contig of other STSs made from BAC and YAC insert ends.
Our results suggest a nonrandom distribution of BAC clone ends that, to our knowledge, has not been described previously, despite the fact that end STSs have been used in the construction of several other human BAC contigs (Boysen et al. 1997; Boycott et al. 1998). In fact, a random distribution of BAC ends across a 1.1-Mb region of the human T cell receptor locus has been reported (Boysen et al. 1997), but a low redundancy of BAC clone coverage may account for a failure to detect identical ends. Nonrandomness in BAC clone end distribution might be expected in light of the long-recognized preference of some restriction enzymes for cleavage at particular sites (Thomas and Davis 1975), including a 14-fold difference in the rates of cleavage ofHindIII sites in λ phage DNA (Nath and Azzolina 1981). A scheme for sequencing the human genome has been proposed that assumes a random distribution of BAC insert ends (Venter et al. 1996). Our experience indicates that, if a random distribution of clone ends is desired, it would be prudent to combine clones from libraries made with different restriction enzymes.
The genomic region corresponding to the contig is contained within large genetic inclusion intervals for three disease loci; thiamine-responsive megaloblastic anemia syndrome (Neufeld et al. 1997), prostate cancer (Smith et al. 1996), and deafness (Fagerheim et al. 1996). A hepatocarcinogenesis susceptibility locus (Hcs7) has also been broadly localized to the corresponding region in mouse (Lee and Drinkwater 1995). The 20 genes and ESTs localized on the contig are candidate genes for these phenotypes.
We have precisely localized two human disease genes within 1q24 by including them in our contig. STSs were created for the 5′ and 3′ ends of MYOC, which corresponds to the GLC1Alocus for open angle glaucoma (Stone et al. 1997). The two ends of the gene are well resolved, indicating that the orientation of transcription of MYOC is from telomere to centromere. We have also localized the FMO3 gene, mutations in which cause trimethylaminuria (Dolphin et al. 1997; Treacy et al. 1998), near to the related genes FMO1, FMO2, and FMO4. Our data demonstrate that the four genes are located within a region of ∼200 kb and constitute a gene array. Interestingly, theFMO5 gene is located more proximal on chromosome 1 at band q21 (Gelb et al. 1997; Lioumi et al. 1998). This probably reflects an ancient duplication event, hypothesized to have occurred during mammalian evolution, which resulted in two blocks of homologous genes that now span the centromere on human chromosome 1 (Mosley and Seldin 1989; Oakey et al. 1992). It will be of interest to determine whether other genes within the 2.2-Mb region are also duplicated on proximal chromosome 1q or 1p.
METHODS
BAC Library Screening
The CITB Human BAC library (Releases I and II, Research Genetics, Inc., Huntsville, AL) was screened by PCR with commercially available DNA pools and DNA pools made at the Stanford Human Genome Center. A total of 5 μl of pool DNA was used in a 10-μl reaction and PCR was carried out in a mixture consisting of 1× buffer C (20 mm Tris-HCl at pH 8.3, 50 mm KCl and 2.5 mm MgCl2), 0.2 mm each dNTP, 1 μm each STS primer and 0.5 units of Taq polymerase with a PTC-200 thermal cycler (MJ Research, Watertown, MA) with the following protocol: 94°C for 15 sec, 62°C for 23 sec, 72°C for 45 sec for 33–35 cycles. The contents of a positive well were streaked for single colonies on agar plates containing 12.5 μg/ml chloramphenicol, and individual clones were confirmed by PCR on bacterial culture.
BAC Insert Sizing
DNA was prepared by alkaline lysis from 1.5 ml of bacterial culture (Luria broth) and used to size BAC inserts and as template for DOP-vector PCR. In most cases, the same miniprep DNA was used for both purposes. A total of 5 μl of 20 μl of DNA was digested in a total volume of 20 μl with 10 units of NotI (New England Biolabs, Inc. Beverly, MA) for 2 hr at 37°C in a buffer supplied by the manufacturer. Samples were loaded on a 1% SeaKem LE (FMC BioProducts, Rockland, ME) agarose gel in a buffer containing 0.5× TBE and subjected to PFGE for 20 hr at 6 V/cm, 15°C, and a switching interval that varied linearly from 1 to 11 sec using a BioRad CHEF DR II apparatus. The gel was stained with 0.5 μg/ml ethidium bromide for 20 min at room temperature and destained with water. Gel images were captured on a IS-1000 Digital Imaging System (Alpha Innotech Corporation, San Leandro, CA).
BAC Insert End Isolation
We used a modification of the DOP-vector PCR method (Wu et al. 1996) to isolate BAC insert ends. A total of 4 μl of a premixed stock solution (1× Sequenase buffer, 40 ng/μl 6-MW primer, and 0.5 mm of each dNTP) was added to the template (1 μl of a 1:100 dilution of BAC miniprep DNA), which was then denatured by heating at 96°C for 3 min and cooling to 30°C. Five microliters of a second premixed stock solution containing 1× Sequenase buffer and 1.3 units of Sequenase version 2.0 (U.S. Biochemical Corporation, Cleveland, OH) were then added to the reaction, and the temperature was ramped to 37°C over a 1 min interval, followed by incubation at 37°C for 3 min. The Sequenase was inactivated by incubation at 72°C for 10 min. Finally, 40 μl of a prewarmed mixture consisting of 1× PCR buffer (50 mm KCl and 20 mmTris-HCl at pH 8.3), 0.2 mm of each dNTP, 0.4 μm for one of two remote vector primers suitable for pBeloBAC11 (T7 side: CGACGTTGTAAAACGACGG; Sp6 side: GTTGTGTGGAATTGTGAGCG), 4 ng/μl of 6-MW primer and 2.5 units of Taq polymerase were added to the reaction. PCR was carried out in a PTC-100 thermal cycler (MJ Research, Watertown, MA) with the following protocol: 94°C for 30 sec, 58°C for 30 sec, 72°C for 1 min for 35 cycles. A 50 μl heminesting PCR was subsequently performed with 2 μl of the initial PCR product as template, 1× buffer C, 1 μm of one of two nested vector primers (T7 side: TGTAAAACGACGGCCAGT; Sp6 side: TACGCCAAGCTATTTAGGTG), 10 ng/μl 6-MW primer, 2.5 units of Taq polymerase and the following conditions: 94°C for 20 sec, 60°C for 30 sec, and 72°C for 1 min. The distance between the remote and nested vector primers is 25 bp for the T7 side and 53 bp for the Sp6 side.
Sequencing and STS Generation
PCR products were precipitated with ethanol and subjected to electrophoresis on a 2% NuSieve GTG (FMC BioProducts, Rockland, ME) agarose gel in 1× TAE buffer. Bands were excised from the gel, melted for 10 min at 70°C, and cooled to 40°C for 10 min. One unit of β-Agarase I (New England Biolabs, Inc. Beverly, MA) was added, mixed, and incubated at 40°C for 15–16 hr. Samples were spun at 14,000 rpm for 10 min in an Eppendorf microfuge and supernatants were transferred to clean tubes. Sample volumes ranging from 1 to 9.8 μl of DNA were sequenced with an ABI Prism Dye Terminator Cycle Sequencing Kit (PE Applied Biosystems, Foster City, CA). The resulting sequences were analyzed for the presence of dispersed repeats and PCR primers were selected from suitable sequences as described previously (Vollrath 1998). End STSs were analyzed by PCR against neighboring BAC clones to detect overlap and to order STSs. All of the BAC end STSs that we generated were amplified under a single set of PCR conditions (see BAC library screening).
EST Mapping
The general locations of ESTs in the 1q21–1q24 region were determined by PCR testing of DNAs from two somatic cell hybrids; a human monochromosomal hybrid containing chromosome 1 (GM13139, Coriell Cell Repositories), and a hybrid containing a human chromosome 1 with a deletion of 1q23–1q25 (Franco et al. 1991). Those ESTs that had potential to map to the region on the basis of the hybrids were then tested against a YAC contig which extended beyond the 2-Mb region. Some ESTs were included and others excluded from the region by this approach (Table 2).
HindIII Fingerprinting
Miniprep DNA was prepared from 1.5 ml of cells grown to saturation in Luria broth and DNA was resuspended in 20 μl of TE. Two microliters of DNA were digested in a total volume of 25 μl with 10 units of HindIII (New England Biolabs, Inc. Beverly, MA) for 2 hr in a buffer supplied by the manufacturer. Digests were mixed with loading dye and an entire sample was loaded into one of 24 wells of a 14 × 20 cm long, 1% LE agarose gel with 1× TAE, and samples were subjected to electrophoresis at 80 V for 15.5 hr at 16°C. Gels were stained in 1× GelStar (FMC BioProducts, Rockland, ME) and visualized with a 310-nm of UV light source. Gel images were photographed with a Polaroid camera and fingerprints were manually inspected to account for as many bands as possible.
Acknowledgments
We thank Mariana Rexan for assistance with BAC library screening and sizing of clone inserts, Hadi Abderrahim for BAC clones from the CEPH library, Chenyan Wu for oligonucleotide sequences for DOP-vector PCR of BAC ends, David Patterson for a somatic cell hybrid, and Richard M. Myers and Richard W. Hyman for comments on the manuscript. This work was supported by grants from the National Institutes of Health (EY11405), and the March of Dimes Birth Defects Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL vollrath{at}genome.stanford.edu; FAX (650) 723-7016.
-
- Received September 29, 1998.
- Accepted December 15, 1998.
- Cold Spring Harbor Laboratory Press








