
Emerging Patterns of Comparative Genome Organization in Some Mammalian Species as Revealed by Zoo-FISH
Abstract
Although gene maps for a variety of evolutionarily diverged mammalian species have expanded rapidly during the past few years, until recently it has been difficult to precisely define chromosomal segments that are homologous between species. A solution to this problem has come from the development of Zoo-FISH, also known as cross-species chromosome painting. The use of Zoo-FISH to identify regions of chromosomal homology has allowed the transfer of information from map-rich species such as human and mouse to a wide variety of other species. From a Zoo-FISH analysis spanning four mammalian orders (Primates, Artiodactyla, Carnivora, and Perissodactyla), and involving eight species (human, pig, cattle, Indian muntjac, cat, American mink, harbor seal, and horse), three distinct classes of synteny conservation have been designated: (1) conservation of whole chromosome synteny, (2) conservation of large chromosomal blocks, and (3) conservation of neighboring segment combinations. This analysis has also made it possible to identify a set of chromosome segments (based on human chromosome equivalents) that probably made up the karyotype of the common ancestor of the four orders. This approach provides a basis for developing a picture of the ancestral mammalian karyotype, but a full understanding will depend on studies encompassing more diverse combinations of mammalian orders.
Gene mapping is currently being carried out in >200 mammalian species. Except for human, mouse, and rat, the only other gene maps that have exhibited exponential growth are those of the domestic animals. The economic implications of the gene maps in the latter species to the farm animal industry have been the driving force behind this progress (e.g., see Georges and Andersson 1996). One of the significant outcomes of the development of gene maps in different species is that they have enabled us to detect those segments of genomes which, beginning from a common ancestor, have remained conserved over several million years of evolution (Wienberg and Stanyon 1995; Comparative Genome Organization 1996; O’Brien et al. 1997b;Wienberg et al. 1997). This branch of genome analysis is referred to as comparative genome analysis. Comparisons between genomes could either be based on conventional and molecular cytogenetic approaches or on the available gene mapping data. Over the past decade, comparative genome analysis has emerged not merely as an off shoot from the main stream gene mapping efforts carried out in various mammalian species, but has evolved as a separate field of research, having practical implications on different aspects of biological/genetical research (O’Brien et al. 1993, 1997a; Comparative Genome Organization 1996; Eppig 1996; Lyons et al. 1997).
Comparative gene maps are generally discussed keeping the human and mouse gene maps at the center. A straightforward explanation for this is that the gene maps of these two species are (and will continue to be) by far the most developed or map rich among mammals (O’Brien et al. 1993; Womack and Kata 1995). Conversely, other mammalian species have poorly developed maps primarily the result of a fewer number of laboratories involved and/or limited funding available. Expansion of gene maps in the latter species could be enhanced if the vast information available from the human and mouse gene maps could be efficiently utilized or transferred. The transfer of mapping information from map rich species to map poor species will be viable only if sufficient and accurate data is available on the comparative organization of any of the two genomes. The significance of detailed comparative information at different levels of resolution (ranging from gross chromosomal to map location of different genes) has been emphasized in the recent past for searching candidate loci governing traits of biological and economic importance, both in humans and farm animals (e.g., Georges et al. 1993; Montgomery et al. 1993; Andersson et al. 1994; Cockett et al. 1994; Charlier et al. 1995; Georges and Andersson 1996; Johansson-Moller et al. 1996; Grobet et al. 1997;Kambadur et al. 1997).
During the past 2–3 years, one of the techniques that has contributed significantly to the identification of conserved chromosomal blocks or syntenic segments between different species is Zoo-FISH or cross species chromosome painting (Scherthan et al. 1994). The application of this technique not only helped in developing a better understanding of comparative organization of the genomes of divergent mammalian species, but has also contributed to identifying potentially ancestral genomic segments. Therefore, in the present review we will focus only on the progress achieved through the generation of comparative chromosome maps using chromosome-specific painting probes among distantly related mammals.
Initial success with the cross-species chromosome painting technique was first reported among the genomes of evolutionarily closely related hominoids (see Wienberg et al. 1990; Jauch et al. 1992). The delineation of homologies between chromosomal complements of various primates by use of whole chromosome-specific paints was facilitated by the high degree of molecular homology among primate genomes (Wienberg et al. 1994; Koehler et al. 1995a,b; Consigliere et al. 1996; Wienberg and Stanyon 1997). Presently, data of comparative chromosome painting with all human chromosome-specific libraries (CSLs) are available for macaque (Wienberg et al. 1992), siamang (Koehler et al. 1995a), gibbon (Jauch et al. 1992; Koehler et al. 1995b), two subspecies of red howler monkey (Consigliere et al. 1996), Capuchin monkey (Richard et al. 1996), marmoset (Sherlock et al. 1996), silvered leaf monkey (Bigoni et al. 1997), and black-handed spider monkey (Morescalchi et al. 1997). Methodological improvements allowed comparative chromosome painting to extend to distantly related species (Zoo-FISH; Scherthan et al. 1994). Subsequent Zoo-FISH studies predominantly used human chromosome-specific plasmid or PCR-generated library DNA probes, because these were the first available (e.g., Collins et al. 1991;Vooijs et al. 1993), and obtaining highly concentrated DNA probes was straightforward. Because the human genome is by far the most densely mapped among mammalian species, its map serves as that of a master mammalian genome (O’Brien et al. 1993). To date, comparative chromosomal maps of eight mammalian species of three different orders (excluding Primates) have been generated by Zoo-FISH. (1) Artiodactyla: pig (Rettenberger et al. 1995a; Frönicke et al. 1996; Goureau et al. 1996; Milan et al. 1996), cattle (Hayes 1995; Solinas-Toldo et al. 1995; Chowdhary et al. 1996), Indian muntjac deer (Scherthan et al. 1994, 1995; Frönicke and Scherthan 1997; Wienberg and Stanyon 1997; Yang et al. 1997). (2) Carnivora: cat (Rettenerger et al. 1995b;Wienberg et al. 1997), American mink (Hameister et al. 1997), and harbor seal (Frönicke et al. 1997). (3) Perissodactyla: horse (Raudsepp et al. 1996, 1997; Rettenberger et al. 1996; Lear and Bailey 1997). Furthermore, fragmentary Zoo-FISH data are available for mouse, fin whale (Scherthan et al. 1994), and sheep (Chowdhary et al. 1996). According to molecular and fossil dating methods, the maximum phylogenetic distance between human and a particular species hitherto investigated by Zoo-FISH is ∼65–100 million years (Arnason et al. 1996).
In this overview, we attempt to provide a comparative analysis of the available Zoo-FISH data in the evolutionary distantly related nonprimate species mentioned above. Where possible, the Zoo-FISH results are discussed with respect to the comparative gene mapping data. The comparisons of the available Zoo-FISH maps allow a preliminary insight into potential rearrangements that led from a putative protoprimate karyotype to that of the extant species analyzed. Therefore, the following discussion refers to human chromosomes (Homo sapiens, HSA) as the reference point, solely because most of the species studied by Zoo-FISH were probed with human CSLs. It should be noted, however, that the human genome should not be confused as representing the ancestral mammalian karyotype (see Fig. 2, below).
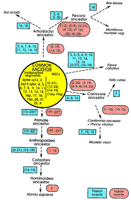
Schematic representation of the proposed karyotypic fission/fusion events in the evolution from the common ancestor to the seven species investigated and the primates (see text for references), which most likely resulted in the formation of the respective karyotypes. The events represent only those that could be identified (or can be proposed) based on the comparative painting data and therefore do not represent other possible rearrangements. Because in each case, correspondence with relation to individual human chromosomes was studied, all of the numbers presented relate to human chromosomes. In the common ancestor (yellow circle), two categories of chromosomes were included. Undisrupted segments represent those human chromosomes or chromosomal arms, the homologs of which were found to be entirely conserved in at least one of the seven species. HSA1p and 1q may also be added to this group; NSCs represented human chromosomes that were found to be contiguously syntenic in at least five species. Beginning from the common ancestor, the probable fission (blue rectangles) and fusion events (red ellipses) leading to the karyotypes of the respective species can be followed along the arrows. Commencing from the proposed ancestral configuration, the comparative painting data indicate very few major rearrangements (2–3 fusion and 3–5 fission events) leading to the formation of the cat, harbor seal, American mink, and human karyotypes; on the other hand, several obvious rearrangements (13–19 fission and 2–5 fusion events) leading to the pig, cattle, Indian muntjac, and horse karyotypes can be observed.
Overall Conservation of Karyotypic Organization
The accumulating gene mapping data across evolutionarily diverged species has progressively indicated that large genomic segments are conserved among mammals (e.g., see Comparative Genome Organization 1996). Zoo-FISH experiments mentioned above have helped to further demarcate physical boundaries of these conserved syntenies and to delineate surprisingly large conserved genomic segments among the species studied. Furthermore, the fractions of individual genomes painted by a particular human chromosome-specific painting probe are similar in size (Chowdhary et al. 1996; Frönicke et al. 1996,1997). Where detected, size differences in the homologous chromosomal segments predominantly relate to the presence of intervening heterochromatin or nucleolar organizing region (NOR) material (Scherthan et al. 1994; Chowdhary et al. 1996; Frönicke et al. 1996, 1997).
Comparison of segment numbers detected in the different genomes by human CSLs range from 31 in cat and harbor seal (Rettenberger et al. 1995b; Frönicke et al. 1997) to 53 in cattle (Hayes 1995). Variations in segment numbers detected for a particular species by different investigators (e.g., cattle; Hayes 1995; Solinas-Toldo et al. 1995; Chowdhary et al. 1996) exclusively concern small segments and most likely reflect variation in complexity and origin of DNA probes applied by different laboratories (e.g., flow sorted or microdissected; see Frönicke et al. 1997; Yang et al. 1997; Chaudhary et al. 1998).
In cattle, in which the highest number of homologous segments was detected (Hayes 1995), most human chromosomes have been conserved as two or three segments. In general, this rule is also apparent in the patchwork genome of the Indian muntjac (2n = 6/7;Frönicke and Scherthan 1997; Yang et al. 1997). A summary of the comparative information obtained so far is presented in Figure1; here, comparative genome organization has been illustrated with respect to the human genome according to whether the conservation of synteny concerns, (1) whole chromosomes, (2) chromosomal arms or large blocks, or (3) neighboring segment combinations (NSCs). Mouse, which has primarily been included as an out group, provides the relative status in a species proposed to have undergone an unusually high number of genomic arrangements per unit of evolutionary time (Graves 1996), as compared with any of the mammalian species included. The comparisons are restricted only to genes mapped in at least one of the nonhuman species analyzed, and provide an insight in comparative genome organization with respect to the mouse (see Fig. 1).
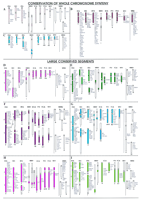
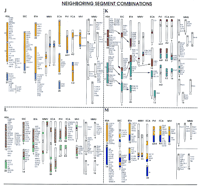
Summary of comparative organization of the human (HSA), pig (SSC), cattle (BTA), Indian muntjac (MMV), horse (ECA), harbor seal (PVI), cat (FCA), American mink (MVI), and mouse (MMU) genomes. Each human chromosome presented herein is denoted with a specific color, which is used for those chromosomes/chromosomal regions in the other seven species; these have been identified as homologous by use of human CSLs as paints. Mouse, the second most densely mapped species, is included solely for comparative purposes. Each chromosome bears a species-specific number below the respective ideogram. A list of hitherto mapped genes (where available) is presented beside each ideogram. The gene mapping data were retrieved from genome databases of the respective species [human: GDB (http://gdbwww.gdb.org); pig: PigBase (http://www.ri.bbsrc.ac.uk/pigmap/pigbase/pigbase.html); cattle: BovMap (http://138.102.88.140/cgi-bin/bovmap/intro.pl); Gao and Womack 1997; Yang et al. 1998; horse: Raudsepp et al. 1996; S. Godard, L. Schibler, A. Oustry, E. Cribiu, and G. Guerin, unpubl.; T. Lear, M.H. Adams, K.J. McDowell, N.D. Sullivan, L. Coogle, E. Ferguson Jr., T.M. Chambers, and E. Bailey, unpubl.; A.T. Bowling and L. Millon, pers. comm.; muntjac: Levy et al. 1992; cat: O’Brien et al. 1997; mouse: MGD (http://www.informatics.jax.org)]. In human and mouse, only those genes mapped in the other seven species have been included. Arrows (e.g., in figures F,G,K) indicate that the detected homology can be further traced for specific arms or regions, with the help of the available gene mapping data. The comparisons are classified into three major categories. (A–C, Conservation of whole chromosome synteny) Chromosomes that tend to be conserved as a single chromosome/block in most of the species studied. These chromosomes correspond to HSA13 (A), HSA17 (B), and HSA20 (C). (D–I, Large conserved segments) Chromosomes that show conservation either as a single chromosome (or arm/segment of a chromosome) or as two major blocks on different chromosomes. These chromosomes correspond to HSA9 (D), HSA11 (E), HSA2 (F), HSA6 (G), HSA4 (H), and HSA5 (I). (J–M, Neighboring segment combinations) Chromosomes that show conserved contiguous synteny in the majority of the species studied but correspond to two chromosomes in the human. Chromosomes corresponding to HSA3 and HSA21 (J), HSA16 and HSA19 (K), HSA12 and HSA22 (L), and HSA14 and HSA15 (M) are included in this category (see text for details). A line in the mouse section separates loci common to individual human chromosomes involved in the combinations.
Conservation of Whole Chromosome Synteny
This type of synteny conservation involves chromosomes that generally show no interchromosomal rearrangements among the species studied through comparative painting. HSA13, HSA17, HSA20, HSAX, and the corresponding homologous segments in the investigated species fall into this category. In the majority of the genomes hitherto studied, these chromosomes are either represented as a single chromosome or as a whole chromosome arm. It should be noted that because of the low chromosome number of the Indian muntjac (2n = 6/7), the regions corresponding to these four human chromosomes are present in a different condition as compared with other species (Fig. 1A–C). The human X chromosome corresponds to an entire chromosome arm, whereas HSA13, HSA20, and HSA17 label a single intrachromosomal segment each, with the exception that the segment painted by HSA20 is disrupted by a small segment from HSA10 (Fig. 1C).
Comparative painting data show complete conservation of the X chromosome in all the species studied hitherto. The mammalian X chromosome is considered to have retained the original genetic constitution of a common ancestor of the eutherian mammals (Graves and Watson 1991). This is evident from the almost identical banding patterns, morphology (with the exception of ruminantia), and gene content (Ohno et al. 1964; O’Brien et al. 1993; Gallagher et al. 1994;Morizot 1994; Hayes 1995; Rettenberger et al. 1995a; Chowdhary et al. 1996). Notwithstanding the high degree of conservation, the rearranged order of genes on the X chromosome suggests intrachromosomal rearrangements (Nadeau 1989; Farr and Goodfellow 1992). Recent studies showing over eight conserved X-chromosomal regions being rearranged between human and mouse (Blair et al. 1994; Carver and Stubbs 1997), and an additional nine just within the distal 12 Mb of HSAXp22.3 (Blaschke and Rappold 1997), indicate a much more complex sequence of events leading to the divergent organization of the X chromosomes of these two species.
HSA13
HSA13 paints a single chromosome in the artiodactyls and horse and an entire chromosome arm in the carnivore complements (Fig. 1A). An extraordinary degree of conservation is still reflected in nearly identical banding patterns detected for HSA13 and its homologs in horse (Rönne 1992), cattle (for review, see Hayes 1995), cat (Rettenberger et al. 1995b), and harbor seal (L. Frönicke and H. Scherthan, unpubl.). A divergent condition is apparent in the mouse in which gene mapping data (see MGD; http://www.informatics.jax.org) indicate dispersal of HSA13 loci on four different chromosomes.
HSA17
HSA17 corresponds to a single chromosome in pig, cattle, horse, and cat karyotypes, whereas in mink and harbor seal, a single chromosome arm is painted (Fig. 1B). In Indian muntjac HSA17 corresponds to one segment, and in Chinese muntjac it corresponds to an entire chromosome (L. Frönicke and H. Scherthan, unpubl.). The conservation of a single HSA17 homologous segment is also observed in sheep (Chowdhary et al. 1996), chimpanzee (Wienberg et al. 1990), and macaque (Wienberg et al. 1992). The comparative gene map in mouse indicates a similar situation, with 115 of the 118 HSA17 loci mapped to MMU11 (note: MMU11 also shares homology with several other human chromosomes). In fin-whale, however, this human chromosome corresponds to two conserved segments (Scherthan et al. 1994). Several inter- and intrachromosomal rearrangements have also been reported for HSA17 homologs in some primate species (Matera and Marks 1993; Koehler et al. 1995a). Furthermore, gene mapping data in cattle suggest that in addition to the Zoo-FISH-based conserved synteny between HSA17 and BTA19, minor HSA17 homologous segments are also present on BTA7 and BTA22 (BovMap). The observations demonstrate that the resolution limit of Zoo-FISH, which is around 7 Mb (Scherthan et al. 1994), will leave small conserved syntenic segments undetected.
HSA20
HSA20 paints a whole chromosome each in pig and horse and a whole chromosome arm each in carnivores (Fig. 1C). Interestingly, even in the mouse genome, this human chromosome has only one homologous counterpart, MMU2. The latter, however, also corresponds to segments on other human chromosomes, for example, HSA9 and HSA15. In cattle and Indian muntjac, two HSA20 homologous segments are present on the same chromosome, and are separated by an HSA10 homologous segment. A fission event within the HSA20 homologous segment, followed by an inversion, most likely inserted the HSA10 homologous portion. It is expected that this occurred prior to divergence of the cervids and bovids.
Large Conserved Segments
In the following discussion we will revisit some of the human chromosomes that are found to be conserved as large segments among the different genomes analyzed.
HSA9
HSA9 most likely corresponds to an ancestral mammalian chromosome because it has been conserved as a single chromosome in human and carnivores and as a single block in pig and cattle (Fig. 1D). In Indian muntjac and horse, this chromosome corresponds to two segments on two chromosomes that most likely have been generated by bifurcation of HSA9 homologous material after the divergence from the common ancestor (Raudsepp et al. 1996; Yang et al. 1997). In the mouse complement, HSA9 genes are distributed over parts of three chromosomes. Of these, the segmental homology with MMU2 is, as yet, restricted only to HSA9q.
HSA2
HSA2 represents a derived condition in the human, because it originated from the fusion of two ancestral primate chromosomes (Dutrillaux 1979), the fusion point being localized in chromosomal band 2q13 (Avarello et al. 1992; Ijdo et al. 1992; Lengauer et al. 1993;Wienberg et al. 1994; Azzalin et al. 1997). Except for horse, which is separately discussed below, all the distantly related mammals analyzed by Zoo-FISH to date showed two distinct segments (represented on separate chromosomes) corresponding to HSA2 (Fig. 1F). On the other hand, comparative gene mapping data in mouse demonstrate that the homology with this human chromosome is shared with parts of six different murine chromosomes.
In most of the species analyzed, HSA2 shows either whole chromosomes or complete chromosomal arm homology. In pig, the synteny on SSC15 is interrupted by an intercalary segment homologous to HSA8 (Fig. 1F). Similarly, three HSA2 homologous segments are present in the Indian muntjac genome. Therefore, it would be of interest to find out whether each of the two segments observed in different species reflect conservation of genetic material corresponding to the respective regions on HSA2, 2q13-pter and 2q13-qter. Reverse and forward chromosome painting between pig and human (Goureau et al. 1996) is not conclusive in this respect. A similar painting strategy in the cat/human species combination suggests that the above-mentioned two segments of HSA2 represent an ancestral mammalian condition (O’Brien et al. 1997a; Wienberg et al. 1997). This interpretation can also be reconciled with the scanty gene-mapping information available in cattle, pig, and cat, which shows that the loci mapped to SSC3p, BTA11, and FCA3q are the same as those located on HSA2q13–pter. IL1A and IL1B loci map in close proximity to the proposed fusion site on HSA2. These loci are also consistently found on the proposed homologous arms of the three species (see Fig. 1F). Furthermore, the majority of the homologous loci mapped to SSC15 and BTA2 correspond to those mapped between HSA2q13-qter (BovMap, PigBase). In spite of scattered homology in mouse, parts of three murine chromosomes share homology only with the short arm, whereas two chromosomes share homology with the long arm of HSA2. One chromosome (MMU6), however, harbors homologous genes from both arms of this human chromosome (see MGD; http://www.bioinformatics.jax.org).
In horse, the HSA2 CSL detected complete correspondence with two acrocentric chromosomes (ECA15 and ECA18) and an additional small segment on ECA1 (Fig. 1F; Raudsepp et al. 1996). However, recent experiments with arm-specific paints from this human chromosome revealed homology with only two segments/chromosomes, each of which was either homologous to HSA2q13-qter or HSA2pter-q13 (Chaudhary et al. 1998). In light of these observations, the additional segment detected on ECA1 by Raudsepp et al. (1996) requires further verification by mapping HSA2 genes in horse.
HSA4
HSA4 paints two large homologous blocks in cattle, Indian muntjac, and horse, and maintains whole chromosome synteny in pig, harbor seal, mink, and cat (Fig. 1H). However, comparative gene mapping between pig and human demonstrates intrachromosomal rearrangements since the divergence of the two species (Johansson et al. 1995). Limited mapping data in cat also indicate intrachromosomal rearrangements with respect to the human genome (O’Brien et al. 1993, 1997a,b). Furthermore, the mouse gene map shows that three chromosomes share synteny conservation with HSA4. Of these, MMU5 contains the maximum number of HSA4 homologous loci, whereas MMU3 and MMU8 share almost equal numbers of loci, all from HSA4q (see MGD; http://www.bioinformatics.jax.org).
HSA6
HSA6 has either been conserved as a single block (e.g., in cat, harbor seal, and American mink) or as two distinct blocks in pigs, horse, cattle, and Indian muntjac (Fig. 1G). Gene mapping data in these species (except for Indian muntjac, in which no data exist) suggest that each of the two homologous blocks corresponds to individual arms of HSA6. Recent Zoo-FISH experiments with HSA6 arm-specific painting probes on pig and horse chromosomes verify this observation (Chaudhary et al. 1998). Thus, there are two possibilities with regard to the evolution/ancestral condition of HSA6. One is that the condition in human, cat, harbor seal, and American mink is of an ancestral type and the synteny breakage in pig, cattle, horse, and Indian muntjac has occurred more recently. However, there are no facts against proposing the reverse possibility, wherein the two syntenic groups were on separate chromosomes in the mammalian ancestral complement that later fused in human and carnivores. Nevertheless, with the carnivore and human karyotypes considered as more closely related to the putative ancestral mammalian complement (Rettenberger et al. 1995b;Frönicke et al. 1997; Wienberg et al. 1997), the possibility of the former process appears more likely. A different scenario with respect to the synteny conservation is again apparent in the mouse in which a high degree of rearrangement with respect to HSA6 material is evident. According to comparative mapping data hitherto available, parts of six murine chromosomes share homology with this human chromosome (see Fig. 1G). Except for one, the five other chromosomes carry homologs mapping to both the short and long arm of HSA6. This could be attributed to the rearrangements introduced during rapid karyotypic evolution of the mouse.
HSA16 and HSA19
The high degree of synteny conservation observed for HSA6 in various species is also noticed for HSA16 and HSA19. In all the species discussed herein (except mouse), each of these human chromosomes corresponds to two distinct segments (Fig. 1K). Available gene mapping and reverse painting data in some of the species combinations, that is, pig–human (Goureau et al. 1996; Milan et al. 1996) and cat–human (only HSA16; Wienberg et al. 1997), as well as comparative arm-specific painting results with HSA16 and HSA19 probes on pig and horse chromosomes (Chaudhary et al. 1998), strongly suggest that each of the conserved segments represent individual arms of these human chromosomes. Recent Zoo-FISH data with HSA16 arm-specific paints in cat and donkey, and HSA19 arm paints in cat (T. Raudsepp and B.P. Chowdhary, unpubl.; T. Raudsepp, R. Chaudhary, X.Y. Guan, H. Zhang, and B.P. Chowdhary, unpubl.), further corroborate these conclusions. Gene mapping data in mouse are partly in agreement with these results. All HSA16q genes hitherto mapped in mouse are located on MMU8. Similarly, of the homologous HSA19q loci mapped in mouse, 95% are located on MMU7. In a species that shows a high degree of rearrangement as compared with human as well as other mammalian species analyzed (Graves 1996), this level of synteny conservation might be significant for drawing conclusions regarding possible karyotypic constitution of the common mammalian ancestor.
It might be of particular interest to discuss HSA21 as a chromosome that displays a general tendency of whole chromosome interspecies synteny conservation (Fig. 1J; except in Indian muntjac). In each case, a single chromosomal segment corresponding to this small human chromosome is present. It is remarkable that an HSA21 homologous segment is present as a component of another chromosome in all nonprimate species examined (except horse; see Fig. 1J; discussed further below). Furthermore, it is also interesting to note that the chromosomal segment painted by HSA21 tends to keep either a centromeric or a telomeric position within the chromosome arms of all of the species reviewed here. Additional studies are needed to assess whether these conditions have any evolutionary or functional significance. In mouse, HSA21 homologs are mapped to three different chromosomes, MMU10, MMU16, and MMU17. Lack of precise physical mapping information for the murine loci precludes inference concerning conservation of their intrachromosomal rearrangement.
Neighboring Segment Combinations
Regions homologous to certain human chromosomes or chromosomal segments tend to be contiguous/syntenic in most of the species hitherto studied. The tendency of these segments to be neighboring in different genomes (see Fig. 1J–M) indicates that these combinations represent ancestral chromosomal conditions. However, because of recent fission events, the likely ancestral combinations probably separated during human karyotype evolution (see below). The possibilities that these combinations could have arisen by the convergent (or de novo) fusion of independent ancestral genomic fragments more recently during evolution is less likely, because neighborhood of particular homologous segments is consistently observed in a number of fairly diverged species.
HSA3 and HSA21
Among the different species studied by Zoo-FISH, all (except horse) show synteny between regions corresponding to these two chromosomes (Fig. 1J). In some primates, for example, lemur, capuchin monkey, and macaque, the two human chromosomes also show synteny conservation (Wienberg et al. 1992; Apiou et al. 1996; Richard et al. 1996; Morescalchi et al. 1997). Even in mouse, synteny between HSA3 and HSA21 is evident on two chromosomes, MMU16 and MMU17 (see MGD;http://www.bioinformatics.jax.org), although each of the human chromosomes also shares homology with other mouse chromosomes. All of these comparative data strongly suggest that HSA21–HSA3 synteny observed in a wide range of diverged species, including the mouse, is ancestral, and its disruption in human and horse can be attributed to independent fission events (Fig. 2).
HSA14 and HSA15
CSLs from these two human chromosomes have demonstrated a variable degree of synteny conservation for the segments painted in different species (Fig. 1M). In pig, the two human chromosomes correspond to SSC1 and SSC7, with the segments not being contiguous in the former (see SSC1 in Fig. 1M). In cattle, both BTA10 and BTA21 harbor contiguous segments corresponding to HSA14 and HSA15. A similar situation is found in the Indian muntjac genome. These observations strongly suggest that the two chromosomes were syntenic in the mammalian ancestor. The noncontiguous synteny on SSC1 in pigs could be the result of an inversion event that probably occurred later in the pig lineage. The equine genome once again demonstrated slight deviation from the general evolutionary trend (as in HSA3–HSA21 synteny breakage) in the sense that part of the HSA14 homologous segment was also detected as a separate chromosome.
Synteny of segments corresponding to HSA14 and HSA15 has also been observed in various primate species like marmoset (Sherlock et al. 1996), macaque (Wienberg et al. 1992), capuchin monkey (Richard et al. 1996), black-handed spider monkey (Morescalchi et al. 1997), and two of the three lemur species hitherto studied (Apiou et al. 1996). These findings provide further support to the hypothesis that the synteny represents an ancestral condition. Furthermore, it can be interpreted that the disruption of synteny in humans, like in siamang and gibbon (Koehler et al. 1995a,b), represents a more recent event. The only indication that HSA14–HSA15 synteny is maintained even in mouse is on MMU7, which harbors one HSA14 locus and twenty-three HSA15 loci (see MGD; http://www.informatics.jax.org). Further expansion of the mouse gene map will provide a more detailed overview of the situation because the mapping data hitherto available shows that the majority of HSA14 loci are located on MMU12 and MMU14, whereas HSA15 loci locate predominantly on MMU2, MMU7, and MMU9 (see Fig. 1M).
HSA12 and HSA22
Segments homologous to these two human chromosomes show synteny in all of the nonprimate species hitherto studied (Fig. 1L). However, there is considerable variation in the arrangement of the syntenic segments in different genomes. Combinations of HSA12–HSA22 can occur in tandem (majority of species) as well as split synteny (e.g., MMV1 and ECA1). Even in mouse, in which only gene mapping data is available, chromosomes 5, 10, 11, and 15 each show conservation of synteny with regard to genes from the two human chromosomes. An overview of the status in various genomes unequivocally suggests that synteny of the segments homologous to HSA12 and HSA22 represent an ancestral condition.
HSA16 and HSA19
Cross-species painting data on all the seven nonprimate species examined show that, except for horse (in which fission probably separated the two segments), the conserved synteny combinations corresponding to the two human chromosomes are maintained (Fig. 1K). In pig, cat, and harbor seal a centromere is present at the facing margin of the two segments. The compiled data suggest that the HSA16–HSA19 synteny combination is also an ancient condition (Rettenberger et al. 1995b; Frönicke et al. 1997). With the help of the available gene mapping data, it is possible to trace the regions corresponding to HSA19q and HSA16q in pig, cattle, and horse (see arrows; Fig. 1K). This analysis indicates that the synteny most likely is restricted to the two human chromosomal arms, which has partly been confirmed by reverse Zoo-FISH in pig (Goureau et al. 1996; Milan et al. 1996) and cat (Wienberg et al. 1997). Recently, Zoo-FISH with HSA16 and HSA19 arm-specific probes to pig chromosomes has further supported this interpretation (Chaudhary et al. 1998). Furthermore, our very recent arm-painting results in cat clearly show that segments corresponding to the long arms of these two human chromosomes are syntenic on FCA E2 (T. Raudsepp and B.P. Chowdhary, unpubl.; T. Raudsepp, R. Chaudhary, X.Y. Guan, H. Zhang, and B.P. Chowdhary, unpubl.). All of these findings strongly suggest that synteny conservation of segments homologous to HSA16q–HSA19q is an evolutionary relic, ancient in nature. In the mouse genome, segments corresponding to HSA16 and HSA19 are syntenic on four chromosomes (MMU7, MMU8, MMU11, and MMU17). The available gene mapping data do not provide evidence of synteny conservation specifically between segments corresponding to HSA16q–HSA19q. It is interesting to note, however, that all HSA16q and the majority of HSA19q genes hitherto mapped in mouse reside on MMU8 and MMU7, respectively.
Concluding Remarks
Despite several decades of intense comparative cytogenetic research, no consensus on the ancestral eutherian karyotype has as yet been reached. This comes as no surprise, because vast information hidden on the genome level is still to be explored to put the evolution jig-saw puzzle together (Eppig 1996). Todd (1970) suggested that the ancestral karyotype consisted of 14 chromosomes, whereas Matthey (1973)considered a chromosome number of 2n = 48 ± 8 as the ancestral eutherian condition. Still others favored an intermediate chromosome number like that of the cat (Rettenberger et al. 1995b). If we analyze the additional information available through various chromosome painting studies hitherto carried out, the different hypotheses mentioned above can be tested, assuming that maximum parsimony should disclose the likely ancestral chromosomes/chromosomal complement. This, in turn, can provide a preliminary pattern of the organization of the ancestral eutherian karyotype (Fig. 2).
On the basis of the set of conserved blocks observed in the eight species analyzed, one can propose a chromosomal pool representing the karyotype of the common ancestor (Fig. 2; Frönicke 1997). As a result of the application of human painting probes in all the species studied, the segments within this pool are denoted according to the corresponding human homologies. The ancestral pool (yellow circle, Fig.2), was generated by selecting those human chromosomes or chromosomal arms that were maintained as a single block in at least one of the nonprimate species studied by Zoo-FISH. In addition, conserved NSCs were also considered as ancestral conditions and included in the pool. Beginning from the putative ancestral karyotype, a minimum number of rearrangements are required to transform the ancestral complement into that of the extant species discussed herein. These rearrangements have been delineated in Figure 2. Assuming that chromosomes that were not conserved as a single block in distantly related mammals were present most likely as two chromosomes in the putative ancestor (e.g., HSA2, HSA16, and HSA19), an ancestral karyotype number can be derived (Frönicke 1997).
The considerations mentioned above suggest that the putative karyotype of the common ancestor of primates, carnivores, perissodactyls, and artiodactyls most likely comprised human chromosome equivalents 1p, 1q, 2pter–q13, 2q13–qter, (3/21), 4, 5, 6, 7, 8, 9, 10, 11, (12/22a), 13, (14/15), (16q/19q), 16p, 17, 18, 19p, 20, 22b, X, and Y. This chromosome number (2n = 48) fits well with the chromosome number suggested by Matthey (1973) for the common ancestor of eutherian mammals. It can be expected that with the availability of more comparative maps/Zoo-FISH karyotypes of species spanning other mammalian orders, and with the refinement of such maps by forward and reverse painting approaches (Goureau et al. 1996; Milan et al. 1996; Wienberg and Stanyon 1997; Wienberg et al. 1997), the presented preliminary view of an ancestral genome organization might sharpen. Because Zoo-FISH does not provide insights in intrachromosomal rearrangements, gene mapping data will have to be incorporated/supplemented to get a more detailed picture. This labor-intensive approach, however, will certainly be restricted to model species or species of commercial interest. Finally, available gene mapping data in mouse provide very limited but interesting comparisons with the Zoo-FISH data of the eight species analyzed. However, the observations are not enough to draw viable conclusions about the ancestral mammalian karyotype. In future investigations, the potential to get further insights into karyotypic evolution by Zoo-FISH comparisons will strongly depend on the inclusion of, for example, more ancestral species like insectivores and marsupials.
Acknowledgments
We thank Professor Leif Andersson for review of the manuscript. The project was financially supported by the Swedish Council for Forestry and Agricultural Research and the Erik Philip-Sörensen Stiftelsen.
Footnotes
-
↵3 Corresponding author. Present address: Division of Animal Genetics, The Royal Veterinary and Agricultural University, 1870-Frederiksberg C, Denmark.
-
E-MAIL bhc{at}kvl.dk; FAX 45-3528-3042.
- Cold Spring Harbor Laboratory Press








