
Alternate Polyadenylation in Human mRNAs: A Large-Scale Analysis by EST Clustering
Abstract
Alternate polyadenylation is an important post-transcriptional regulatory process now open to large-scale analysis by use of cDNA databases. We clustered 164,000 expressed sequence tags (ESTs) into ∼15,000 groups and aligned each group to a putative mRNA 3′ end. By use of stringent criteria to discard artifactual mRNA extremities, clear evidence for alternate polyadenylation was obtained in 189 of the 1000 EST clusters studied. A number of previously unreported polyadenylation sites were identified, together with possible instances of tissue-specific differential polyadenylation. This study demonstrates that, besides quantitative aspects of gene expression, the distribution of alternate mRNA forms can be analyzed through EST sampling.
Expressed sequence tags (ESTs), the short sequences produced from randomly selected cDNA clones (Adams et al. 1991; Okubo et al. 1992; Hillier et al. 1996), are widely exploited in gene identification (Banfi et al. 1996; Schuler et al. 1996) and in establishing extensive gene catalogs (Aaronson et al. 1996). A now classical use of EST sampling is also the production of transcript profiles, where the redundancy of EST sequences is used to quantify tissue-specific gene expression (Okubo et al. 1992; Lee et al. 1995;Kuska 1996; Audic and Claverie 1997; O’Brien 1997). It is expected that the wealth of information contained in EST databases can be used to investigate more qualitative aspect of mRNA expression, such as the frequency of alternative forms (e.g., cap sites, splicing, and polyadenylation variants). This article presents the first extensive survey of alternate mRNA polyadenylation from an EST database.
Eukaryotic genes have long 3′-untranslated regions (UTRs) that often contain several polyadenylation sites [poly(A)] sites. Alternate poly(A) sites can be used as a means to produce mRNAs with specific properties, which is now recognized as a major post-transcriptional regulation mechanism in eukaryotes (Wahle and Keller 1996). Cleavage and polyadenylation of mammalian mRNAs require several sequence signals, the most conserved of which is the AAUAAA motif, 10–30 bases upstream of the poly(A) site itself (O’Hare 1995;Manley and Takagaki 1996). This hexamer, however, may well occur randomly, and its presence alone does not warrant the existence of a poly(A) site. A reliable identification of bona fide poly(A) sites is achieved only through the experimental isolation of mature mRNAs. ESTs sequenced from cDNA 3′ ends (3′ ESTs) are expected to provide multiple experimental examples of this variable 3′-end processing.
We compared about 164,000 human 3′ ESTs from the Washington University–Merck project and clustered them into homogeneous groups, each corresponding to a putative gene. Clusters of overlapping/redundant ESTs were analyzed for the use of distinct poly(A) sites. Alternate polyadenylation events were clearly identified in as many as 189 of the 1000 EST clusters studied. Alignments of alternatively cleaved ESTs provide a striking view of what may be a widespread regulatory mechanism.
RESULTS
EST Clusters and Contigs
EST clustering procedures must deal with several obstacles leading to invalid cluster merges or breakdowns, notably sequencing errors, alternate splicing, and the presence of chimeric ESTs and paralogous genes. The clustering procedure now gaining acceptance in the field (Hillier et al. 1996; Schuler et al. 1996) is twofold. ESTs are first submitted to a fast pairwise sequence comparison, such as Blast (Altschul et al. 1990), to build up rough clusters and then to a more accurate, indel-permitting local alignment such as Fasta (Pearson and Lipman 1988) or Smith–Waterman (Smith and Waterman 1981). The latter step is designed to retain in each cluster ESTs having an uninterrupted, highly significant overlap (typically >95% similarity). We applied a similar procedure to classify the 3′ ESTs from the Washington University–Merck project (see Methods). The EST classification procedure produced 15325 clusters containing two sequences or more. The 1000 largest clusters ranged in size from 1413 to 17 sequences, illustrating the high redundancy of the original EST database (Table 1). EST clusters were visualized in the form of alignments to putative mRNA 3′ ends, as shown in Figure1. Putative mRNA 3′ ends were produced from EST clusters by use of the contig assembly program CAP (Huang 1992). Because of sequence discrepancies remaining in some clusters, CAP often produced several alternative contigs, with a mean number of 1.6 contigs per cluster (Table 2). Comparisons of contigs obtained from the first 1000 clusters with GenBank primate sequences yield significant BLAST scores (score >150, i.e.,P ≤ 0.02) for 72% of the contigs (Table 2). The majority of these highly expressed sequences can thus be related to known mRNAs.
General Characteristic of 3′ EST Clusters

Clusters of 3′ ESTs aligned with their respective contigs (top line of each cluster). Contigs annotated with a GenBank entry name can be considered as identical to the corresponding mRNA (BLAST2 score ⩾ 2000, 97%–99% identity over highest scoring segment). (Unknown mRNA) Contigs do not show any significant resemblance (BLAST score ⩾150 or P ⩾ 0.02) to a human non-EST sequence in GenBank release 104. mRNA extensions are shown with broken lines. Contigs that do not extend corresponding mRNAs are numbered from the mRNA 5′ end; other contigs are numbered from position 1. Thicker segments in contigs indicate possible internal priming sites (see Methods). Potential destabilization signals are shown with blue and dark blue dots, corresponding to sequences AUUUA and UUAUUUA(U/A)(U/A), respectively. ESTs are colored according to their source library, as indicated at bottom right. Numbers in parentheses indicate the total number of ESTs in each library. Red lines on contigs indicate coding sequences. Vertical red and yellow lines give the positions of all AAUAAA and AUUAAA sequences, among which are the actual polyadenylation signals (see text). Only ESTs that fully match their respective contig are shown. Clusters are numbered according to the number of ESTs they contain (1 is largest).
Characteristics of the 1000 Largest Clusters
To assess the quality of contig sequences, their nucleotide compositions were compared to those of actual human mRNA 3′ UTRs obtained from the UTRDB database (Pesole et al. 1996; Table3). Only the last 30 positions, those supposed to contain polyadenylation signals, were analyzed to minimize differences caused by gene-specific sequences. Base compositions are remarkably similar, and the most abundant hexamers correspond in both cases to the known mammalian polyadenylation signals, AAUAAA and AUUAAA. Either of these two signals is present in 59% of the EST contigs, vs. 70.9% of the real UTRs, indicating that at least 83% of the contigs probably do contain bona fide mRNA 3′ ends.
Base Composition and Hexamer Frequencies in the Last 30 Positions of Human mRNA 3‘ UTRs (UTRDB) and of Contigs Obtained from the 1000 Largest 3‘ EST Clusters
Characterization of Alternate Polyadenylation
Members of 3′ EST clusters were aligned with their respective contigs by use of the Fasta program (Pearson and Lipman 1988). ESTs that could not be fully aligned to their contig from 3′ to 5′ (i.e., with >10 mismatched positions at either extremity) were discarded as possible alternatively spliced or chimeric products. A sample of these alignments is shown in Figure 1. Although 3′ ESTs are, in theory, sequenced from mRNA poly(A) tails, it is readily apparent that ESTs matching the same mRNA do not all share the same 3′ end. These variations, however, are not necessarily attributable to alternate mRNA 3′ ends. ESTs may also result from internal priming, that is, primers hybridizing to internal poly(A) stretches instead of the expected poly(A) tail. Looking for adenine stretches in contig sequences flanking EST extremities (see Methods), we estimated conservatively that about 14% of ESTs in the first 1000 clusters could be attributable to internal priming (Table 2). The discrepancy with the previously reported rate of 2.5% (Aaronson et al. 1996) can be attributed to the particular sample of highly redundant ESTs studied here.
To consider an EST as an actual mRNA 3′ end, we required that is was clearly not attributable to internal priming and that it contained an AAUAAA or AUUAAA polyadenylation signal in the last 30 positions. These two signals are marked by red and yellow vertical lines, respectively, in Figure 1. This constraint is knowingly conservative, considering that a large fraction (∼30%) of the available mRNA sequences do not contain canonical polyadenylation signals (Table 3), even though these are reputedly ubiquitous (Manley and Takagaki 1996;Wahle and Keller 1996). Other elements of mammalian poly(A) signals, namely a CA dinucleotide at the poly(A) site followed by a GU-rich region, appear even less conserved than the canonical hexameric signals. Therefore, we did not require their presence.
ESTs drawn with thick lines in Figure 1 meet all the above criteria and are thus very likely to represent polyadenylated mRNA 3′ ends. According to our rules, 189 of the 1000 largest clusters show evidence for two or more poly(A) sites (Table 2). These include several human mRNAs already known to be alternatively polyadenylated, such as mRNAs for cytosolic aspartate aminotransferase (Bousquet-Lemercier et al. 1990; cluster 158, Fig. 1), calcium ATPase (Lytton and Maclennan 1988; cluster 758, Fig. 1), or calmodulin-I (Senterre-Lesenfants et al. 1995; cluster 2276, Fig. 1). Most of the 189 putative alternate polyadenylation patterns, however, are undocumented, either in the corresponding GenBank entries (for clusters matching known sequences) or in the literature. Those processed EST clusters are thus a unique source of novel information on mRNA 3′ end formation.
Novel Alternate Poly(A) Sites
Of the 720 EST contigs exhibiting significant similarities with GenBank primate sequences, 359 go beyond the 3′ end of the GenBank sequence by 20 bp or more, strongly suggesting that these published mRNA sequences are incomplete. Figure 1 presents examples of 3′ end extensions in which additional 3′ segments are confirmed by multiple ESTs and contain new putative polyadenylation sites. Furthermore, we verified that these 3′ extensions did not significantly match any GenBank sequence. The largest extension occurs with cluster 147, adding 630 nucleotides and at least two poly(A) sites (around positions 1450 and 1550) to the mRNA for placental protein PP5, a serine proteinase inhibitor (Miyagi et al. 1994). Cluster 448 extends the 3′ UTR of the signal recognition particle subunit 14 mRNA (GenBank accession no. X73459) by 300 bp, adding a new poly(A) site, cluster 208 extends the reported myosin regulatory light chain mRNA by ∼100 positions, introducing two novel poly(A) sites, and cluster 529 extends the α-catenin mRNA (Rimm et al. 1994) by 230 positions, introducing a new poly(A) site. Cluster 85 (Fig. 1) matches a genomic segment comprising the 3′ UTR of the rhoH12 mRNA (up to position 987) and into a region that is not marked as mRNA in the GenBank entry (accession no. M83094). Interestingly, the article accompanying that entry (Moscow et al. 1992) reports the isolation of a large cDNA encompassing both rhoH12 and the 721-bp 3′ extension that we observed in cluster 85, which independently confirms the alternate polyadenylation pattern.
Signals Lying between Alternate Poly(A) Sites
An obvious interest of collecting alternatively polyadenylated mRNAs is the identification of new functional sequences located between poly(A) sites that could thus act as regulatory elements. Among the well-characterized functional sequences in 3′ UTRs are the AU-rich elements (ARE) AUUUA and UUAUUUA(U/A)(U/A) that mediate mRNA destabilization (Shaw and Kamen 1986; Zubiaga et al. 1995). Occurrences of these 5-mer and 9-mer patterns are shown in Figure 1 with blue and dark blue dots, respectively. AREs are often clustered within 100 nucleotides upstream of polyadenylation signals. Their presence in the mature mRNA then depends on the poly(A) site used. Clusters 108, 147, 529, 758, and 2276 (Fig. 1) provide clear examples of this pattern, where cleavage at specific poly(A) sites determines whether or not mRNAs contain AREs in their 3′ extremities. In clusters 108, 147, and 529, most AREs are found near the distal poly(A) site and would thus mediate destabilization of the longer mRNAs only. This could suggest a common way of producing additional mRNAs of lesser stability under specific conditions. On the other hand, cDNAs in cluster 758 would contain AREs whatever polyadenylation signal is used. Although not all AREs actually function as destabilization elements, these patterns obviously deserve further experimental consideration.
Differential Polyadenylation
EST sampling is generally expected to provide significant new data on expression variations in response to environment, cell differentiation, or disease (Adams et al. 1991; Okubo et al. 1992;Audic and Claverie 1997). Alternate polyadenylation may also depend on the environment or tissue (O’Hare 1995). In this case, we will use the term differential polyadenylation. Novel instances of differential polyadenylation can be inferred from the observation of biased uses of poly(A) sites in certain EST libraries.
An evaluation of the statistical significance of an observed bias is possible through use of Fisher’s 2 × 2 exact test (Siegel 1956;Agresti 1992). This test computes the probability of a given 2 × 2 occurrence table for two independent categorical variables (here, one variable is the library, the other the mRNA form). Consider for instance the distribution observed for cluster 2422 (Fig. 1): Two forms of mRNA (short and long) are observed. mRNA originating from the multiple sclerosis library are only found in the short form (four times), while mRNA from the melanocyte library are only found in the long form (four times). The probability of such a bias to occur without the mRNA form and tissue type being correlated is only 0.028. Similarly, the significance levels corresponding to bias observed in clusters 974 (brain vs. other tissues) and 147 (placenta vs. other tissues) are 0.003 and 0.035, respectively. These P values are consistent with the differential use of poly(A) sites in certain tissues. This type of analysis is another way of looking at tissue-specific expression to point out targets of biomedical or biotechnological interest, irrespective of our knowledge of the gene functions. Actual mRNA levels, however, might differ from the observed values because of library normalization. The last say in this matter will thus be left to experiment, EST analysis mostly acting as a very efficient means to unveil the most striking expression profiles (i.e., that have not been flattened out by the normalization procedure).
The mRNA regions lying between alternate poly(A) sites constitute obvious hot spots for post-transcriptional regulatory elements. Besides AREs, a variety of RNA functional elements probably remain to be discovered in these regions. The present analysis of EST clusters is thus a good starting point for the systematic search of new functions in mRNA 3′ UTRs. It should also remind us that, besides mere transcription levels, other important aspects of gene expression might be sought after when constructing and analyzing EST databases.
METHODS
The complete dbEST database was downloaded from NCBI (ftp://ncbi.nlm.nih.gov). Washington University–Merck ESTs were extracted by use of identifications provided by the Washington University ftp server (ftp://genome.wustl.edu). Library/tissue names and numbers of ESTs are shown at the bottom of Figure 1. ESTs were scanned for the presence of contaminating sequences such as vectors, PCR primers, microsatellites, and human repeated sequences Alu, Line, LTRs, etc. Any Blast (Altschul et al. 1990) match with these elements at a score above 150 (110 for microsatellites) was masked in subsequent analyses (Claverie 1996). The first step of sequence classification, binning, involved a pairwise BLAST comparison of all 164,704 3′ ESTs, run in parallel on a cluster of 10 Silicon Graphics Indy R4400 workstations. For each EST sequence, all matching ESTs with a BLAST score >150 were retained. In the next step, each pair of matching ESTs was realigned by use of FASTA (Pearson and Lipman 1988), under the same parallel computing environment. Pairs of ESTs were considered as matching when the Fasta alignment had <10 mismatches at each extremity and >95% base identity overall (Fig.2). Mismatches involving undefined nucleotides (letter N) were not considered in these calculations. To cluster all ESTs corresponding to a given cDNA, any pair of matching ESTs was grouped, even when this grouping eventually placed nonmatching ESTs into the same cluster (A matches B, B matchesC, A does not match C).
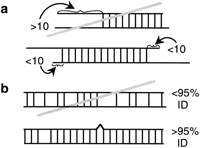
Criteria used for EST clustering. Each pair of EST sequences related by a BLAST (Altschul et al. 1990) score higher than 150 was further aligned with the FASTA program (Pearson and Lipman 1988). Any pair of EST sequences with <10 mismatched positions at either extremity (a) and >95% identity (b) was clustered.
Contigs were generated from each of the 15,325 clusters by use of the CAP program (Huang 1992) with default parameters. To avoid mismatch problems during the alignment of polyadenylated and nonpolyadenylated RNAs during contig construction, poly(A) tails (actually poly(T) caps) were deleted prior to contig building. ESTs in each cluster were aligned to their contig by use of the Fasta program (Pearson and Lipman 1988). ESTs that could not be fully aligned to their contig (from 5′ to 3′ of EST, with an authorized 10-base mismatch at each extremity) were discarded. Internal priming was assessed by looking for adenine stretches in the contig sequences flanking the 3′ extremity of an EST. Six or more consecutive adenines, or seven adenines in a 10-nucleotide window were considered as a possible source of internal priming (thickened lines on contigs in Fig. 1).
Fisher’s exact test calculations were performed with the WWW interface created by Oyvind Langsrud (http://www.nr.no/~langsrud/fisher.htm).
The set of contigs and 3′ EST clusters generated in this study with the corresponding similarity information (GenBank hits) is available from our anonymous ftp server (ftp://igs-server.cnrs-mrs.fr/pub/Polya-EST/).
Acknowledgments
We thank Incyte Pharmaceutical, Inc., for its financial support, including the salaries of O.P., F.L., and S.A.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL gauthere{at}igs.cnrs-mrs.fr; FAX (33) 4 91 16 45 49.
-
- Received November 17, 1997.
- Accepted February 17, 1998.
- Cold Spring Harbor Laboratory Press








