
Abstract
Sequencing of large clones or small genomes is generally done by the shotgun approach (Anderson et al. 1982). This has two phases: (1) a shotgun phase in which a number of reads are generated from random subclones and assembled into contigs, followed by (2) a directed, or finishing phase in which the assembly is inspected for correctness and for various kinds of data anomalies (such as contaminant reads, unremoved vector sequence, and chimeric or deleted reads), additional data are collected to close gaps and resolve low quality regions, and editing is performed to correct assembly or base-calling errors. Finishing is currently a bottleneck in large-scale sequencing efforts, and throughput gains will depend both on reducing the need for human intervention and making it as efficient as possible. We have developed a finishing tool, consed, which attempts to implement these principles. A distinguishing feature relative to other programs is the use of error probabilities from our programs phred andphrap as an objective criterion to guide the entire finishing process. More information is available athttp://www.genome.washington.edu/consed/consed.html.
Although complete automation of data processing in shotgun sequencing is clearly desirable and may be feasible in the near future, at present finishing still requires extensive human intervention. This is customarily done by use of an interactive computer program. The program (which is usually called asequence editor) must, at a minimum, display the aligned sequences of the assembled reads and allow the user to access underlying raw data (e.g., the fluorescence trace profiles from automated sequencers) and other information that may be useful in evaluating the base calls and assembly. It should also facilitate the detection of regions where additional data are needed, help in determining reagents (e.g., sequencing primers and templates) needed to obtain these data, and allow editing to correct errors.
A good editor makes the finishing process as efficient and painless as possible. The display should indicate, with appropriate size and color emphases, the most important information about the assembly, with less important information being easily accessible with a minimum of effort, and the user should have the ability to change which information is shown, on the basis of the task at hand. Locations requiring human inspection should be efficiently pinpointed. The user manipulations required to accomplish a given task should be as natural and efficient as possible. The program should allow customization to suit individual preferences, facilitate quick detection and correction of user mistakes, and be easy to learn. It should have a quick response time and allow recovery from hardware and software problems on the users’s computer.
A number of editors are available commercially or from academic developers. The pioneering work in both assembly and editing was done by Staden, and his gap4 program (Dear and Staden 1991; Bonfield et al. 1995) remains among the best. Commercially available programs include Sequencher, DNA-Star Seqman (Swindell and Plasterer 1997), and ABI AutoAssemble.
We have developed an editor consed that is intended to be used in conjunction with several other sequence data processing programs developed by our group, including the base-calling programphred (Ewing et al. 1998), the assembler phrap (P. Green, in prep.), and the high-level assembly viewerphrapview. A key feature of these programs is their emphasis on objective criteria to measure the accuracy of sequences and assemblies. In particular, phred uses trace parameters to produce error probabilities associated to each called read base, andphrap uses these together with the read alignments to attach an error probability to each base of the inferred underlying sequence (consensus sequence) of the clone. These error probabilities (or log-transformed versions of them, which we call qualities) have proven extremely useful in increasing the accuracy of assembly and of the consensus sequence, and in avoiding the need to trim less accurate (but still useful) data from the ends of reads.
The error probabilities also provide an objective criterion to guide finishing. In our view, all sequencing should have a predefined accuracy target for the final sequence, and finishing should be directed at attaining that target. Specifically, one should start by specifying a target expected number of errors (e.g., four errors for a 40-kb cosmid, if the target error rate is one error in 10,000 bases). Following assembly of the shotgun reads, the expected number of errors in the consensus sequence is determined as the sum over all bases of the per-base error probabilities. Finishing then proceeds by identifying sequence regions that make a relatively high contribution to the expected number of errors (including gaps, which count as one error per missing base), and obtaining additional data or editing in such regions to reduce the expected number of errors in the consensus sequence, ending when it drops below the target level.
Consed is intended to implement the above finishing strategy. Its major distinctive feature relative to other editors is the use of error probabilities as the primary tool for guiding the entire finishing process. Other features include a minimalist editing philosophy; because, ultimately, all that is needed is an accurate consensus sequence, our view is that almost no read editing should be done, any errors in the consensus sequence instead being corrected by forcing it to agree with the highest quality read. The generally high accuracy of the assembly and consensus sequence produced byphrap and the discriminating ability of the error probabilities tend to make finishing in this manner quite efficient.
In view of the parallel goal of full automation, it is important that the editor play to the strengths of humans vis a vis computers. It should discourage operations by the human user that are error-prone and better carried out by computer. Human editing is generally most effective at a local level (inspection of raw data, correction of base calls, evaluation of read discrepancies). Global issues, involving the data set as a whole, are generally more appropriately handled algorithmically. A human can easily and reliably determine that two reads should not overlap because of discrepancies; however, it is much more difficult to be confident that two reads do overlap, because this requires knowledge of the entire set of reads to rule out the possibility that other joins should have been made instead. Consequently, consed implements a philosophy in which the human finisher corrects a misassembly by indicating that certain reads have been incorrectly assembled together; reassembly to correct this error is then carried out by phrap.
Finishing in Consed
The process of finishing can, in principle, be divided into three stages: viewing the assembly and data for the purpose of deciding where additional data or editing are necessary or identifying other anomalies; obtaining additional read data; and editing to correct errors in the assembly or consensus sequence. It is most efficient to carry these out in that order, although in practice several iterations of the process may be required (particularly with large projects).
As indicated above, our preferred finishing strategy is focused on the goal of producing a final sequence for which the expected number of errors is below a predetermined target level. Consed has been designed with this approach in mind, but has evolved into a flexible tool that permits a variety of other strategies. In fact, it can and is being used for purposes other than finishing, such as polymorphism detection in regions sequenced from multiple individuals (Nickerson et al. 1997).
Finishing, Stage 1: Viewing and Navigating Assemblies and Traces
INPUT FILES
Consed requires three main types of data input files: chromatogram files (one for each read), containing the fluorescence trace profiles; phd files (one for each read), created initially byphred or another base-calling program and containing the base calls, quality values, and trace peak positions for the read bases, and any tags attached to the read; and an .ace file, created initially byphrap or another assembly program and containing assembly information including the contig sequences and quality values, tags attached to the contig sequences, and read alignment information. It is assumed that the quality values are related to error probabilities by the transformationq = −10 × log10(p), in whichq is the quality value and p is the estimated error probability for a read or contig base. Tags are annotations (e.g., sequencing vector) relevant to the assembly, which may have been generated either automatically by the assembly or base-calling program or in a previous editing session, that are attached to segments of the read or contig sequence. Information about read chemistry and template identifiers may be recorded either in the read name, or in the form of tags.
Consed retains multiple versions of the phd files and .ace file, with version 1 being the original phred- orphrap-generated file, and subsequent versions incorporating the edits made in particular consed sessions. Chromatogram files may be in ABI format or standard chromatogram format (SCF) (Dear and Staden 1992), either compressed or uncompressed.
VIEWING ASSEMBLIES
On startup, consed displays a popup window indicating the available .ace files. The user selects one of these, whereuponconsed reads it and the associated phd files, and displays acontig selection window containing a list of contigs and a list of reads. Double clicking on a contig or read name brings up the corresponding contig in the aligned reads window (Fig.1), showing the sequences of the contig and of the aligned reads. Clicking on a base with the left mouse button sets the cursor and causes the numeric quality value of a base to be displayed.
Aligned reads window, in color means quality and tags color mode. The top line gives the contig sequence, and below it are the read sequences for the top strand (right-pointing arrows) and bottom strand (left-pointing arrows). Read names are in yellow. The background gray scale indicates base quality, with the highest quality being white and the lowest quality black. Red indicates a character (such as the x or *) that disagrees with the contig sequence. (x) A base that has been masked by cross-match as being vector. (*) A pad that is inserted by phrap to align reads that have insertions and deletions. Tags are indicated by colored bars covering the bottom half of the background square for each base. The blue tag represents sequencing vector, and the orange tag indicates compressions. An edited base has a green tag attached. Gray letters on a black background indicate that phrap clipped these bases off because of low quality.
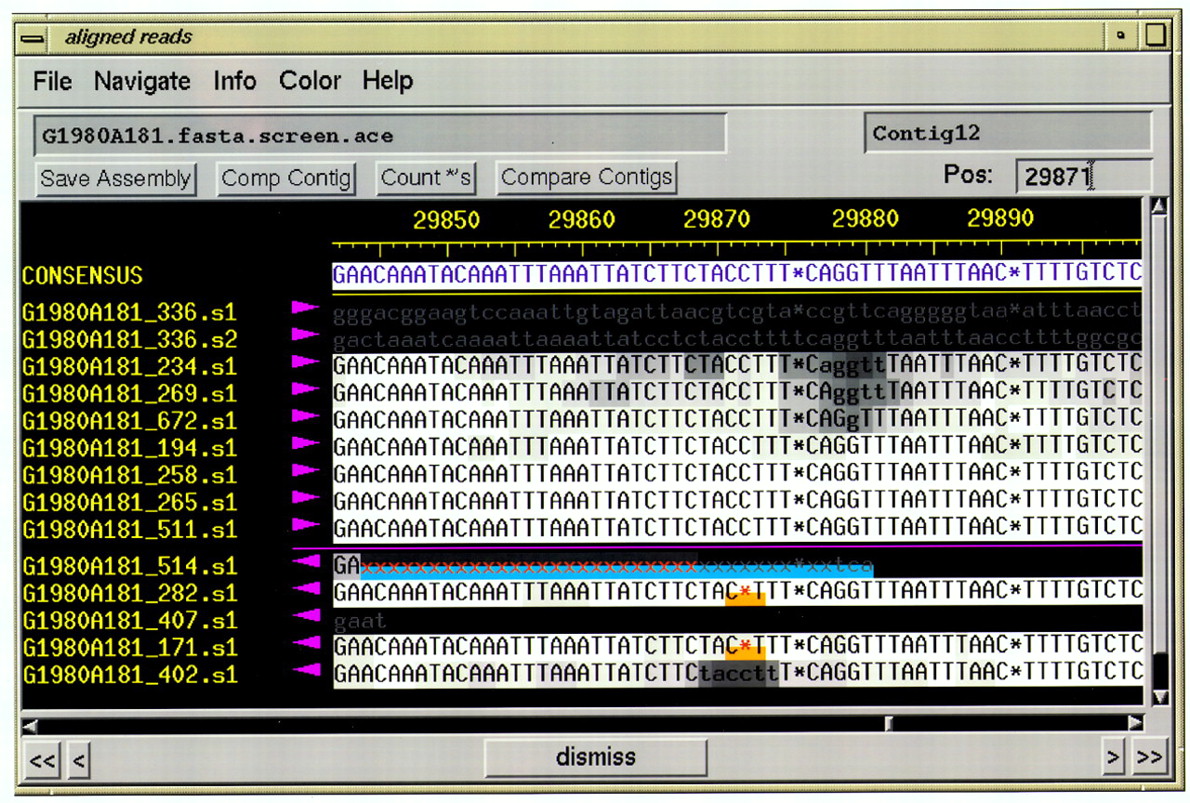
The aligned reads window can be shown in any of several possible color modes, each of which emphasizes different kinds of information. In the color means quality and tags color mode (Fig. 1), color is used to indicate base qualities, discrepancies between read and contig bases, tags, and the low quality or unaligned tails of reads (as determined by phrap).
In the color means match color mode, color indicates read/contig discrepancies, read bases used by phrap to create the contig sequence, and bases trimmed by phrap. Other color modes include color means quality (like color means quality and tags but omitting tags) and color means edits (which indicates edits and read/consensus discrepancies). The user can easily switch between color modes using a menu button and change the criteria used for graying the unaligned or low-quality tails of reads.
NAVIGATION
The aligned reads window is initially positioned at the beginning of the contig (if a contig was double-clicked in the contig selection window) or at the beginning or end of the high-quality part of a read (if a read was double-clicked). From there the user can move to other contig locations by scrolling, by typing in the base number of a particular location, by using another program that communicates withconsed and sends it locations to scroll to (such as thephrapview graphical assembly viewer), or by using thenavigate window (Fig. 2). The latter is very useful for directing the user’s attention to regions requiring inspection because of potential misassemblies, low quality data, or other problems. A list of positions of features of any of several different types may be generated automatically by consed, or by another program and read in by consed; the user can then either double click on a list item to move to that location, or move to each item in succession by clicking on Next or Previous buttons. The list can be saved to a file for printing or filtering by another program.
Navigation window. Each line contains the contig name, the read name, the range of consensus positions, and an indication of the problem. The Go, Prev, and Next buttons cause the associated aligned reads window to scroll to the location on the currently highlighted line, the line above it, or the line below it, respectively. All items can be visited in order by repeatedly clicking Next. The Save button creates a file containing the list.
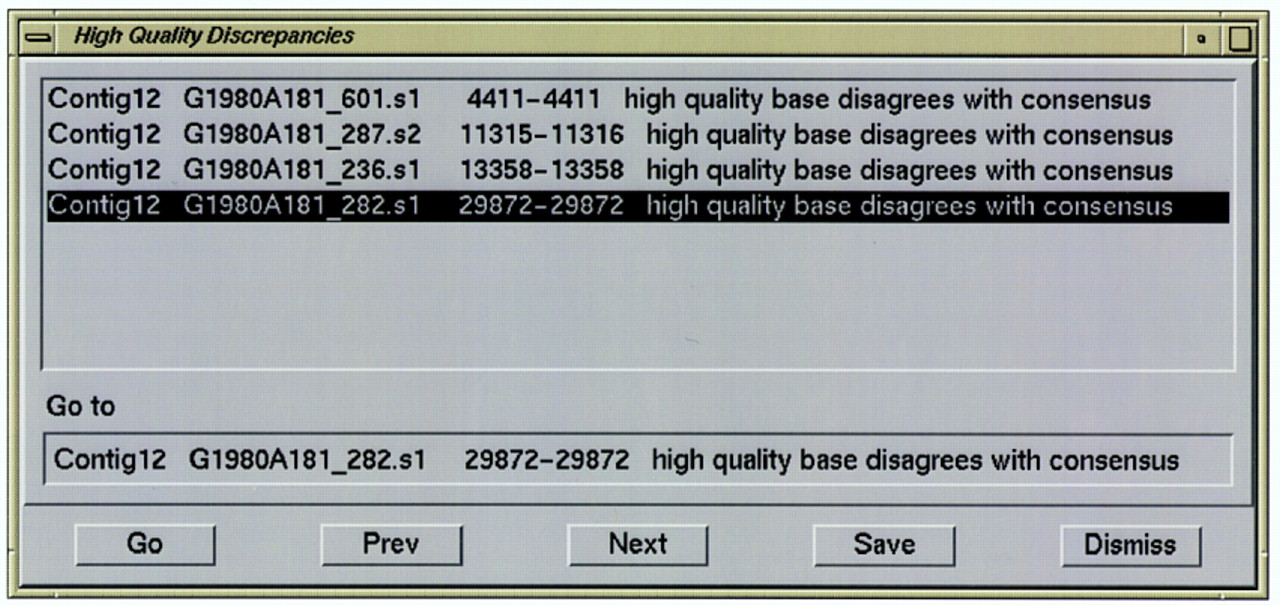
The types of features that can currently be listed and navigated include
1.
Low-quality regions in the contig sequence, defined as regions having a high expected number of errors. This is the preferred method for finding regions requiring additional data.
2.
High-quality read bases that are aligned with the contig sequence but disagree with it and are not tagged as chimeric, sequencingVector, or contaminant. This is the preferred method for finding regions requiring editing (because of misassembly or contig sequence errors).
3.
Unaligned regions of reads that are not contained in the low-quality read tail.
4.
Occurrences of a particular string of bases, either in the entire set of reads, or in the contig sequences.
5.
Occurrences of a particular tag type.
6.
Bases that have been edited.
VIEWING TRACES
Clicking on a read base with the middle mouse button displays the trace of the read in the trace window (Fig.3); the cursor blinks in both the trace window and aligned read window at corresponding locations. Clicking on a different read causes its trace to be brought up and the previously displayed traces to be aligned to it. Clicking on a different base of a read for which the trace is already displayed causes the trace window to scroll to the new position. At the user’s option, the traces of different reads will either scroll together or separately. To make it easy to compare traces from different reads at the same position, a silver vertical line in each trace indicates the position of the peak that corresponds to the cursor location in the aligned reads window.
Trace window. The lines in each panel above the trace chromatogram indicate the following: (con) consensus position; (rd) read position; (con) consensus bases; (edt) editable read bases; (phd) phredbase calls; and (ABI) ABI base calls. The H and V scale bars change the horizontal and vertical magnification of the traces. (Scroll together Yes/No buttons) Allows the user to scroll the traces individually or locked together. (Remove) Removes this trace panel from the window. (Undo) Undo the last edit operation on that read.
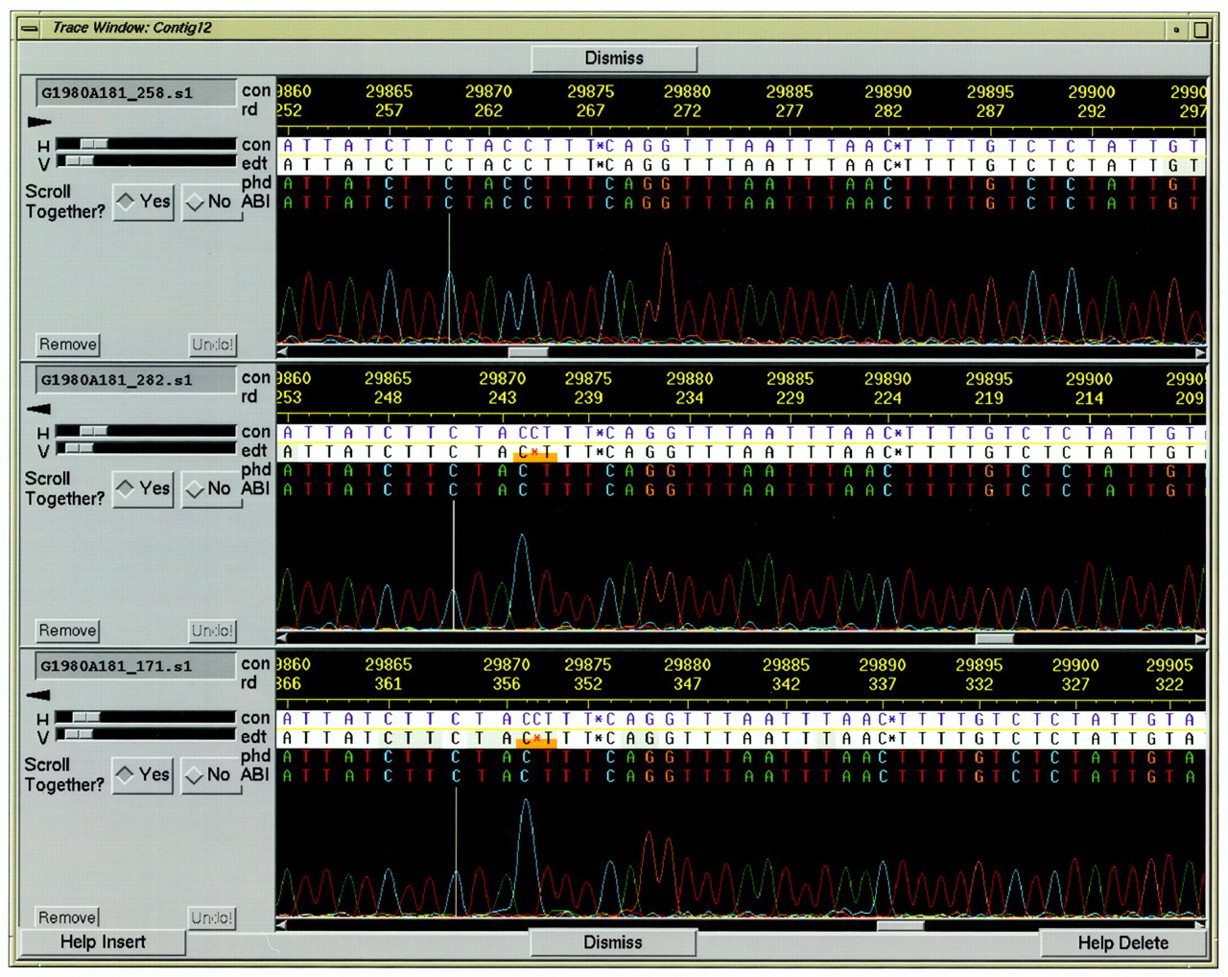
Clicking the middle mouse button on a contig sequence base brings up four traces at once: the highest quality top strand and bottom strand reads that agree with the contig sequence, and the highest quality top and bottom strand reads that disagree with the contig sequence. These are generally the most relevant traces for making a decision about the correctness of the contig sequence at that location.
COMPARING CONTIGS
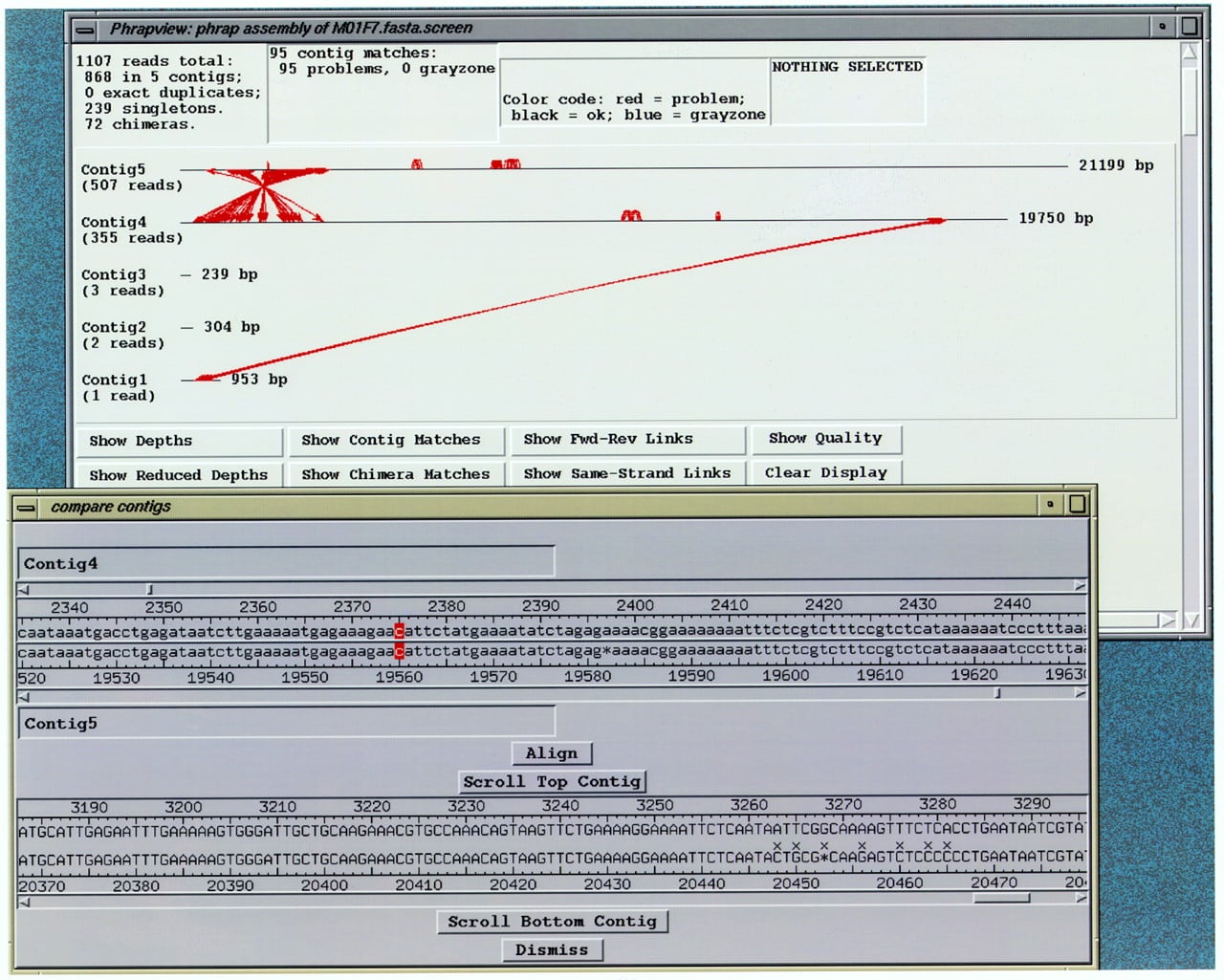
The user may select two regions in the same or different contigs, to be aligned by a pinned Smith–Waterman algorithm (Smith and Waterman 1981) and shown in the compare contigs window (Fig.4). Traces for the reads in those regions can then be displayed. This is useful for investigating possible joins not made by the assembly program.
Compare contigs window, indicating an alignment selected to investigate a contig match indicated in phrapview. (Top): Aphrapview window showing matches between contigs as red lines. (Bottom) consed compare contigs window showing the sequence alignment of one match from the phrapview window. The two rows of bases in lowercase are the unaligned contigs, which can be scrolled relative to each other. The two rows of bases in uppercase are the aligned contigs, which are locked together if they are scrolled. (x) A mismatch. Red cursors indicate the bases to be pinned together. (Align button) Click this to show alignment. (Scroll Top Contig/Scroll Bottom Contig buttons) After clicking on a base, this causes the Aligned reads window to scroll to the appropriate location.
TAGS FOR ANNOTATING PROBLEMS
During inspection of the assembly, the user may wish to tag certain regions of reads or of the contig sequence to indicate problems that have been identified and/or places where additional data are needed. Read tags currently available for this purpose includechimera, cloningVector, sequencingVector, compression,and contaminant. Tags that can be attached to the contig sequence include repeat, cloningVector, dataNeeded,and polymorphism.
Finishing, Stage 2: Obtaining More Data
In consed’s approach to finishing, additional data collection involves the specification of one or more experiments. Each experiment corresponds to a sequencing reaction that will be carried out to obtain new read data and includes a chemistry (dye primer or dye terminator), a template (which currently must be the full clone), and a priming site in the template sequence.
In accordance with the philosophy of guiding finishing by the objective criterion of reducing the expected number of errors below a target level, consed attempts to determine a minimal set of experiments most likely to significantly reduce the number of errors, and presents these to the user for approval or revision. A key assumption here is that if a read through a particular site has already been obtained, then an additional read through that site is not likely to reduce the expected number of errors unless it is on the opposite strand, or uses a different chemistry; this is because reads with the same strand and chemistry tend to have similar error profiles, whereas opposite-strand or opposite-chemistry reads tend to have independent error profiles.
For each potential experiment, the expected error reduction(or simply error count) is defined to be the expected number of errors in that part of the contig sequence that (1) would be covered by the potential high quality part of the (intended) read, and (2) are not already covered by the potential high quality part of an existing read of the same chemistry and strand. By default, the potential high quality part consists of positions 50 to 300 of the read. Each potential high-quality read base extending into a gap contributes one to the error count.
All potential experiments whose error count exceeds 0.1 (by default) are considered. First, the primer criteria below are applied and the experiment is eliminated if the thresholds are not met. The remaining experiments are then processed as follows, either by presentation to the finisher for consideration, or (optionally) in automatic mode. The experiment with the greatest error count is presented first. If accepted, the following occur: (1) the expected number of errors for the contig is reduced by the error count of the selected experiment; (2) the selected experiment is deleted from the list and printed; (3) any experiment whose potential high-quality part overlaps that of the selected experiment has its error count adjusted downward; and (4) the experiment with the greatest (revised) error count is found. This entire process is iterated until the expected number of errors for the contig drops below the target level (default contig length times 0.0001). If the list of experiments is exhausted before the target level of contig errors is reached, a second list of experiments is made in which the expected error reduction no longer discounts experiments of the same chemistry and strand as an existing read, and this list is processed as before.
The criteria used to evaluate possible primers are as follows:
1.
Primer melting temperature is in an acceptable range (default 50°C–55°C). Melting temperature is computed with the nearest-neighbor formula of Breslauer (1986) and Rychlik (1990).
2.
Every primer base has a quality value at or above the threshold (default 30).
3.
The primer has low propensity to anneal elsewhere on the sequencing template. The user must specify a template type, either subclone, in which case consed checks for possible annealing within 3000 bases (by default) from the site of the primer, or clone, in which caseconsed checks for possible annealing anywhere in the entire assembly (all contigs). Potential annealing within the appropriate clone or subclone vector sequence (provided by the user in an appropriate file) is also tested.
4.
The algorithm used to test potential false annealing aligns the primer against each potential priming site in the permitted range other than the correct site, and scores the site as follows: Starting at the 3′ end, +2 is awarded for each A/T pair, +3 for a G/C pair, and −6 for a mismatch (gaps are not considered). The maximum attained score (over all primer subsequences ending at the 3′ end) is taken to be the score of the site. A primer is rejected if some site other than the correct one has a score exceeding 17 (by default). In practice it is this screen that eliminates the largest number of candidate primers.
5.
The primer has low self-annealing propensity. For each possible ungapped alignment of the primer and its complement, annealing propensity is scored with an algorithm that is identical to the one above with the exception that −1 is added for each base at the 3′ end of the primer that extends past the end of the complement. A primer is rejected if it anneals to itself with a score exceeding 3 (default).
Primers may also be selected by specifying a particular region in which additional read data are desired, by using the cursor to indicate the region. Consed then considers all primers that could be used to obtain reads spanning part of the region, and displays as above a list of such primers.
The user can select a primer from the list returned byconsed by clicking an accept primer button, causing a primer tag containing all relevant information about that experiment to be automatically created and named, attached to the contig sequence, and (like other contig sequence tags) written to the .ace file where the information is accessible to other programs (e.g., a script that submits an e-mail order for an oligonucleotide or creates a work schedule for a technician). To test the effectiveness of the primer picking criteria, we tracked the first 98 primers that were chosen for cosmid sequencing reactions using consed at the University of Washington Genome Center. All were successful.
Finishing, Stage 3: Editing
Edits are used to correct base-calling errors and assembly errors, and create or change tags. Errors in the contig sequence can usually be corrected by finding a read that appears to have the correct sequence and marking it with a becomeConsensus tag to force the contig sequence to agree with it. If no read appears correct, but one of them has adequate trace quality to infer the correct sequence, then the base-calling errors in that read can be corrected by the customary operations (overstrike, insert, and deletion of bases) before marking it with the becomeConsensus tag. Other reads may be edited if desired to bring them into conformity with the consensus, but this is not required. An edit tag is created for each edit that is performed.
Assembly errors are corrected by marking reads in the region of the error with one of the following assembler directive tags, and reassembling by use of phrap. ThesignificantDiscrepancy tag is used to break an incorrect join. Two reads that have regions marked with this tag and have a discrepancy within the region will not be joined on reassembly withphrap. The ignoreMismatches tag directsphrap to ignore mismatches with other reads in this region, thus allowing joins that might otherwise be rejected. TheignoreMatches tag directs phrap to ignore matches with other reads in this region, causing it to reject joins that would involve this region alone.
After reassembly, contig tags are transferred to the new assembly by use of a script that first runs cross_match(P. Green, in prep.) to generate a map relating the old contigs to the new contigs, and then creates an image of each old tag in the appropriate new contig. This is useful in cases where it is necessary to start annotating the sequence before it is finished.
Other Program Features
CUSTOMIZATION
Extensive customization of program features is possible inconsed. During the editing session, users can modify various parameters, such as quality thresholds or primer selection parameters, through pop-up windows. A number of features can be modified prior to the session by supplying appropriate files. As noted above, custom navigation of a list of sites generated by another program can be performed; similarly a custom list of experiments can be provided for review within consed. The user may define his own tag types and/or automatically add tags to the .ace or phd files using another program. For example, the program Polyphred (Nickerson et al. 1997) annotates polymorphism sites in this way.
In addition, a large number of features can be modified by means of a user-configurable X resource file that is read at program startup. Fonts and colors can be set for most graphical items in each colormode, including window backgrounds, tags, traces, and bases. Default values for a number of parameters can be set here as well, including the thresholds for low consensus quality and the threshold for high quality discrepancy. A list of all configurable X resources is obtainable using the Show X Resources menu item from the Info menu in the main window. There are hot keys to make some operations more efficient.
The Aligned reads window in consed can be made to scroll to particular locations specified by a separate program, by use of a message sent via a Berkeley socket connection. This feature has been used with phrapview, for example. It does not interfere with normal consed response to user actions.
Consed can be used for applications other than genome sequence assembly that require inspection of alignments and traces, for example, polymorphism detection and genotype determination. For the latter purpose, several additional tag types are available, includinghomozygote and heterozygote tags.
CRASH RECOVERY/DATA INTEGRITY
To allow for recovery of the edits in the event of hardware or software failures or operator errors, consed maintains a log file of edits up to the instant before the crash, which can optionally be replayed automatically when the program is restarted. In cases of extensive misediting, the use of multiple versions for phd and .ace files makes it possible to go back to the results of earlier editing sessions. The user can also undo single and multiple edits made since the last “save.”
PROGRAM DEVELOPMENT AND AVAILABILITY
The original version of consed was written by Chris Abajian and David Gordon, with subsequent development by David Gordon, who currently maintains and distributes it. It is written in object-oriented style in the C++ computer language, using the Rogue Wave tools.h++ class library (http://www.roguewave.com), and the Motif and X11 graphics libraries. Executables are available for the following platforms: DEC Alpha (Digital Unix, Linux/Alpha), Silicon Graphics (Irix), Hewlett-Packard (HP-UX), and SUN (Solaris, SunOS).Consed is available at no charge to academic users for research purposes, and by commercial license from the University of Washington to other users; contact David Gordon at[email protected]. More information is available athttp://www.genome.washington.edu/consed/consed.html.
Consed is now in active use at over 80 sites in 20 countries, including biotech, chemical, pharmaceutical, and agricultural companies; genome centers; small academic laboratories; and government laboratories.
This work was partly supported by grants from the Department of Energy and the National Human Genome Research Institute. We are grateful to many users for contributing ideas and suggestions and particularly thank Lee Rowen, Pat Minx, and Bob Waterston.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Notes
[1] Present address: Geospiza, Inc., Seattle, Washington 98107 USA.
[2] Corresponding authors.
Notes
[3] E-MAIL [email protected]; [email protected]; FAX (206) 685-7344.
REFERENCES
- ↵S. AndersonM.H. de BruijnA.R. CoulsonI.C. EperonF. SangerI.G. Young (1982) Complete sequence of bovine mitochondrial DNA. Conserved features of the mammalian mitochondrial genome. J. Mol. Biol. 156:683–717.
- ↵J.K. BonfieldK.F. SmithR. Staden (1995) A new DNA sequence assembly program. Nucleic Acids Res. 23:4992–4999.
- ↵K.J. BreslauerR. FrankH. BlockerL.A. Marky (1986) Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. 83:3746–3750.
- ↵S. DearR. Staden (1991) A sequence assembly and editing program for efficient management of large projects. Nucleic Acids Res. 19:3907–3911.
- ↵S. DearR. Staden (1992) A standard file format for data from DNA sequencing instruments. DNA Sequence 3:107–110.
- ↵Ewing, B., L. Hillier, M. Wendl, and P. Green. 1998. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. (this issue).
- ↵D.A. NickersonV.O. TobeS.L. Taylor (1997) PolyPhred: Automating the detection and genotyping of single nucleotide substitutions using fluorescense-based resequencing. Nucleic Acids Res. 25:2745–2751.
- ↵W. RychlikW.J. SpencerR.E. Rhoads (1990) Optimization of the annealing temperature for DNA amplification in vitro. Nucleic Acids Res. 18:6409–6412.
- ↵T.F. SmithM.S. Waterman (1981) Identification of common molecular subsequences. J. Mol. Biol. 147:195–197.
- ↵S.R. SwindellT.N. Plasterer (1997) SEQMAN: Contig assembly. Meth. Mol. Biol. 70:75–89.