
Functional Genomics in the Mouse: Phenotype-Based Mutagenesis Screens
Abstract
Significant progress has been made in sequencing the genomes of several model organisms, and efforts are now underway to complete the sequencing of the human genome. In parallel with this effort, new approaches are being developed for the elucidation of the functional content of the human genome. The mouse will have an important role in this phase of the genome project as a model system. In this review we discuss and compare classical genetic approaches to gene function—phenotype-based mutagenesis screens aimed at the establishment of a large collection of single gene mutations affecting a wide range of phenotypic traits in the mouse. Whereas large scale genome-wide screens that are directed at the identification of all loci contributing to a specific phenotype may be impractical, region-specific saturation screens that provide mutations within a delimited chromosomal region are a feasible alternative. Region-specific screens in the mouse can be performed in only two generations by combining high-efficiency chemical mutagenesis with deletion complexes generated using embryonic stem (ES) cells. The ability to create and analyze deletion complexes rapidly, as well as to map novel chemically-induced mutations within these complexes, will facilitate systematic functional analysis of the mouse genome and corresponding gene sequences in humans. Furthermore, as the extent of the mouse genome sequencing effort is still uncertain, we underscore a necessity to direct sequencing efforts to those chromosomal regions that are targets for extensive mutagenesis screens.
The end of the first decade of the Human Genome Project is marked by the transition from the physical mapping of genomes to the characterization of their functional content. The sequencing of expressed sequence tags (ESTs) is relentlessly moving toward the identification of nearly all transcribed genes in mice and humans (Adams et al. 1995; Hillier et al. 1996). These ESTs are being placed onto the genetic and physical maps, further facilitating the process of positional cloning (Schuler et al. 1996). Recently developed technologies enable the simultaneous analysis of the expression patterns of many genes on a large scale (Schena et al. 1995; Chee et al. 1996; DeRisi et al. 1997). The final endeavor in genome characterization—the sequencing of the human genome—is now under way.
The most difficult challenge now lies in devising ways to use the vast amount of information gathered in the Genome Project, such as physical/genetic mapping data and complete nucleotide sequence, to understand the complexity of life; with a complete transcriptional map of the genome and the entire genomic sequence known, the function of the genes will still remain unclear (Miklos and Rubin 1996; Oliver 1996). The problem of elucidating function for all of the 100,000 or so genes in humans/mammals has spawned a new area of research that is being called “functional genomics” or “new genomics” (Lander 1996; Hieter and Boguski 1997). The goal of the functional genomics effort is to determine the biological function of a genome using strategies that will ultimately coalesce with the genetic maps, physical maps, DNA sequence, and gene transcription patterns.
Although the function of many genes can be characterized with in vitro culture systems, or inferred from the function of their orthologs in a model organism such as yeast, it is widely agreed that analysis of human gene function, in the context of a whole organism, will rely heavily on use of the mouse as a model (Paigen 1995; Meisler 1996;Bedell et al. 1997). Transgenesis or targeted mutagenesis in the mouse has provided biologists with an extremely powerful tool to define the in vivo function of mammalian genes (Capecchi 1989; Brandon et al. 1995a,b,c). Nevertheless, it is impractical to use targeted mutagenesis on a case-by-case basis to mutate all of the genes in mice. Clearly, a more global, efficient, and integrated technology is needed.
In any endeavor, it serves well to learn from history. Until recently, the first step in studying gene function was phenotypic characterization of mutants or natural variants. In flies and nematodes, whole genome mutagenesis screens continue to be a major tool in the identification of gene function (Brenner 1974; Hirsh and Vanderslice 1976; Nusslein-Volhard and Wieschaus 1980; Wieschaus et al. 1984). These screens have been performed on a scale that is sufficiently large to ensure recovery of an array of mutations that elicit the desired phenotype. Collections of such phenotype-selected mutants in flies have had a major impact in the elucidation of gene function in developmental pathways. Importantly, the hierarchy of gene (inter)actions or ordering of genes in functional pathways could be established without prior knowledge of the molecular nature of the gene product (Avery and Wasserman 1992; Huang and Sternberg 1995). Also notable is that saturation screens tend to yield multiple mutations within identical genes. Collections of alleles, aside from those that are strictly loss-of-function, often enable better insight into the function of a gene.
The experimental obstacles posed by mammalian model systems have made it impractical to efficiently pursue mutagenesis strategies as in invertebrates. That has not stopped us from trying, however, on a scale far from the “saturation” level. Radiation has been used to generate mutations in mice for quite some time, ultimately to create “deletion complexes” around several visible loci (Lyon and Morris 1966; Holdener-Kenny et al. 1992; Rinchik 1994; Russell et al. 1982,1995; O’Brien et al. 1996). To ensure that the observed mutant phenotype is caused by a single gene mutation, however, other mutagens, namely those that induce small intragenic lesions, are employed. InDrosophila melanogaster and Caenorhabditis elegans,the chemical ethylmethane sulfonate (EMS) is used routinely to create single gene mutations or new alleles of previously mutated genes, whereas in the mouse and zebrafish, the most potent and commonly used chemical mutagen is N-ethyl-N-nitrosourea (ENU) (Russell et al. 1979; Ashburner 1889; Mullins et al. 1994;Solnica-Kerzel et al. 1994; Anderson 1995). Finally, the advance in molecular biology and interest in the molecular identification of genes causing mutant phenotypes has influenced mutagenesis techniques. Transposons (Tc1 in nematodes and P elements in flies) and transgenes are used to functionally alter or inactivate genes, whereas DNA flanking the transposon/transgene provides a molecular tag for the gene isolation (Soriano et al. 1987; Spradling et al. 1995). In the mouse, insertional mutagenesis has never reached the efficiency needed for large phenotypic screens.
The ability to manipulate mammalian genes in vitro in embryonic stem (ES) cells, combined with the generation of transgenic mice harboring DNA of these ES cell in their germ cells, has advanced the mutagenesis of specific genes (Capecchi 1989; Joyner 1993). Similarly, the application of gene traps to ES cells has enabled insertional mutagenesis akin to transposon tagging in flies, bacteria, and plants. These approaches have endowed the mouse with a gene-based means of mutagenesis—that is, it has become relatively simple to generate a mutation of any known molecularly identified gene. A recently reported library of ES cells (Omnibank) with >5000 sequenced tagged mutations provides a powerful functional genomics resource (Zambrowicz et al. 1998; A.T. Sands, pers. comm.). The mouse field, however, has seriously lagged in one major respect—the ability to perform systematic, phenotype-based screens on a scale large enough to reach the level of saturation. The nature of flies enables the application of chemical, deletional, and insertional mutagenesis approaches to identify mutations in genes that cause specific phenotypes in the whole organism. Although the mouse will never be as amenable to comprehensive phenotype screens as flies, recent progress has nevertheless provided the tools to perform in vivo mutagenesis that is efficient enough to enable phenotype screens. In this review, we discuss the combination of two particular mutagenesis tools—deletions and chemical mutagenesis and strategies to exploit them in phenotypic screens.
ENU Mutagenesis
ENU has proven to be a highly effective mutagen in several model organisms, including the mouse (Russell et al. 1979; Ashburner 1989;Mullins et al. 1994; Anderson 1995). It primarily induces point mutations, and therefore results in a variety of alleles (Vogel and Natarajan 1979; Skopek et al. 1992; Provost and Short 1994; Schumacher et al. 1996; Marker et al. 1997). These include hypomorphs, recessive nulls, and dominant gain-of-function mutations. A series of alleles at the same locus is extremely useful in discerning the function of genes and can facilitate the positional cloning of the altered gene. ENU induces mutations in mice and zebrafish at a frequency of greater than 1:750/locus/gamete, which enables highly efficient screening of mutagenized animals for aberrant phenotypes in subsequent breeding (Hitotsumachi et al. 1985; Mullins et al. 1994; Solnica-Kerzel et al. 1994). Another feature of ENU is that it mutagenizes stem cell spermatogonia. Therefore, treated males will produce progeny carrying ENU-induced mutations for the rest of their reproductive lives.
A major concern of any mutagenesis screen is the efficiency of recovering mutations of interest, and to genetically analyze them once they are identified. In this context, we will compare recessive versus dominant versus region-specific mutagenesis screens. The conclusions of these comparisons are summarized in Table 1.
Mutagenesis Statistics for Saturation Screens
Two extensive genetic screens for recessive developmental mutations in the zebrafish Danio rerio were accomplished by ENU mutagenesis (Mullins et al. 1994; Solnica-Kerzel et al. 1994; entire December 1996 issue of Development). Over a million and a half embryos were examined for morphological anomalies, resulting in >6000 mutants, of which >400 define genes essential for the development in vertebrates. These screens were feasible because of the nature of the organism—mutations are easily identified because the zebrafish embryo is transparent and develops outside the mother’s body (enabling detection of lethals), zebrafish reproduce prolifically, and thousands of offspring can be raised at relatively low cost.
Random saturation mutagenesis of the entire mouse genome for all classes of recessive mutations faces logistical problems relative to zebrafish. Consequently, ENU mutagenesis projects in mice have been conducted on a small scale. These efforts were focused as follows (1) to genomic subregions (Bode 1984; Shedlovsky et al. 1986; Rinchik and Russell 1990); (2) to isolate recessive visible or biochemical defects in a known gene by noncomplementation tests (Chapman et al. 1989;Cordes and Barsh 1994); (3) to identify recessive mutations causing a metabolic defect detected by a simple biochemical assay (Bode et al. 1988); and (4) to isolate mutations affecting a particular developmental process or morphological feature (K. Anderson; A. Peterson; both pers. comm.).
In the mouse, the whole genome approach has major shortcomings when it comes to screening for recessive mutations. There are two slightly different breeding schemes that can be used in a whole-genome screen for recessive mutations. Both schemes involve the mating of mutagenized males (G0) with wild-type females, followed by breeding of their F1 progeny (G1) to wild-type partners to establish families of siblings (G2) sharing the same set of mutations (Fig. 1). In scheme A of Figure 1, random matings are performed among the G2 progeny, giving G3 progeny. In this scheme, if a family carries a recessive mutation, only 25% of G2 matings will represent matings between two heterozygotes and will show a mutant phenotype in 25% of their progeny. In the zebrafish screen, to increase the recovery of mutations, this breeding scheme has been modified by mating two independently derived G1 progeny, both heterozygous for a different mutagenized genome (Driever et al. 1996; Haffter et al. 1996). In the second scheme (Fig. 1B), commonly used in the mouse, G3 progeny for screening are generated by backcrossing G2 females to their G1 fathers. In this scheme, a mutant phenotype can be observed in 25% of progeny from 50% of matings. Although less than in the former scheme, a tremendous number of mice still need to be raised, maintained, and screened to identify mutations for any given phenotypic trait. Another confounding limitation of such screens concerns initial characterization or classification of novel mutations. This is particularly relevant for recessive lethal mutations, and mutations with a late onset or subtle phenotypes, such as behavioral anomalies. Furthermore, phenotypes observed in the G3 might be a consequence of several mutations, which may need to be dissected by several rounds of backcrossing to a wild-type strain.
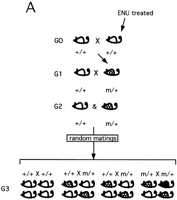
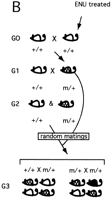
Breeding schemes for a genome-wide phenotype-based mutagenesis in the mouse. Recessive ENU-induced visible mutations (m) can be identified by using two breeding schemes. Both schemes involve the mating of mutagenized males (G0) with wild-type females, followed by breeding of their F1 progeny (G1) to wild-type (+/+) partners to establish families of siblings (G2) sharing the same set of mutations. (A) Random intercrosses are performed among the G2 progeny, giving G3 progeny. (B) G3 progeny for screening are generated by random matings between G2 females to their G1 fathers (backcross). (Shaded mice) Carriers of the recessive mutation (m/+); (black mice) recessive mutant phenotype (m/m).
With respect to the efficiency and logistics, a screen of about 3000 gametes for recessive mutations in a genome-wide recessive screen has the potential of uncovering a mutation in every one of the 75,000 or so genes in the genome at 98% confidence, assuming a mutation rate of 1:750/locus/gamete (Table 1). A three-generation, genome-wide screen (Fig. 1B) would require the generation and maintenance of 166,140 animals to detect at least one animal homozygous for each newly induced inherited mutation with high confidence (see Table 1 footnote). Considering that an eventual goal would be to clone the gene behind the new mutations, we could estimate that at least 100 animals per mutation would be needed for genetic mapping. Therefore, the total number of animals needed to generate, identify, and map mutations in 75,000 genes (assuming that every gene in the genome will have a phenotype) is 7,666,140.
Identification of Clock, the first circadian rhythm mutation in the mouse (Vitaterna et al. 1994), illustrates that screens for mutations with dominant phenotype offer a simple alternative, enabling whole-genome scans by analysis of first generation progeny (Pickard et al. 1995; for review, see Takahashi et al. 1994; Nolan et al. 1997). The ability to observe the mutant phenotype in heterozygotes not only simplifies the screen but also the maintenance of stocks, as well as the genetic and phenotypic characterization of the mutation. Dominant mutations, however, are not as frequent as recessives, and many genes may not be recoverable as dominants at all. Nevertheless, if we assume that dominant mutations arise at 10% the rate as recessives, then the progeny of a mutagenized male will contain on average 10 mutations, assuming 75,000 genes and a dominant mutation rate of 1:7500/locus/gamete. Therefore a minimum of 7500 G1animals would be needed, or 29,338 to have a 98% chance of obtaining a dominant mutation in any given gene (see Table 1).
With the potential drawbacks of classical recessive and dominant screens in mind, we discuss here the relative merits of “region-specific” mutagenesis in mice, which exploits chromosomal deletions to conduct systematic and efficient mutagenesis of specific regions of the genome, in conjunction with chemical (ENU) mutagenesis.
Region-Specific Saturation Mutagenesis
The general strategy of region-specific saturation mutagenesis presented in this review has been used extensively inDrosophila (Ashburner 1989). This approach provides a tool for identifying and characterizing genes in a particular chromosomal subregion in a simple, two-generation breeding scheme. The basic principle is to cross flies that have been mutagenized by means such as a chemical agent, P-element mobilization, etc., to flies that bear a known chromosomal deletion (in flies, called a “deficiency”) (Fig. 2). The offspring that inherit the deletion from one parent and an altered/mutated gene from the other parent will display a mutant phenotype. This simple breeding scheme allows phenotypic characterization of a large number of mutagenized gametes—sufficient to reach theoretical saturation, such that a mutation in every gene in the region should have been produced and scored for a novel phenotype. Finally, an attractive aspect of a region-specific mutagenesis is that a panel of nested deficiencies, when available, can be used for fine mapping of a newly induced mutation by complementation analysis. Such classical mutagenesis strategies, in part, were responsible for making D. melanogaster a premier model for developmental and molecular genetic analyses of higher organisms.
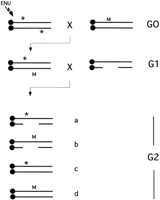
Region-specific saturation mutagenesis using ENU and deletions. Mutagenized mice (G0) with ENU-induced point mutations (*) are mated with female mice that harbor a dominant visible marker (M) and preferably a chromosomal inversion of the target region. The dominant marker allows tracking of the ENU-mutagenized chromosome and the inversion prevents recombination between the mutagenized and nonmutagenized chromosome. The offspring carrying a copy of the ENU mutagenized genome and the marker/inversion chromosome (G1) are mated with deletion heterozygotes. Four classes of (G2) progeny are generated in this cross: (a) Mice with the deletion in trans to the mutagenized chromosome; (b) mice with the deletion in trans to the marker/inversion chromosome; (c) mice that are heterozygous for the mutagenized chromosome; (d) mice heterozygous for the marker/inversion chromosome. Mice in class a are observed for novel recessive mutations, whereas the absence of this class indicates the induction of a recessive lethal mutation within the target region. In this case, G1 parent and/or class c siblings can be used to recover novel lethal mutation.
The critical genetic reagent for region-specific mutagenesis is the chromosomal deletion. The nature of Drosophila makes it amenable to induce and select for deletions in certain regions of the genome. Two institutions in the world—the Radiobiology Unit in Harwell, UK, and the Oak Ridge National Laboratory (TN)—have been responsible for generation of chromosomal deletions by whole-mouse irradiation. Deletions at the albino (c), brown (b), Brachyury (T), pink-eyed dilution (p), short ear (se), non-agouti (a), dilute (d), and piebald (s) loci have been exploited to perform a systematic characterization of functional units along these chromosomal regions, and to identify genes important in mammalian development (Russell et al. 1982; Cattanach et al. 1993; Rinchik et al. 1993; Metallinos et al. 1994; Rinchik 1994; Bell et al. 1995; Holdener et al. 1995; Johnson et al. 1995; O’Brien et al. 1996). Furthermore, Rinchik and colleagues at Oak Ridge National Laboratory have demonstrated the feasibility of a region-specific saturation mutagenesis approach in the albino deletion complex (Rinchik et al. 1990).
With the advent of targeted mutagenesis in the mouse, it is now possible to create chromosomal rearrangements suitable for a region-specific saturation screen. Two procedures have been developed recently to create large chromosomal deletions in ES cells. One method uses Cre–LoxP site-specific recombination to remove sequences between two targeted loxP sites, thereby resulting in a single, precise deletion (Ramirez-Solis et al. 1995). The other method is designed to produce deletion complexes by irradiation of ES cells containing a targeted thymidine kinase gene (You et al. 1997a,b). This makes it possible to derive mice bearing sets of nested deletions anywhere in the genome.
Now that techniques are in place for the efficient creation of deletions via ES-cell technology, and for “point mutagenesis” using ENU, saturation screens are possible in any defined chromosomal region. This approach, which we also call combined mutagenesis, allows phenotype-driven screens that provide several advantages over genome-wide random screens. (1) As illustrated in Figure 1B, in a random screen for recessive mutations in the mouse, backcrosses of G2 animals to G1 parents are typically performed. Only 25% of resulting progeny from half of the crosses are homozygous for a given mutation. Because there is no way to genetically distinguish potential homozygotes for any given mutation, phenotypic characterization must be conducted on all offspring. Another complication is that because the ENU-induced mutation rate is so high, there is a good chance that several mutations will be manifested in the backcross, threatening the reliability of the phenotypic characterization. (2) The majority of mutagenesis screens in the mouse have been aimed at the identification of mutations causing visible and viable phenotypic traits. Recessive lethal mutations, particularly those that are not “marked” by a dominant visible trait, are rarely detected. In the case of region-specific screens, these developmental mutations are detectable on the basis of failure to generate progeny that carry a mutagenized chromosome over the deletion (Fig. 2). (3) A significant advantage of the combined approaches is that the locations of the induced point (ENU) mutations are known by virtue of their failure to complement deletions used in the screen. This enables one to bypass the labor-intensive steps of narrowing down the chromosomal location of the mutant locus based on the genetic linkage analysis in a mapping cross. Moreover, establishment of allelic relationship between newly generated mutations is facilitated by having information about map position. Finally, fine mapping and positional cloning of the novel mutant locus, within the region corresponding to noncomplementing deletion, can also be facilitated by availability of a deletion complex in which the breakpoints of multiple deletions are nested randomly across the interval in which mutations are being selected. For example, if an ENU-induced mutation is uncovered by a deletion spanning 5 cM, and 10 other deletions are available with breakpoints in that interval, then the mutation can be mapped to an average resolution of 0.5 cM, or ∼750 kb.
In a scenario to recover recessive mutations in all genes using region-specific mutagenesis, it would be necessary to have about 300 deletions of 5 cM in length to span the entire genome. Each of the deletion-bearing mice would have to be mated with 3000 F1progeny (as mentioned above, this number of gametes would contain mutations in all genes at 98% confidence) of mutagenized mice, in a screen similar to that diagrammed in Figure 2 to uncover every newly generated mutation. Although 7,743,480 mice need to be generated, the use of genetic markers may allow identification of only those progeny that have to be screened for novel recessive mutations (progeny that carry a mutagenized chromosome in trans to the deletion). Whereas any genome-wide recessive screens require the use of additional mice to determine chromosomal localization, in the case of mutations identified in a region-specific effort, the chromosomal location is defined by the extent of the deletion used in the screen. Overall, the total number of mice needed for region-specific screens compared with random recessive screens is very similar. Obviously, if deletions larger than 5 cM are used, the number of animals required would drop proportionately.
Target Region: Proximal Portion of the Mouse Chromosome 5
As a model for a region-specific mutagenesis screen, we chose a proximal portion of mouse chromosome 5 (syntenic to human 4p16–4q12 and 7q36). This chromosomal region encompasses a large segment of syntenic conservation with human chromosome 4 (4p16–4q12), as well as smaller segment of homology with the distal portion of human chromosome 7 (7q36) at the most proximal end of the target region (Carver and Stubbs 1997 and on-line resources listed therein). Deletion complexes are being generated around well-defined loci (Dpp6; Hdh; Qdpr; Gabrb1) by irradiation of targeted ES cells, and a set of five to seven large deletions spanning the 30-cM segment will be initially used in crosses with ENU-mutagenized males (Fig. 3A). This screen will involve a search for a wide range of visible and developmental mutations, including those that cause embryonic lethality and meiotic defects. Moreover, to test the feasibility of a more comprehensive large-scale screen, 6000 progeny from these crosses will be examined for several nonvisible phenotypic traits such as hearing, vision, abnormal rest-activity behavior, and anomalies in sensorimotor gating.
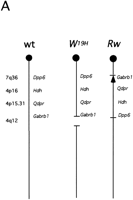
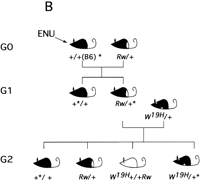
Region-specific mutagenesis screen for novel mutations in the proximal portion of mouse chromosome 5. (A) Schematic representation of the wild-type chromosome 5 and the two marker chromosomes Rwand W19H. The approximate position of four loci that serve as starting points for the generation of deletion complexes (dipeptidyl amino peptidase like protein 6, Dpp6; mouse homolog of the Huntington disease gene, Hdh; quininoid dihydropteridine reductase, Qdpr; and GABAAreceptor, β-1 subunit, Gabrb1) are indicated. Atleft chromosomal locations of these four loci in the human genome are indicated. The Rw chromosome is associated with a 30-cM chromosomal inversion (Stephenson et al. 1994; R.B. Hough, A. Lengeling, and M. Bucan, unpubl.). The W19H mutation is associated with a 4-Mb chromosomal deletion, which includes several known genes, such as a cluster of receptor tyrosine kinases, Kit, Pdgfra, and Flk1, cytoplasmic kinases Txk andTec, and the Clock gene (King et al. 1997a,b). (B) The breeding scheme of the pilot screen usingW19H. In this screen, ENU-treated males are crossed to Rw/+ females and the Rw/+ G1progeny are used for matings with the W19H /+ mice to generate G2 families that are screened for novel (recessive) mutations within the chromosomal region uncovered by theW19H deletion. Four classes of (G2) progeny are generated in this cross. (1) Mice with theW19H deletion in trans to the mutagenized chromosome; (2) mice compound heterozygous for W19H deletion and Rw chromosome; (3) normally pigmented mice that are heterozygous for mutagenized chromosome; and (4) mice heterozygous for the Rw chromosome. W19H /+* mice are observed for novel recessive mutations; failure to recoverW9H /+* mice in this cross implies that a recessive lethal mutation exists in this region.
Our choice of the proximal portion of mouse chromosome 5 as a target permits use of several existing dominant visible mutations (Tht; Ht; W; Rw) as genetic markers (Fig. 3A). Figure 3B illustrates a pilot screen being performed using W19H, an existing 4-Mb deletion located in the central portion of mouse chromosome 5 (Lyon et al. 1984; Nagle et al. 1994; King et al. 1997a) (Fig. 3B). The use of the Rw mutation serves two functions—the “white rump” of G1 progeny marks the nonmutagenized chromosome, whereas the large inversion associated with this mutation suppresses recombination between the mutagenized and nonmutagenized chromosome. In addition, the pigmentation defect associated withW19H further simplifies the identification of obligate carriers of the mutagenized chromosome in trans to the deletion. Although the current mutagenesis screen for novel mutations in the central portion of the mouse chromosome 5 uses existing mutations as genetic markers and balancers, transgenesis and loxP/cre technology may be used to create these tools in any portion of the mouse genome (Ramirez-Solis et al. 1995; Justice et al. 1997).
How many genes may reside in the target region of our region-specific mutagenesis screen? The Rw inversion, which defines the target of this screen, covers 30 cM (Stephenson et al. 1994; R.B. Hough, A. Lengeling, and M. Bucan, unpubl.). Thirty centimorgans in the mouse genome corresponds, on average, to 60 Mb or 2% of the genome. Therefore, this region may contain 1000–2000 genes. How many of these genes can mutate to recessive phenotypes or cause embryonic lethality? Based on previously published estimates, this region may contain 100–200 genes with functions that are essential for survival (Carter 1957; Shedlovsky et al. 1986). As addressed by Miklos and Rubin (1996), however, current estimates for a number of lethal genes in the mouse may not be reliable; they range from 5000–26,000 genes and are based entirely on small scale experiments. Although only a large-scale random (saturation) screen may finally resolve this issue, results of several independent region-specific screens for developmental anomalies may also reveal a more accurate number of essential genes.
Large Mutagenesis Screens: Collaborative Efforts
A mutation rate of 1:750/locus/gamete for ENU-induced mutations has been established based on the detection of visible (pigmentation) mutations in noncomplementation tests (specific locus test; SLT) (Russell et al. 1979). Future experiments, by us and other groups, will show whether other classes of mutations, such as embryonic lethals or behavioral anomalies, will be obtained at the same mutation rate. In terms of region-specific mutagenesis, this means that by a phenotypic characterization of 750 G2 progeny (Fig. 2), one has a 50% chance of detecting a recessive mutation in every gene “uncovered” by the deletion. Whether or not the mutation will be detected depends on the mutability of the individual genes. Factors that influence mutability are not known even in organisms amenable to saturation screens. These factors may, however, include locus/gene size or number of nucleotide changes within the gene that will disrupt or alter the protein structure and cause a mutant phenotype. Finally, a novel mutation will be detected only if phenotypic characterization includes scoring of the particular phenotype affected by the mutated gene. Therefore, to effectively use the considerable investment in resources, a region-specific screen should seek to identify diverse physiological, behavioral and developmental abnormalities, including subtle phenotypes. We believe it may be incumbent on researchers who conduct region-specific screens to make efforts to identify all mutants in that region, so that the effort is as near to saturation as possible. This would preclude the need for subsequent researchers to repeat the effort. In practice, this would necessitate a collaboration of researchers from many disciplines. To this end, efficiently integrated assays that can uncover anomalies in several distinct phenotypic traits have been developed for large-scale mutagenesis screens [Justice et al. 1997; web information and personal communications by Rudi Balling (http://www.gsf.de/isg/ENU.index.html) and Steve Brown (http://www.mgu.har.mrc.ac.uk/mutabase/)]. Of course, comprehensive screens must be performed so that initial tests do not compromise subsequent tests, that is, they must be noninvasive. For example, blood from the offspring of mutagenized animals can be subjected to a wide range of biochemical (clinical-like) tests before more invasive phenotypic characterization. Tests that require the presence of the mouse, however, such as those for behavior or adult-onset diseases, dictate the necessity of a central facility that is either staffed by personnel capable of carrying out a series of tests, or that the mutagenesis screening is conducted in such a fashion that experts can visit periodically to evaluate groups of animals. An important consideration in a large scale screen is that mutants uncovered should be preserved in some fashion, ideally by sperm freezing.
Limitations and Prospects
New and Improved Protocols for ENU Treatment
At present, mutagenesis of male germ cells with ENU is the most efficient method available for mutagenizing the mouse genome. There are certain technical drawbacks, however, that warrant efforts to improve ENU mutagenesis or to develop alternate technologies. First, the process of generating mutagenized males that contain re-populated germ lines with viable, highly mutagenized spermatogonia is rather lengthy and variable. After the last injection of ENU, males will not become fertile for about 4 months. During that time, a significant proportion of these males may die, and others may not become fully fertile. The results of any mutagenesis experiment may vary from lab to lab and experiment to experiment because of factors related to the batch of ENU, state of the mice, their genetic background, or other still unknown factors. This issue is difficult to overcome, as the regeneration of the seminiferous epithelium occurs at a fixed rate. We urge caution in the administration and quantitation of the ENU stocks, and suggest that investigators conduct potency trials to assess sterility/fertility over a range of doses for a given strain background (Nolan et al. 1997).
Second, for any given mutagenized male or set of males, it is not easy to determine the efficiency of the ENU treatment. Current estimates are based on mutation rates in the specific locus test. Clearly, in a small-scale screen, it is not possible to generate sufficiently high numbers of mice to monitor mutation rate by means such as noncomplementation tests with visible marker genes. In a large-scale screen, however, this SLT experiment is warranted, perhaps combined with the molecular testing of ENU-induced nucleotide changes by methods such as SSCP or sequencing, or transgenic reporters. This control experiment may at the same time serve as a “prescreen” for mutations in a battery of genes of particular interest, before the phenotypic characterization of progeny. The assessment of the mutagenic effects of the treatment is a critical issue in a mutagenesis screen in that a minor decrease in efficiency could translate into tens of thousands of additional mice required to produce a particular number of mutations in a given screen.
Third, the last step in the molecular identification of the gene disrupted in an ENU-induced mutation may be difficult, because of the fact that this alkylating agent induces point mutations. Small intragenic deletions that have a higher chance to induce a loss-of-function phenotype would be simpler to detect in molecular assays. Of the chemicals known to induce such lesions (such as EMS and chlorambucil) in the SLT, however, none have demonstrated the efficiency of ENU with respect to mutagenizing germ cells (Ashburner 1989; Mullins et al. 1994). It is conceivable, however, that mutagenesis might be done outside the mouse, in ES cells for example, whereby mutation rates could be measured in advance to creating mice, and a wider range of chemical mutagens or transposable elements can be tested in a shorter period of time.
Deletion Resources
The major limiting factor in region-specific mutagenesis is the availability of deletions. Both the Cre–loxP and radiation approaches require initial homologous recombination events into a specific locus or loci (Ramirez-Solis et al. 1995; You et al. 1997a). This remains a nontrivial exercise that requires the construction of a targeting vector and selection of transfected clones for desired recombination events. In the absence of mice containing deletions spanning the genome, the next best alternative would be a bank of ES cells that either contain deletions or vectors integrated throughout the genome that can be used for subsequent generation of deletions. Toward this end, the Merck Genome Research Institute (West Point, PA) is funding the creation of a bank of F1 hybrid ES cell lines that contain TK–neo insertions mapped throughout the genome at ∼5-cM intervals. These lines can be used to induce deletions by radiation as described (You et al. 1997a). A description of this project and a list of available lines is available athttp://lena.jax.org/∼jcs/Delbank.html.
Improved Physical Maps of the Target Regions
Ideally, a region-specific mutagenesis screen should be implemented in parallel with the fine-mapping and molecular characterization of the target region (Brown and Peters 1996). As described earlier, the first step in region-specific mutagenesis is the generation of a series of ES cells with nested deletions around the targeted locus. To select ES cell lines with the appropriate deletions for generation of transgenic mice to be used in the screening and subsequent fine mapping of new mutations, the extent of the deletions must be determined. Although deletion breakpoints in F1hybrid ES cells can be characterized through the use of polymorphic simple sequence repeats (SSR) before the creation of mice (You et al. 1997a; Thomas et al. 1998), there is substantial room for improvement in the resolution of breakpoint mapping. First, the current collection of 6000 SSRs may not be sufficient (Dietrich et al. 1992, 1994) for high-resolution mapping. Whereas this collection translates into an average of one marker every 500 kb, these markers are not equally distributed throughout the genome. Finally, not all of the SSRs are polymorphic between the two parental ES-cell strains. The issue of polymorphism between the strains of F1 hybrid ES cells should be lessened through the use of cells in which one parent was derived from the evolutionary distant strain Mus castaneus (Thomas et al. 1998).
There are several solutions to this problem, short of sequencing the entire genome. One is to create a new generation of markers. There is currently much discussion of single nucleotide polymorphisms (SNPs), which we agree would be a welcome new reagent. It is important to note, however, that the utility of any new form of marker, no matter how plentiful, will be limited by the resolution to which they are genetically or physically mapped. Ideally, a contig of bacterial artificial chromosomes (BACs) across the genome, annotated by their SSR and SNP content, would provide a satisfactory resource that would lay out the relative locations of all these markers and enable the mapping of deletion breakpoints to within ∼100 kb. In the absence of a genome-wide resource, however, it is prudent to selectively establish BAC contigs in parallel with particular region-specific mutagenesis efforts. Furthermore, as the extent of the mouse genome sequencing effort is still uncertain pending completion of the sequencing of the human genome (∼2005), it is desirable to direct any limited sequencing efforts to those particular chromosomal regions that are targets for extensive mutagenesis screens.
Placement of ESTs on the Physical Map
The mapping of all expressed genes in the mouse genome would be of wide benefit to genetics and medical research, as well as for new rounds of mutagenesis projects. Several projects to develop a large collection of ESTs for the mouse have been initiated (Washington University, St. Louis, MO) and efforts are underway to develop high-resolution maps by analysis of radiation hybrids (RH) (McCarthy et al. 1997; Flaherty and Herron 1998; L. Rowe, pers. comm.). With respect to mutagenesis, this effort would bring two benefits. First, the act of mapping ESTs in an RH panel, which involves identification of polymorphisms within the 3′UTR, would provide a new set of markers useful for mapping deletion breakpoints. More importantly, for a new mutation localized to a deletion interval, a list of candidate genes could be deduced readily, particularly if expression profiles were available on all the ESTs. “The joy of screening” will come not only from finding new interesting phenotypes, but from the knowledge that the genes responsible for these phenotypes have been isolated already.
Acknowledgments
We thank members of our laboratories and many colleagues for insightful conversations and correspondance about mutagenesis in general as well as a collaborative project; for access to unpublished studies; and for critical comments on the manuscript. M.B. thanks Scott Poethig for pointing out the usefulness of Rw as a balancer long before described screens where initiated. These studies were supported by grants from the National Institutes of Health HD 28410 (M.B); MH 57855 (M.B.); HD 24374 and HD35984 (J.S.).
Footnotes
-
↵3 Corresponding author.
-
E-MAIL bucan{at}pobox.upenn.edu; FAX (215) 573-2041.
- Cold Spring Harbor Laboratory Press








