
Estimation of Distances and Map Construction Using Radiation Hybrids
Abstract
A method of estimating distances between pairs of genetic markers is described that directly uses their observed joint frequency distribution in a panel of radiation hybrids (RHs). The distance measure is based on the strength of association between marker pairs, which is high for close markers and decays with distance. These distances are then submitted to a previous method that generates linear coordinates for the markers directly from the intermarker distance matrix. This method of map building from RH data is simpler than others, because it uses only the observed joint frequency distributions of markers in the panel, and does not attempt to model unobserved quantities such as the retention of different sized fragments that contain the markers. It also incorporates directly the observed variation in retention of different markers, without needing a model for differential fragment retention dependent on chromosomal location, which is generally not known. Only small, precise distances are used in map construction, thereby reducing any effects of different fragment retention frequencies and local variations in X-ray sensitivity. The method is tested by simulation, and known marker distances and locations are successfully recovered from RH raw data. The method is also applied to publicly available data sets related to the recent transcript map of the human genome.
Radiation hybrid (RH) mapping is a flexible and powerful technique for mapping unique DNA sequences on genomes. It was originally developed by Goss and Harris (1975), who irradiated human fibroblasts and fused the resulting fragments with recipient rodent cells. The observed patterns of coretention of markers in a collection of hybrid cells allowed markers to be ordered on the X chromosome and chromosome 1 (Goss and Harris 1977a,b) by assuming that markers far apart are more likely to be separated by irradiation than closer markers, and therefore segregate independently in the component hybrid cells of the panel. The technique was extended by Cox et al. (1990) who irradiated donor somatic cell hybrids, which contained just a single copy of one human chromosome, and fused the fragments with rodent cells. The technique was used to map markers on the proximal (Burmeister et al. 1991) and distal (Cox et al. 1990) regions of chromosome 21. Subsequently, it has been used to map markers around disease loci in localized areas of the genome, for example, BRCA1 (Abel et al. 1993; Black et al. 1993; O’Connell et al. 1994), neurofibromatosis type 2 (Frazer et al. 1992), incontinentia pigmenti 1 (Gorski et al. 1992), and Huntington’s Disease (Altherr et al. 1992). A summary of chromosome-specific RH panels has been compiled (Leach and O’Connell 1995). Whole genome RH panels have been constructed recently (Walter et al. 1994; Gyapay et al. 1996), and genome maps containing hundreds (Gyapay et al. 1996) or thousands (Hudson et al. 1995; Stewart et al. 1997) of markers have been reported. Data gathered by an international consortium have been used recently to construct a human gene map (Schuler et al. 1996) that located ∼19,000 expressed sequence tags (ESTs) to intervals bounded by ∼1000 polymorphic framework markers in the final genetic map (Dib et al. 1996) consisting of ∼5000 polymorphic markers.
Many different methods have been proposed for the analysis of RH data (Boehnke et al. 1991; Falk 1991, 1992; Chakravarti and Reefer 1992;Guerra et al. 1992; Lange and Boehnke 1992; Lawrence and Morton 1992;Lunetta and Boehnke 1994; Lange et al. 1995; Lunetta et al. 1995a,b). Currently there are four main software packages that have been used to construct complete maps from RH data: RHMAP (Boehnke et al. 1991; Lange et al. 1995), RHMAPPER (Stein 1996), SAMAPPER (Stewart et al. 1997), and MultiMap (Matise et al. 1994). The first method uses a combination of minimizing the obligate number of breaks required to explain the observed retention patterns together with maximum-likelihood analysis, and has been used to map Genethon markers and ESTs across the genome (Gyapay et al. 1996). RHMAPPER uses a Markov model applied to genetic analysis (Lander and Green 1987; Boehnke et al. 1991) to obtain optimal positions for the loci on a linear map, and was used to construct an STS-based map of the human genome (Hudson et al. 1995). SAMAPPER has been used to construct an STS-based RH map of the human genome containing >5000 unique markers (Stewart et al. 1997). Each of these methods model the retention of different chromosomal fragments in the different clones comprising an RH panel (Boehnke et al. 1991). The parameters in these models are the breakage frequencies between all pairs of markers, and the retention frequencies of different fragments (which may depend on chromosomal location), which are adjusted to maximize the likelihood of the data in the context of the current model. Different models of fragment retention can be specified, such as equal retention, centromeric retention, and the left end-point model (Boehnke et al. 1991).
An alternative approach is adopted here, by first estimating the distances and their precision between all pairs of markers given a simple model for the retention of genetic markers in an RH panel that can be confirmed by direct observation, and subsequently building a map from these distances by use of an earlier method (Newell et al. 1995). The panel represents a sample of the donor genome, specifically a sample of the radiation events that have occurred in a number of donor chromosomes to give rise to the observed marker retention patterns in the RH panel. This sample can be used to estimate the distances between all pairs of markers, if several assumptions are made about the irradiation process, allowing quantitative interpretation of the observed patterns. The basic assumptions generally made about the RH experiment (Cox et al. 1990; Boehnke et al. 1991; Walter and Goodfellow 1993) are as follows. (1) A recipient rodent cell is paired with one donor cell, whose chromosomes are fragmented with X-rays. The donor cell may be haploid (one genome equivalent) or polyploid (more than one genome equivalent). The recipient chromosomal fragments of the donor cell are then available to the recipient cell for incorporation into its own genome. (2) According to a Poisson process, chromosome breaks occur randomly and independently of each other. Breakage results in a series of fragments, which together comprise the donor DNA. The process is assumed to be homogeneous, therefore, equal, nonoverlapping intervals along the chromosome have the same chance of a break occurring within them. This assumption was borne out by an experiment (Catcheside 1938) that showed that the number of X-ray induced breaks in Drosophila chromosomes had the Poisson distribution. A more recent study of chromosome 21 (Teague et al. 1996) indicates that radiosensitivity might have some dependence on chromatin content. Whether this is general across the whole genome is not yet known. (3) The probability that one or more breaks have ocurred between two markers is denoted υ. Its complement, the probability of no breaks occurring, is P 0 and is related to physical distanceD in units of mean number of breaks (Rays, R) byP 0 = e −D from the Poisson model of chromosome breakage. (4) Fragments are taken up by the host cell independently of each other, until a certain amount of donor DNA is incorporated, after which the remaining fragments are lost to the surrounding medium. Cox et al. (1990) observed that the number of fragments retained by RH cells had the Poisson distribution, implying random and independent retention of fragments. (5) The resulting hybrid cells are then typed for the presence or absence of a number of markers; the actual number of retained copies of a marker, in the case of a diploid or polyploid donor cell, is not determined.
To measure the degree of linkage and, hence, the distance between pairs of markers, here we use methods developed in the field of information and communications theory (Hartley 1928; Shannon 1948; Kullback and Leibler 1951; Fano 1961) to obtain a measure of the codependence between pairs of markers. The two measures used are the statistical entropy of the distribution of a single variable, and the mutual information (MI) between two different variables measured from their joint distribution. Shannon (1948) equated the entropy of a random process with the amount of information in that process. The MI is a measure of the information contained in one process about another process, and is also a convenient measure of the statistical codependence of two random variables. In the context of RH mapping, it measures the amount of information conveyed in the retention of one marker about the retention of a second marker, and can be quantified directly from experimental data. The relation between statistical entropy and MI is shown diagrammatically in Figure 1.
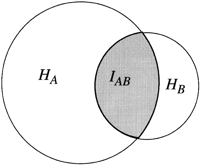
Relation between individual marker retention entropies,HA and HB, and the mutual information, IAB. HA andHB lie in the range 0 < H < log2, and 0 < IAB < min (HA, HB ).
To use the MI to estimate distances between pairs of markers, a simple model of X-ray breakage and fragment retention is used to estimate the expected numbers of the four classes AB, Ab, aB, and ab in terms of the breakage frequency between A and B, υAB. The model assumes that X-rays fragment the chromosomes at random and independent locations, and a subset of the resulting fragments are retained randomly and independently of each other in cells of the panel. The MI obtained from the observed frequencies of the four classes is compared with that from the expected frequencies, given a value of υAB, and this value is updated until the observed MI and the expected MI are equal. The model and algorithms are described in detail in Methods. Having obtained υAB, it is converted to linear distance in units of mean number of breaks, DAB, and distances between all pairs of markers are obtained in the same way. Coordinates in the principal axis are then calculated from the distance matrix as before.
To test the methods proposed here, simulated data were constructed as described in Methods and maps compared with the (known) locations of markers, and new maps constructed from the raw data related to the recent transcript map (Schuler et al. 1996) from the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov), and the Radiation Hybrid Database (RHdb, http://ebi.ac.uk/RHdb) at the European Bioinformatics Institute (EBI, http://ebi.ac.uk).
RESULTS
Simulated Data
Analysis of simulated data enables validation of the method, because all marker positions are predefined, and the conditions of the experiment (e.g., number of hybrids and density of markers) can be controlled. To validate the statistical model used in the estimate of distance (random and independent chromosome breaks and random and independent fragment retention), we check that the model reproduces the observed frequencies of the four classes AB, Ab, aB, and ab, given the estimate of υ and the observed retention frequencies. The observed and expected distributions are compared by the χ2 test for each pair of markers whose distances were used directly in map construction. In all cases, the observed and predicted frequencies were indistinguishable (P>0.9, the probability that any discrepancy between the observed and estimated frequencies would be this large or even larger by chance). The mutual information measured from simulated data (100 markers randomly placed on a chromosome of length 3R typed on a panel of 100 hybrids) is plotted versus known physical distance in Figure 2. As expected, the MI decays with distance, as pairs of markers become randomized in the RH panel with more frequent breaks between them. The distances estimated from the measured and estimated values for the MI are plotted versus known physical distance in Figure 3. The estimate of distances from marker coretention patterns after chromosome fragmentation and retention is shown to accurately reproduce the real distances for small real distances, but fluctuations of the estimated distances about the true distances increases with real distance. As the number of hybrids used in the panel increases, so the quality of the fit at larger distances increases, as expected, and consequently, so does the largest measurable distance.
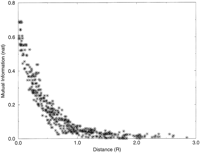
MI between pairs of markers vs. known physical distance between them. The MI is close to its maximum value of log2 (0.693) for very close markers and decays with distance. Total of 100 hybrids, 100 markers, chromosome length 3 R.
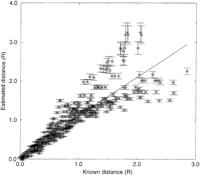
Estimated vs. known distances for a panel of 100 hybrids, 100 markers, chromosome length 3 R.
The distance matrix from Figure 3 was then submitted to the map-building procedure, on the basis of the techniques of distance geometry, and the map generated here is shown in Figure4. Optimal coordinates in the largest two dimensions are shown on the left, coordinates in the principal axis projected in the middle, and the original marker locations on the right. The principal axis coordinates are the optimal linear map locations for the markers obtained by the method described here. However, if there is more than one principal dimension, the distances on the two-dimensional map are closer to the measured distances as a result of experimental typing error, and the two-dimensional map can indicate particularly error-prone markers that can be removed and the remaining marker set remapped. The simulated panel with 100 hybrids locates all markers to the correct chromosomal region. Discrepancies between the map and the actual locations are all the result of the lack of any breaks between some groups of close adjacent markers, giving identical retention patterns in the component cells of the panel (raw data, Fig.5). Examples of sets of unresolved markers are 1–3, 33–40, and 92–95, and the name order of such clusters is arbitrary (they are all at the same location). Close markers can be resolved by increasing the size of the panel, thereby effectively increasing the sample size of radiation events for the same dose. A map of 100 markers, with a panel of 500 hybrids (not shown) correctly places and resolves all markers. The simulation demonstrates that the method accurately regains known marker positions from raw RH data, even for small RH panels.
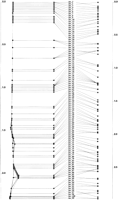
Map constructed from the distances in Fig. 3. The optimal order is to the left of the names, the two-dimensional configuration at the extreme left, and the original (known) positions at theright.

Raw data used for the map in Fig. 4. (Black) Present; (white) absent. The retention frequencies are plotted to the left.(Bottom) Relative numbers of markers and breaks observed in each hybrid cell of the panel.
The simulation allows the three main parameters of the experiment to be altered. These are (1) the density of breaks (a function of the X-ray dose), (2) the density of markers, and (3) the number of hybrids in the panel. These parameters were altered to investigate the resolution and range of panels that can be expected from these three quantities. The resolution we define as the minimum distance (>0) that can be distinguished by the panel, in units of mean number of breaks, R. A distance of zero is obtained when the retention patterns of Aand B are identical (i.e., only concordant hybrids are observed, no +− or −+ classes). The smallest distance that can be observed is the smallest difference in the retention patterns of markers A and B, which is one discordant class (+− or −+). By use of the expressions for the expected frequencies of the four different cell types, values for D and the error in D were calculated assuming all markers are retained with an average retention frequency of 0.5, for different numbers of hybrids in the panel (Table 1). As the number of hybrids in the panel increases, so the smallest resolvable distance decreases. ForN = 10 (where N is the number of hybrids in the panel), the smallest resolvable distance is 0.17 R, forN = 100, it is 0.017 R, and for N = 1000, it is 0.002 R. Also predicted were the minimum resolvable distances for the G3 and G4 panels, by use of the observed average retention frequencies of 0.16 and 0.32 for these panels. The G3 panel can resolve markers >0.038 breaks apart. The G4 panel can resolve markers >0.021 breaks apart. The TNG panel can resolve markers >0.032 breaks apart.
Resolution (Minimum Resolvable Distance) and Range [Maximum Distance That Can Be Determined with Some Confidence (Z > 3)] for Panels Differing in the Number of Hybrids
The complementary property, the physical range of a panel, is defined as the maximum distance that can be measured between two markers with some confidence, before intermarker breakage becomes so frequent as to remove all linkage. The largest significant distance [log-of-odds (lod) > 3] increases with the number of hybrids in the panel. For a panel of 50 hybrids and an average retention frequency of 0.5, the largest measurable distance is 0.654 R; for 100 hybrids, it is 1.022 R. For the G3 panel, with a retention frequency of 0.16, the largest distance is 0.985 R, for the G4 panel (retention frequency = 0.32) it is 1.036 R, and for the TNG panel (retention frequency = 0.18) it is 0.968 R. The calibration of R to Mb is examined below.
Application to the Gene Map EST Data Set
The method was applied to the EST mapping data from the recent Gene Map (Schuler et al. 1996), which integrated data accumulated on two different RH panels, differing greatly in the dose used to construct them. The method shown here is unable to mix data from different panels to make a composite map, because scores for markers typed on one panel cannot be compared with scores for markers typed on a different panel; cells in different panels contain an entirely different set of fragments. This problem was addressed by the RH mapping consortium (Schuler et al. 1996) by linking the two data sets by use of the independent Genethon genetic map, the final gene map being locations of ESTs in intervals defined by two Genethon markers, and the units being genetic map units of centimorgans. Distances on this map may be inflated or deflated in recombination hot- and cold-spots.
Here, the method was applied to build new framework maps from the raw RH data for the Genethon markers. This has the advantage that EST maps built subsequently can be directly compared with framework maps, because they are in the same units. The framework data from Gene Map were split by panel and chromosome, to give separate raw data files for each chromosome on each panel. Each data set was then mapped separately. The numbers of framework (fw) markers and ESTs mapped on the two panels were G3fw 521, G3EST 2728, G3total 3249, G4fw 1207, G4EST 15913, and G4total 17120. The density of framework markers on the G3 panel is 0.17/Mb, by use of the physical size estimates of Morton (1991), fw markers occurring every 5.7 Mb on average, and the framework density on the G4 panel is 0.4/Mb. Because the majority of ESTs were mapped on the G4 panel, attention was directed towards G4-typed markers.
The largest framework dataset (Chr 1) contained 110 markers, and a map was built in ∼45 sec (SG Indy). In most cases, the initial framework maps revealed two major linkage groups, corresponding to the pand q arms, separated by a weak link spanning the centromere. The largest centromeric gap was observed in chromosome 1. No large gap was seen in the acrocentric chromosomes 13, 14, 15, 21, and 22, and the framework markers formed complete linkage groups. Where large centromeric gaps were observed, the data for the two arms were analyzed separately, to give two more strongly-linked maps. Error-prone markers were then identified as lying far from the major axis, and removed. The remaining markers were remapped, and the process continued until the map was close to linear. The p and q arm maps were then joined by estimating the distance between the most centromeric markers on the two maps. The framework map of chromosome 2 generated here is shown in Figure 6. The gap across the centromere is obvious and the link is weak, but within each arm, the markers are strongly linked, and both maps are near to linear. There is good agreement with the order on the Genethon map, the only discrepancies being local reversals. None of the markers are placed far away from their locations on the Genethon map. The raw data for this map are shown in Figure 7, the markers in the order obtained in the map. The centromeric gap is obvious as a discontinuity between the raw data for D2S2181 (p cen) and D2S373 (q cen). Comparison of Figures 6 and 7 might also explain some of the discrepancies of the RH map with the Genethon map. For instance, the markers D2S148, D2S326, and D2S364 are at different locations on the two maps. Examination of the RH raw data (Fig. 7) shows a range of hybrid cells (nos. 28–57) typed negative for all or most of these markers, whereas neighboring markers are typed positive. This might indicate that PCR is inefficient for these markers in this range of hybrid cells. Other markers that seem to have an unusually high negative rate are D2S145, D2S286 (p arm), and D2S331, D2S2338 (q tel), and these markers are also inverted with respect to their locations on the genetic map. Such comparison of the map with the raw data might reveal similar PCR anomalies in different regions of the genome.
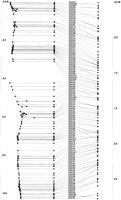
Framework map of Chromosome 2 containing 108 framework Genethon markers. Units on the RH maps are R (1 R ≅ 25 Mb) and M (1 M ≅ 100 Mb) on the genetic map.

Raw data for Fig. 6. (Black) Present; (white) absent; (gray) ambiguous/undetermined. Loci are rows from p telomere (top) to q telomere (bottom). Retention frequencies are plotted to the left. Relative numbers of markers and breaks observed in each hybrid cell of the panel are plotted below.
The framework maps were used to calibrate the RH maps to Mb units, by use of the physical length estimates of Morton (1991), and to compare the distance estimates with those in the previous G4 map (Hudson et al. 1995). The maps generated here indicate that breaks occur on average every 24 Mb (on the basis of the separate p and q arm maps). This estimate agrees closely with the rate of breaks for the STS map (Hudson et al. 1995) of one break every 25 Mb, omitting the centromeric intervals. Because of the inability to map G3-typed framework markers with confidence, no equivalent independent estimate of the density of breaks on the G3 panel could be made here. However, an earlier result of Goss and Harris (1975) showed that the number of X-ray induced breaks has a dose dependency of d 1.6. The expected frequency of breaks on the G3 panel (constructed with a dose of 10,000 Rads; Stewart et al. 1997) is then obtained from the frequency of breaks on the G4 panel (3000 Rads; Walter and Goodfellow 1995) as (10/3)1.6 × 0.04 = 0.27/Mb, that is, one break every 3.6 Mb. For the original panel used by Cox et al. (1990)the dose used was 8000 Rad, giving a breakage frequency of 0.19/Mb, that is, one break every 5.2 Mb. This agrees almost exactly with the original estimate of fragment sizes as 5.3 Mb (Cox et al. 1990), and these results confirm the dose dependency determined by Goss and Harris (1975). It was shown above that the maximum distance that could be directly measured on the G3 panel is ∼1R, which is 3.6 Mb for the most recent G3 panel (dose 10,000 Rads; Stewart et al. 1997). However, the density of framework markers on the G3 panel is only one every 5.9 Mb. This explains why no maps could be obtained for the G3 panel: The fragment size is too small for the density of framework markers. This problem will be alleviated by typing more markers on the G3 panel. The frequency of breaks on the new TNG4 panel (dose 50,000 Rads; Beasley et al. 1997) is similarly obtained as one beak every 280 kb (3.6/Mb).
The RH framework maps are then used as the starting point for constructing EST maps in local chromosomal regions. This approach is preferred to constructing whole chromosome EST maps, because local maps with fewer markers contain only relevant data, are optimal for that region, and are not skewed by error-prone data outside the region of interest. Local maps are constructed by first selecting the closest markers around a seed marker. This seed can be specified as either a framework marker, or the name of an EST. Closest markers are then selected rapidly from the RHdb raw data set by use of a Hamming distance measure. The resulting raw data subset is then submitted to the map-building procedure. This means of building maps allows greater accuracy in local regions, and positional cloning projects rarely need to consider the whole chromosome. An example is shown in Figure8, in which markers close to A002520 have been selected and mapped; a snapshot is shown from Java tools being developed as an interface to the current method. Three maps are shown. At the left is the map constructed here from the raw data; in the middle the markers are placed at the midpoint of their framework interval as determined in Gene Map (Schuler et al. 1996); at the right are the Genethon framework markers at their locations in the genetic map (Dib et al. 1996). Lines between the mid-point Gene Map locations and the Genethon framework markers indicate the range of the assigned interval. Although all 35 markers are resolved in the DG map, these are localized to just 10 unique intervals in Gene Map, some of which are quite large (the interval DXS997-DXS1058 to which seven markers were assigned is 9.4 cM).
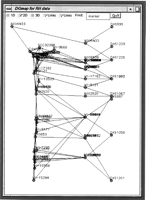
Printout of a Java applet showing an EST map from the X chromosome. (Left) Map generated here; (middle) markers are placed at the midpoint of their assigned interval in Gene Map (Schuler et al. 1996); (right) the locations of the framework markers on the Genethon genetic map (Dib et al. 1996). Lines between themiddle and right columns indicate the range of the interval to which ESTs have been assigned in Gene Map.
DISCUSSION
Estimation of Distances
Demonstrated here is the use of a measure of association, the MI, which can be used to estimate distances between loci, in units of average number of breaks between the loci. The MI measures the amount of the concordant classes that arise from retention or loss of a pair of markers on the same fragment (correlated retention) and retention or loss on different fragments (uncorrelated retention). This measure can be calculated directly from the observed patterns of retention of two markers across the RH panel, and distances obtained from a simple model of marker retention that has υ as the only estimated parameter, the other parameters (the marker retention frequenciesrA and rB ) being estimated directly from the data. Therefore, it takes account of the observed differences in marker retention frequency in the panel, which might arise from different chromosomal locations (Lawrence et al. 1991), without requiring prior knowledge or estimation of those locations. This method is therefore considerably simpler and requires less computation than other methods that model fragment retention. This cannot be measured directly and must instead be modeled in terms of the estimated fragment location with respect to the centromere. This requires either prior knowledge, or arbitrary assignment, of the location of fragments with respect to the centromere. The distance estimate used here is also independent of the ploidy of the donor cell. Missing data (e.g., a hybrid typed for marker A but not for B) are not considered because they provide no distance information. The distances are then submitted to a distance geometry routine (Newell et al. 1995), from which map positions are obtained from the intermarker distance matrix.
The model used here is general and uses only the experimental observations of marker retention to estimate a single parameter υ, the probability of breakage, for all pairs of markers. The only assumptions are that X-ray breaks occur randomly and independently, and that fragments are retained randomly and independently in the hybrid cells. There is experimental support for both assumptions (Cox et al. 1990). The lengths of fragments have the exponential distribution, in support of a Poisson model for chromosome breaks, and the number of fragments retained has the Poisson distribution as expected if fragments are retained randomly and independently of each other. This model gives estimates of υ that accurately predict the observed patterns of coretention of markers in simulated and real RH panels. The complication that markers are retained at different frequencies that implies preferential incorporation of the centromere is largely circumvented in this method by use of only very short (precise) distances to construct the map, and equal retention of a pair of markers becomes more accurate the closer they are together. The same technique also removes some of the possible dependence of breakage frequency on chromatin content (Teague et al. 1996). Therefore, the significance of chromosomal location (which is generally not known prior to such analyses) is minimized, and is not considered a parameter in this method. The observed retention frequencies are, however, still incorporated directly into the model. Any unequal retention caused by location is revealed after the map has been constructed.
Map Construction
The use of the techniques of distance geometry for map construction has been demonstrated previously (Newell et al. 1995), and the major advantage is its scaling properties. The generation of coordinates from a distance matrix has scalingO(N 3). Distance weights are incorporated before the coordinate generation, by smoothing the distance matrix, which here involves substituting large imprecise distances with sums of smaller, more precise distances. Previously, we optimized the coordinates in the principal axis using conjugate gradient methods as used in coordinate refinement for 3D model construction from NMR data (Havel et al. 1983), but here the improvement was negligible. In the current investigation, a major advantage of the DG technique in initial map building, by use of all the available data, was the identification of linkage subgroups that could subsequently be analyzed separately. The similarity between the DG technique (or the more general principle component analysis) and general clustering methods has been discussed previously (Shepard 1980). Another useful feature is the rapid identification of error-prone probes that lie far away from the other markers, and indicates that such markers should not be included in the analysis, and retyped.
Gene Map
Gene Map (Schuler et al. 1996) comprises the locations of ESTs to intervals between pairs of markers on the genetic map of Genethon. The assignment to different intervals is achieved by observing linkage by use of common retention patterns on the G3 and G4 panels. However, no further map information was computed, because the G3 and G4 panels differ so greatly in their properties. The units of the Gene Map are centimorgans, and locations are defined by the Genethon map (Dib et al. 1996). The distances on this map are therefore not physical, and may be inflated or deflated in recombination hot and cold spots. The framework maps shown here have been constructed from the RH raw data by use of the distance measure discussed, and distances are assumed to be proportional to physical distances in all regions of the genome. Another feature of Gene Map is that not all ESTs are resolved, because of the binning procedure. There are only 1740 unique locations defined on the G4 map, although there are 16,404 unique score vectors (July 97 version of RHdb). These ESTs have here been assigned separate map locations. The suggested method of making EST maps in local regions of a chromosome, around a known marker of interest, has the advantage of accuracy and speed. By initially selecting markers that are known to be close (by their similar retention patterns across the cells of the hybrid panel), error-prone data are automatically excluded. This estimate is general for any panel, incorporating the number of hybrids, and the average marker retention frequency, and is independent of the ploidy of the donor cell. It could be used to extract markers close to a defined marker from any data set. The defined marker could be specified by name, or by an experimental RH typing result for a novel marker. Once a sufficient number of markers has been retrieved in the region of interest, or a sufficiently large genomic region has been covered, the selected markers could then be mapped to give an accurate local map in a limited region of a chromosome. This will be useful in positional cloning projects.
Resolution and Range of Different Panels
The RH experiment is potentially very flexible, because all parameters are under experimental control. Only three parameters determine the results of an RH mapping experiment: The radiation dose that determines the density of breaks in unit distance intervals (μ), the density of typed markers in unit distance intervals (λ), and the number of hybrids in the panel (N) that determines the significance of any detected linkage and the precision of the resulting distance estimate. The first two parameters determine the mean density of breaks between adjacent markers, μ* = μ/λ. The probability that adjacent markers are not separated by X-ray breaks isP 0 = e−μ*, and its complement the breakage frequency is υ = 1 − e−μ*. The average breakage frequency between adjacent markers, therefore, increases with μ*, and as μ increases and λ decreases, the linkage between adjacent markers becomes less significant. The absolute significance of association (equivalently, the lod score for linkage) can be calculated by use of the expressions described here. The three quantities μ, λ, and N are known or can be estimated for the G3 and G4 panels in relation to the Gene Map data set, and can be used to compare them and assess their suitability for long-range (chromosomal) mapping. For the G4 panel, μG4 = 0.04/Mb, λG4 = 0.36/Mb, μ*G4 = 0.11/Mb, and N = 93. The breakage frequency between adjacent markers on the G4 panel is υ = 1 − e −0.11 = 0.10, which, with an average retention frequency of 0.32 and 93 hybrids, gives an average intermarker lod score of 17.5. For the G3 panel, λG3 = 0.16/Mb, N = 83, we estimated μG3 = 0.27/Mb, giving μ*G3 = 1.7. The intermarker breakage frequency is then 0.82, with a lod score of 1.0. This very low significance of linkage between adjacent markers, which will be the most strongly linked, explains why no long-range framework maps of whole chromosomes could be built by use of the G3 raw data. The development of new markers and their typing data on the G3 panel (Stewart et al. 1997) will resolve this problem. By use of similar arguments, we predict that the frequency of breaks on the new TNG3 panel (50,000 Rad) is μTNG3 = (50/3)1.6 × 0.04 = 3.6/Mb. The estimated resolution and range of the G3, G4, TNG, and simulated panels are shown in Table 1.
Success and Future of RH Mapping
RH mapping is currently the preferred technique for localizing new markers to localized regions of genomes. It uses sampling of irradiation-induced fragmentation of chromosomes originally devised byGoss and Harris (1975), and brought to practical large-scale application by Cox et al. (1990) and Walter et al. (1994). The power of this technique has been demonstrated frequently, and is accentuated by good maps obtained without the need to consider physical processes occurring during RH panel construction. Specifically, none of the methods of analysis need consider the physical rearrangements of chromosomes upon irradiation. These were investigated inTradescantia by Catcheside et al. (1946) who observed many different rearrangements (rather than simple breaks). Similar studies on human cells might improve the models used for estimating relative locations of markers. For example, the conditions (temperature, stage in cell cycle, etc.) that determine the efficiency of breaking chromosomes, and the types of changes that result, such as chromatid exchanges, isochromatid exchanges, inversions, etc., are not well understood. It is also likely that X-irradiation will cause some changes that will prevent transfer of human DNA into the rodent genome. These changes could be quantified in different conditions and incorporated into more accurate stochastic models. It is possible that many of the previously classified errors might be consequences of these highly nonlinear events. The second stage in RH panel construction is the nonselective incorporation of human DNA fragments in rodent cells. This process gives rise to 16% of the genome being retained in the G3 panel, and twice as much in the G4 panel, and is therefore highly variable. The underlying physical processes will become better understood as the demands on the RH mapping method increase and as more unique sites on the human genome are discovered and mapped.
METHODS
Notation
The symbol r represents the observed frequency of a particular class of hybrids in the panel, and the symbol prepresents the expected frequency in terms of the breakage frequency υ and the observed marker retention probabilities given the model described below, uppercase/lowercase subscripts represent the presence/absence of a marker (or pair of markers) in a panel; for example, rAB is the observed frequency of hybrids retaining both markers A and B(rAB = NAB /N),ra = 1 − rA, pAb is the estimated frequency (in terms of υ,rA, and rB ) of hybrids in the panel containing marker A but not B, etc.
Scheme
- 1.
- Estimate the significance and degree of association between all pairs of markers, from the observed frequencies of retention and coretention of pairs of markers in the hybrid panel, that is, the frequencies of the classes AB, Ab, aB, and ab for all pairs of markers A and B, as the observed MI in the joint frequency distribution IAB in terms of the observed frequencies rAB, rAb, raB, andrab.
- 2.
- Formulate expressions for the expected frequencies of the four classes,pAB, and so forth; hence, the expected MI in terms of the observed frequencies rA andrB and an additional model parameter υ,IAB, υ, based on the most general and least restrictive assumptions held to be true concerning the RH experiment.
- 3.
- Find that value of υ for which the observed and expected values of the MI are equal, convert to distance (mean number of breaks), and estimate the variance of the distance estimate.
- 4.
- Build a full N × N distance matrix, using only the most accurate distances required for complete linkage.
- 5.
- Generate coordinates from the distance matrix.
Data
Simulated and public data were used to test the methods of map construction described below. Public data were obtained from the FTP sites at the Whitehead Institute (http://www.genome.wi.mit.edu/ftp/distribution/human_STS_releases/july97/rhmap/), the Stanford Human Genome Center (SHGC:ftp://shgc.stanford.edu/pub/hgmc/RH_data/), and the RHdb (ftp://ftp.ebi.ac.uk/pub/databases/RHdb/). All data were converted to a common format: a flat file consisting of consecutive lines, one per marker, each of which has the format “UNIQid, score vector, reported position, aliases.”
Significance of Association between Two Markers
The significance of association can be measured from the value of χ2 for a 2 × 2 table,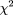 where nxy
=NxNy/N is the expected number of hybrids in each group, in the null hypothesis of no association. (
where nxy
=NxNy/N is the expected number of hybrids in each group, in the null hypothesis of no association. (
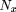 are the row and column marginal totals). The probability that the two variables are not associated is given by the χ2 probability function with one degree of freedom, p(χ2, 1), which gives the probability that the observed value of χ2should exceed the value by chance (Press et al. 1988). In the context of RH mapping, it is the probability that the observed joint retention pattern of a pair of markers could
have arisen with each marker always on a separate fragment to the other (random segregation of the two markers across the
panel). Small values indicate that it is unlikely that the two markers are independent. It is equivalent to a likelihood (odds)
ratio of nonlinkage versus linkage. The negative logarithm is a lod score for linkage versus nonlinkage. The distribution
used to compute the significance here is just the χ2 distribution, rather than a distribution expected from a more complicated model with a larger number of parameters, as in
Boehnke et al. (1991).
are the row and column marginal totals). The probability that the two variables are not associated is given by the χ2 probability function with one degree of freedom, p(χ2, 1), which gives the probability that the observed value of χ2should exceed the value by chance (Press et al. 1988). In the context of RH mapping, it is the probability that the observed joint retention pattern of a pair of markers could
have arisen with each marker always on a separate fragment to the other (random segregation of the two markers across the
panel). Small values indicate that it is unlikely that the two markers are independent. It is equivalent to a likelihood (odds)
ratio of nonlinkage versus linkage. The negative logarithm is a lod score for linkage versus nonlinkage. The distribution
used to compute the significance here is just the χ2 distribution, rather than a distribution expected from a more complicated model with a larger number of parameters, as in
Boehnke et al. (1991).
Degree of Association Between Two Markers from Their Observed Joint Distribution
Methods from communications theory (Shannon 1948; Kullback and Leibler 1951; Fano 1961) are used to measure the degree of dependence between pairs of variables. The measures used are the statistical entropy and
the MI. In the case of RH mapping, the experimental observation consists of the presence or absence of markers in a hybrid
panel. The presence or absence of each marker is considered a random variable, which is nominal taking one of two possible
values, present or absent. Ambiguous results are treated as missing data in this analysis and are ignored. The entropy of
the distribution (or variability of retention) of a marker A in a panel of radiation hybrids is given by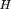 where A denotes presence and a absence of the marker. Its maximum value is log2, obtained when the marker is retained in exactly half of the hybrids in
the panel (rA
=ra
= 0.5), and its minimum value is 0, when it is retained in all, or none, of the hybrids.
where A denotes presence and a absence of the marker. Its maximum value is log2, obtained when the marker is retained in exactly half of the hybrids in
the panel (rA
=ra
= 0.5), and its minimum value is 0, when it is retained in all, or none, of the hybrids.
The entropy of the joint distribution of two different markersA and B in the hybrid panel (the variability of the joint retention of A and B) is given by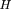 and has a maximum attainable value of H(A) +H(B), when A and B are retained independently of each other (they are always on separate fragments following irradiation). If the joint entropy
is less than the sum of the individual entropies, there is some dependence between the two markers: The variability in the
joint distribution is less than that expected if A and B are independent due to an excess of concordant (AB, ab) over discordant (Ab, aB) types. The minimum value of the joint entropy is the smaller of the two individual entropies.
and has a maximum attainable value of H(A) +H(B), when A and B are retained independently of each other (they are always on separate fragments following irradiation). If the joint entropy
is less than the sum of the individual entropies, there is some dependence between the two markers: The variability in the
joint distribution is less than that expected if A and B are independent due to an excess of concordant (AB, ab) over discordant (Ab, aB) types. The minimum value of the joint entropy is the smaller of the two individual entropies.
The mutual information is the difference between the sum of the individual entropies and the joint entropy and measures the
degree of statistical codependence between or association between A and B,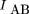 and can also be expressed in terms of the observed frequencies as
and can also be expressed in terms of the observed frequencies as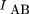 The mutual information is therefore equivalent to the relative entropy of the two distributions rxy (dependent retention) and rxry
(independent retention), and to the Kullback-Leibler distance between them. Its minimum value is 0 whenA and B are retained independently on separate fragments (θ = 1, rXY
=rXrY
) and is >0 if there is some dependence (θ < 1, rXY ≄′ rXrY
). Its maximum value is the smaller of H(A) and H(B) (Fig. 1).
The mutual information is therefore equivalent to the relative entropy of the two distributions rxy (dependent retention) and rxry
(independent retention), and to the Kullback-Leibler distance between them. Its minimum value is 0 whenA and B are retained independently on separate fragments (θ = 1, rXY
=rXrY
) and is >0 if there is some dependence (θ < 1, rXY ≄′ rXrY
). Its maximum value is the smaller of H(A) and H(B) (Fig. 1).
Estimate of Breakage Frequency υ from the Mutual Information
The expression for the mutual information above depends on only the observed data, with no dependence on υ, the probability that two markers have been broken apart by radiation, which is the parameter to be determined from the experiment. To find this dependence ofIAB on υ, and, hence, to estimate υ fromIAB, some assumptions about the physical processes occurring in the RH experiment must be made to explain the observed distribution of markers in the panel in terms of υ. These are used to estimate the expected joint frequencies of a pair of markers (pAB, pAb, paB, pab ) by use of a simple model of fragment breakage and retention in the panel, in terms of the observed frequencies of retention (rA, rB ) and the single model parameter υ. The approach adopted here is most similar to that of Chakravarti and Reefer (1991) who assumed equal retention frequencies of fragments containing markers A and B to give expected frequencies of the four classes AB, Ab, aB, and ab.Here, the observed retention frequencies of markers A andB, which in general are not equal, are used directly.
Estimation of the Expected Frequencies of Discordant TypesAb and aB
The estimated frequency of cells retaining A but notB across the panel, pAb, is estimated in terms of rA
and rB
as follows. For a hybrid typed Ab, marker A is necessarily on a different fragment to B. The probability of retaining a fragment containing A but not B is the product of (1) the probability of retaining marker A, estimated from the data as rA, and (2) the probability that the retained fragment contains just marker A (not B) which is υ, because the fraction of all fragments containing A but not B is υ, by definition of υ. This fraction is independent of the ploidy of the donor cell. The probability of a cell retaining
A on a separate fragment to B is therefore υrA, because υ and rA
are independent random variables. The probability of not retaining B in any single cell in the panel is estimated from the
data as 1 − rB
= rb. Assuming independent retention of fragments, the expression for the expected frequency of the class Ab in terms of the observed marker retention frequencies and the single model parameter υ is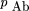 If θ = 1, then pAb is simply the product of the frequencies of two independent events, retention of Aand nonretention of B (independent because A andB are always on separate fragments). If θ < 1, the observed numbers of the discordant type Ab is less than that expected if A and B were independent, and if θ = 0, discordant types are never observed in the panel.
If θ = 1, then pAb is simply the product of the frequencies of two independent events, retention of Aand nonretention of B (independent because A andB are always on separate fragments). If θ < 1, the observed numbers of the discordant type Ab is less than that expected if A and B were independent, and if θ = 0, discordant types are never observed in the panel.
Similarly for the expected frequency of retention of B but not A in the component cells in the panel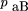 These expressions allow the retention frequencies of A andB to differ, as is generally the case, and they are incorporated directly in the model. The total frequency of the discordant
types in terms of θ, rA
andrB
is
These expressions allow the retention frequencies of A andB to differ, as is generally the case, and they are incorporated directly in the model. The total frequency of the discordant
types in terms of θ, rA
andrB
is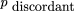 which is 0 when A and B are never broken apart (θ = 0).
which is 0 when A and B are never broken apart (θ = 0).
Estimation of the Expected Frequencies of Concordant TypesAB and ab
Extimates of the frequencies of the concordant classes (AB, ab) in terms of θ is complicated by retention (absence) of both markers in the same hybrid arising from dependent retention
(or absence) of both markers on the same fragment, and independent retention (or absence) of both markers on separate fragments
(Cox et al. 1990). The frequency of the positive concordant types in terms of θ (pAB
) can be estimated as either (1)pAB = rA
− pAb = rA
(1 − θrb
), or (2)pAB
= rB
−paB
= rB
(1 − θra
). Setting these two estimates equal would imply rA
= rB. However, it has been observed that markers nearer the centromere can be retained at a higher frequency than other markers.
To take account of unequal marker retention (rA
≄′ rB
) using the observed marker retention frequencies,pAB
is estimated as the average of the two estimates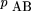 Similarly, the estimate of the frequency of absence of A andB in terms of θ is given by
Similarly, the estimate of the frequency of absence of A andB in terms of θ is given by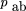 The sum of the concordant type frequencies is
The sum of the concordant type frequencies is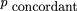 which is 1 when θ is 0 (only concordant classes are observed whenA and B are never broken apart).
which is 1 when θ is 0 (only concordant classes are observed whenA and B are never broken apart).
Use of the Probability Model to Estimate θ
To estimate θ using all the available experimental data, the expected MI in terms of rA, rB, and θ, denoted I
AB,θ, is given by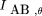 and θ is found as the root of I
AB,θ −IAB
= 0 by bisection search in the interval [0,1]. The search is terminated when ɛ ⩽ 10−6. This estimate of θ uses all the observed and estimated frequencies of the classes AB, Ab, aB, and ab.
and θ is found as the root of I
AB,θ −IAB
= 0 by bisection search in the interval [0,1]. The search is terminated when ɛ ⩽ 10−6. This estimate of θ uses all the observed and estimated frequencies of the classes AB, Ab, aB, and ab.
Precision of θ and Physical Distance
The breakage frequency θ is a binomial random variable (the probability that two markers are broken apart), its complementP
0 being the probability of no breaks betweenA and B. It therefore has variance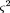 where N is the number of cells in the panel typed for both markers. The relation of θ to physical distance is θ = 1 - e-D, assuming random and independent breaks (a Poisson process); hence, D = −ln (1 − θ). The variance ofD is obtained from the law of errors of functions (Topping 1962) as
where N is the number of cells in the panel typed for both markers. The relation of θ to physical distance is θ = 1 - e-D, assuming random and independent breaks (a Poisson process); hence, D = −ln (1 − θ). The variance ofD is obtained from the law of errors of functions (Topping 1962) as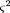
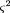 The variance of D is 0 for θ = 0 (D = 0), and ∞ for θ = 1 (D = ∞). Distances are given a weight equal to the inverse variance, so that smaller distances (small θ) are given greater weight
than longer distances, and arepreferred where possible.
The variance of D is 0 for θ = 0 (D = 0), and ∞ for θ = 1 (D = ∞). Distances are given a weight equal to the inverse variance, so that smaller distances (small θ) are given greater weight
than longer distances, and arepreferred where possible.
Linear Map Construction
Linear maps of markers were built from theN × N distance and weight matrices by use of an earlier method (Newell et al. 1995), which uses the techniques of distance geometry for embedding points in a limited number of dimensions (Havel et al. 1983). It was shown (Havel et al. 1985) that coordinates generated in this way are optimal, in the sense of reducing the rotation error between the lower-dimensional
conformations, and the completely accurate (N-1)-dimensional conformation. A preliminary round of data smoothing was applied
by first identifying the weakest (least accurate) interlocus distance required for complete linkage, and substituting all
weaker links with sums of intermediate (shorter and more accurate) links. This has the effect of building a map from small
distances between close clusters of markers, which damps the long-range effects of preferential retention of markers near
the centromere and possible local areas of particular X-ray sensitivity. The maximum distance variance that is required for
complete linkage was found by bisection search in the interval 0 < max required variance < max observed variance. All distances
with larger variance were substituted, increasing the dominance of the major axis by reducing the variance in higher dimensions.
The metric matrixG was then constructed from the distance matrix. Gcontains the dot products of the vector from locus i to the origin and the vector from locus j to the origin,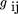 D
2
i0is the squared distance of point i to the origin, which can be measured without knowledge of the coordinates (Crippen and Havel 1978), according to
D
2
i0is the squared distance of point i to the origin, which can be measured without knowledge of the coordinates (Crippen and Havel 1978), according to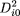 Coordinates are then obtained from X = √λ · W, where X is the matrix of coordinates in N dimensions, λ is the vector of eigenvalues of G and W is the vector of eigenvectors of G. Coordinates for point i in the principal axis were obtained from xi
= √λ1
·w
1i
, where λ1 is the largest eigenvalue, and w
1i is the corresponding eigenvector. Error-prone markers are identified as lying far from the major axis (λ2 is significant) and removed in further analysis. The final error in the location of the points was estimated as
Coordinates are then obtained from X = √λ · W, where X is the matrix of coordinates in N dimensions, λ is the vector of eigenvalues of G and W is the vector of eigenvectors of G. Coordinates for point i in the principal axis were obtained from xi
= √λ1
·w
1i
, where λ1 is the largest eigenvalue, and w
1i is the corresponding eigenvector. Error-prone markers are identified as lying far from the major axis (λ2 is significant) and removed in further analysis. The final error in the location of the points was estimated as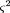 if ς2(Dij
) is less than the maximum required distance variance. The goodness-of-fit of the map to the estimated distances was estimated
as before (Newell et al. 1995).
if ς2(Dij
) is less than the maximum required distance variance. The goodness-of-fit of the map to the estimated distances was estimated
as before (Newell et al. 1995).
Simulation of a RH Panel
RH panels containing an arbitrary number of hybrids (e.g., 50–1000) containing random fragments from X-irradiated chromosomes were built according to the following procedures. Markers were placed randomly on identical donor chromosomes with a defined length. Each cell in the panel was paired with one donor cell, which containsc copies of the donor chromosome, in which c is the ploidy of the donor cell. Each of chromosomes was independently fragmented, by placing a number of breakpoints at random and independent locations. The average number of breaks μ defines the length of the chromosome as μRay (R), and the number of breaks placed on individual chromosomes was selected from a Poisson distribution with mean μ, modeling random and independent occurrence of breaks. A subset of the fragments generated from the donor cell chromosomes was then incoporated into the recipient rodent cell, the number of fragments from each donor cell taken up by the recipient cell chosen to give an average marker retention frequency of 0.5 (or another specified value). This number was determined as follows. The average number of chromosomal fragments generated from the donor cell chromosomes is c(μ + 1), where cis the ploidy of the donor cell. The number of copies of each unique marker in the total of fragments is c. The probability that a fragment chosen at random contains a particular marker isp(fA ) = c/c(μ + 1) = (μ + 1)−1, and is independent of the ploidy of the donor cell. The number of fragments that should be taken up by the cell to give a retention frequency r isN = r/p(fA ) = r(μ + 1). This value is in general noninteger, but the number of fragments is necessarily integer. The number of fragments taken up by each hybrid is therefore taken from a Poisson distribution with mean N,thereby modeling random and independent retention of fragments (Cox et al. 1990). Each hybrid was then typed for the presence or absence of each marker. For each pair of markers, a 2 × 2 table was constructed containing the number of hybrids of each of the four classes AB, Ab, aB, and ab, and submitted to the method described above.
Application to Public Human Transcript Mapping Data
RH data for the framework markers (Gyapay et al. 1996) used to locate ESTs to framework intervals in the recent Gene Map of the human genome (Schuler et al. 1996) were submitted to the same analysis. The raw data for the Gene Map were obtained from NCBI, comprising EST name, mapping lab, mapping panel (G3/G4), RHdb identifier, chromosome, and closest linked anchor markers from Genethon, RHdb (Rodriguez-Toméand Lijnzaad 1997) for the RH raw data for framework markers and ESTs, and Genethon for the genetic map locations for the framework markers for comparison of the positions obtained here with the reported positions. The data were split into chromosome-specific files, two for each chromosome for data measured on the G3 and G4 panels. Framework marker data were treated in the same way as above. First, the complete data set was mapped. If this showed a large centromeric gap, the data set was split, and the two arms mapped separately. Error-prone markers were then identified, removed, and the remaining markers remapped. The two maps (p and q arms) were then joined by estimating the distance between the most centromeric markers on thep and q arms, to give a complete framework map of each chromosome.
The framework maps are the main interface to mapping ESTs. Because large data sets often contain error-prone markers that
will skew otherwise good maps, a different approach was adopted for mapping EST data sets. First, the user specifies either
a framework or an EST seed marker around which a map of the neighboring ESTs is required. This is achieved by first obtaining
the RH score vector for the specified seed marker, then the chromosome-specific RH data set is scanned for the closest markers.
Closeness is measured as the Hamming distance between two score vectors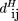 wheretik
= {0,1} is the absence of presence of marker i in hybrid K. The nearest Nmarkers then form a new RH raw data set that is submitted to the map-building procedures described. This approach allows greater
flexibility and accuracy, because selecting only close vectors automatically filters inconsistent or error-prone markers.
It also allows for possible errors in the framework maps, because scanning the raw data file for near ESTs does not rely on
the framework maps. Framework maps are used only as an initial zone locator.
wheretik
= {0,1} is the absence of presence of marker i in hybrid K. The nearest Nmarkers then form a new RH raw data set that is submitted to the map-building procedures described. This approach allows greater
flexibility and accuracy, because selecting only close vectors automatically filters inconsistent or error-prone markers.
It also allows for possible errors in the framework maps, because scanning the raw data file for near ESTs does not rely on
the framework maps. Framework maps are used only as an initial zone locator.
Availability
The maps are viewable over the Web in Java applets athttp://www.oxmol.com/OM-GW-Lib/dgmap/. Source code for building maps from raw RH data is written in C and is available on request. The code makes use of routines from Numerical Recipes in C (Press et al. 1988), available from http://cfatab.harvard.edu/nr/.
Acknowledgments
We acknowledge valuable discussions of different aspects of this work with Gillian Amphlett, Mike Arnautov, Frank Pennycook, John Riley, and Philippe Sanseau. This work was started at the Imperial Cancer Research Fund, London, UK, under a BioMed1 grant (PL930075).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
Present addresses: 3The Sanger Centre, Hinxton, Cambridge CB10 1SA, UK; 4Max-Planck Institute for Molecular Genetics, D-14195 Berlin (Dahlem) Germany.
-
↵6 Corresponding author. Glaxo Wellcome Medicine Research Centre, Stevenage SG1 2NY, UK.
-
E-MAIL gxx33447{at}glaxowellcome.co.uk; FAX 44 01438 763082.
-
- Received July 16, 1997.
- Accepted February 27, 1998.
- Cold Spring Harbor Laboratory Press








