
Base-Calling of Automated Sequencer Traces UsingPhred. I. Accuracy Assessment
Abstract
The availability of massive amounts of DNA sequence information has begun to revolutionize the practice of biology. As a result, current large-scale sequencing output, while impressive, is not adequate to keep pace with growing demand and, in particular, is far short of what will be required to obtain the 3-billion-base human genome sequence by the target date of 2005. To reach this goal, improved automation will be essential, and it is particularly important that human involvement in sequence data processing be significantly reduced or eliminated. Progress in this respect will require both improved accuracy of the data processing software and reliable accuracy measures to reduce the need for human involvement in error correction and make human review more efficient. Here, we describe one step toward that goal: a base-calling program for automated sequencer traces,phred, with improved accuracy. phred appears to be the first base-calling program to achieve a lower error rate than the ABI software, averaging 40%–50% fewer errors in the data sets examined independent of position in read, machine running conditions, or sequencing chemistry.
Overview of Sequence Data Processing
At present, nearly all DNA sequencing is done using the enzymatic dideoxy chain-termination method of Sanger (Sanger et al. 1977). One starts with a purified DNA template of interest (usually in single-stranded form) and an oligonucleotide primer complementary to a specific site on the template strand. For each of the 4 bases (A, C, G, T), a reaction is carried out in which DNA polymerase synthesizes a population of labeled single-stranded fragments of varying lengths, each of which is complementary to a segment of the template strand and extends from the primer to an occurrence of that base. These fragments are then separated according to length by gel electrophoresis, whereupon their relative sizes, together with the identity of the final base of each fragment, allow the base sequence of the template to be inferred.
In automated sequencing (Smith et al. 1986), the fragments are labeled with fluorescent dyes attached either to the primer (dye primer chemistry) or to the dideoxy chain-terminating nucleotide (dye terminator chemistry) (Prober et al. 1987). Typically a different dye is used for each of the four reactions, so that they can be combined and run in a single gel lane (in the case of dye–terminator chemistry, this also allows all four reactions to be carried out in a single tube). Multiple templates (36 or more at a time) are analyzed in separate lanes on the same gel. At the bottom of the gel, a laser excites the fluorescent dyes in the fragments as they pass, and detectors collect the emission intensities at four different wavelengths. The laser and detectors scan the bottom of the gel continuously during electrophoresis in order to build a gel image in which each lane has a ladder-like pattern of bands of four different colors, each band corresponding to the fragments of a particular length.
Computer analysis is then used to convert the gel image to an inferred base sequence (or read) for each template. Typically this analysis consists of four distinct steps: lane tracking, in which the gel lane boundaries are identified; lane profiling, in which each of the four signals is summed across the lane width to create a profile, or trace, consisting of a set of four arrays indicating signal intensities at several thousand uniformly spaced time points during the gel run; trace processing, in which signal processing methods are used to deconvolve and smooth the signal estimates, reduce noise, and correct for dye effects on fragment mobility and for long-range electrophoretic trends; and base-calling, in which the processed trace is translated into a sequence of bases. This paper focuses only on the last step.
The processed traces are usually displayed in the form of chromatograms consisting of four curves of different colors, each curve representing the signal for one of the four bases and drawn left to right in the direction of increasing time to detection (increasing fragment size). An idealized trace would consist of evenly spaced, nonoverlapping peaks, each corresponding to the labeled fragments that terminate at a particular base in the sequenced strand. Real traces deviate from this ideal for a variety of reasons having to do with imperfections of the sequencing reactions, of gel electrophoresis, and of trace processing. Because of anomalous migration of very short fragments (caused by relatively greater effects of the dye and specific base sequence on mobility) and unreacted dye–primer or dye–terminator molecules, the first 50 or so peaks of a trace are noisy and unevenly spaced. Toward the end of the trace, the peaks become progressively less evenly spaced as a result of less accurate trace processing, less well resolved as diffusion effects increase and the relative mass difference between successive fragments decreases, and more difficult to distinguish from noise as the number of labeled fragment molecules of a given size decreases. In particular, poorly resolved peaks for the same base may yield a single broad, often lumpy peak.
In better resolved regions of the trace, the most commonly seen electrophoretic anomalies are compressions (Sanger and Coulson 1975;Sanger et al. 1977), which occur when bases near the end of a single-stranded fragment bind to a complementary upstream region, creating a hairpin-like structure that migrates through the gel more rapidly than expected from its length, thus causing a peak to be shifted left of its expected position. This can result in one peak being beneath another or in two successive peaks for the same base being merged into one. GC-rich sequence tends to be more compression prone than AT-rich sequence because of the greater likelihood of stable hairpins. Dye–terminator chemistry appears to resolve most compressions (Lee et al. 1992), possibly because the dye on the terminal nucleotide interferes with base-pairing or because of the use of deoxyinosine triphosphate in place of deoxyguanosine triphosphate; but this chemistry has its own data quality problems caused by reduced polymerase affinity for the dye-labeled terminal nucleotide, one particular problem being a substantial decrease in the signal for a G base following an A (Lee et al. 1992; Parker et al. 1996).
Other frequently seen problems include weak or variable signal strength and noise peaks not corresponding to a base. These can result from variation in the efficiency of chain elongation or termination by the polymerase because of sequence context effects or inefficient incorporation of the dideoxy nucleotide, from impure or degraded reagents, or from laboratory protocol errors. Secondary structure in the template can produce polymerase stops, resulting in a pileup of peaks at one point in the trace. The signal downstream of a run of mononucleotide or dinucleotide repeats frequently is degraded, possibly because of strand slippage during copying by the polymerase.
The goal of base-calling software is to produce a sequence as accurate as possible in the face of the above data problems. Some of the earliest base-calling software was part of the processing software installed on the first ABI sequencing machines (Connell et al. 1987). That software achieves impressive accuracy and remains the standard against which other methods must be judged. Although full algorithmic details have not been published, according to a recent ABI description of its base-calling software (ABI 1996), the program uses mobility curves to predict the peak spacing, and identifies the most likely peak in intervals of the nominal peak spacing, assigning an N in the absence of a good choice. Subsequently, it adds and removes bases using a criterion involving the uniformity of peak spacing.
The advent of high volume sequencing has prompted development of other programs (Giddings et al. 1993; Golden et al. 1993, 1995; Berno 1996). These all perform multiple gel image processing steps including base-calling and have the merit of allowing efficient centralized processing of the data on a computer independent of the sequencing machine; however, none of them report equaling the accuracy of the ABI software.
This paper and the following one describe a computer programphred that performs base-calling and assigns an error probability to each called base. Accuracy is shown to exceed that of the ABI base-caller.
METHODS
Base-Calling Algorithm
Overview
The phred base-caller uses a four-phase procedure to determine a sequence of base-calls from the processed trace. In the first phase, idealized peak locations (predicted peaks) are determined; the idea is to use the fact that fragments are locally relatively evenly spaced, on average, in most regions of the gel, to determine the correct number of bases and their idealized evenly spaced locations in regions where the peaks are not well resolved, noisy, or displaced (as in compressions). In the second phase, observed peaks are identified in the trace. In the third phase, observed peaks are matched to the predicted peak locations, omitting some peaks and splitting others; as each observed peak comes from a specific array and is thus associated with 1 of the 4 bases, the ordered list of matched observed peaks determines a base sequence for the trace. In the final phase, the uncalled (i.e., unmatched) observed peaks are checked for any peak that appears to represent a base but could not be assigned to a predicted peak in the third phase, and, if found, the corresponding base is inserted into the read sequence. The entire procedure is rapid, taking less than half a second per trace on typical workstations.
Below, we describe in greater detail the ideas involved in the above four phases. Although the basic ideas are simple, the full implementation is a complex, somewhat inelegant rule-based procedure, that has been arrived at empirically by progressively refining the algorithms on the basis of examining performance on particular cosmid data sets [generated early in theCaenorhabditis elegans sequencing project (Sulston et al. 1992) and distinct from the ones used for the accuracy testing in this paper]. Specific parameter values and algorithmic details not provided here may be found in the source code, which is available from the authors.
Phred takes as input chromatogram files, in ABI format or Standard Chromatogram Format (SCF) (Dear and Staden 1992), containing the processed trace data; currently traces produced either by the ABI analysis software, or by our own lane processing programplan (B. Ewing and P. Green, in prep.) may be used. Prior to peak prediction, simple additional processing of the trace is performed. The trace array amplitudes are first normalized by summing the dominant peak areas of each array from point 1500 to 2500 and scaling to the smallest of the four average peak areas. From the four normalized trace arrays, a skyline projection is then created by taking the maximum of the four array values at each point in the trace.
Phase 1: Locating Predicted Peaks
Peak prediction attempts to find the idealized locations of the base peaks, using simple Fourier methods. This, in effect, supplies a peak spacing criterion that, in conjunction with peak size considerations, can help discriminate noise peaks from true ones and resolve groups of merged peaks.
Peak prediction begins by examining the four trace arrays to detect peaks. (These are used only as a temporary tool for peak prediction and do not necessarily correspond to the observed peaks defined in the next section.) A detected peak is identified as the location of the maximum value, or, if the maximum does not exist, the midpoint, between a pair of inflection points. The peak is retained only if its height exceeds 10% of the height of the previous peak and is greater than the heights of the other three arrays at the same position. To minimize the effects of varying peak heights for the Fourier analyses described below, a synthetic trace is then constructed as a frequency modulated symmetric square wave with values of 1.0 and −1.0, such that each positive peak of the square wave is centered on a detected peak location and has width equal to one fourth of the peak-to-peak spacing; in particular the synthetic trace has the same peak locations as the original (processed) trace.
The processed trace is then scanned to find regions of uniform peak spacing, where a region is defined to be a window of 200 trace points. For each region centered on a detected peak, the peak period (peak-to-peak spacing) values are determined for all pairs of adjacent detected peaks within the region, and the mean and standard deviation of these periods are determined. Any region for which the mean-scaled standard deviation is <0.45 is designated as having well-defined spacing. The region with the lowest mean-scaled standard deviation is assumed to have the most uniformly spaced peaks and is selected as the starting region (S).
Finding the set of predicted peaks for the trace relies on repeated application of an algorithm that, given a region (R) and a set of permitted periods, finds a predicted peak locationpeakR near the center of the region together with an estimated period θ R chosen from the permitted set. This is done as follows. First a damped synthetic trace is constructed by multiplying the value at each point in the synthetic trace by a symmetric triangular filter that has value 1.0 at the region midpoint and 0 outside the region, and is linear between the midpoint and each edge of the region; this effectively weights trace points according to their distance from the midpoint, with points near the midpoint getting the highest weight. Then, among all sine waves having a period in the permitted set, the one having the largest inner product with the damped trace is found, using simple Fourier methods (Press et al. 1988). This sine wave can be thought of as the one that best approximates the damped trace. The location of the peak in this sine wave that is nearest to the center point of the region is then taken as the predicted peak peakR, and the period of the sine wave is taken as the estimated periodθ R.
The predicted peaks for the trace are found by iteratively applying the above algorithm, proceeding first from the starting region toward the end of the trace, and then from the starting region toward the beginning of the trace. Specifically:
- 1.
- Find the predicted peak peakS and periodθS associated to the starting regionS, using the above procedure (in this case the set of permitted periods consists of all possible periods, and the fast Fourier transform is computed to find the optimal periodθS).
- 2.
- Set the region R to S, and the direction dto “rightward”.
- 3.
- Shift R in the direction d by an amountθR, and relabel this new region asR. If R has well-defined spacing and d is “rightward,” take the set of permitted periods to be {θR − 0.03,θR,θR + 0.03}; if Rhas well-defined spacing and d is “leftward,” take the set of permitted periods to be {θR − 0.25,θR − 0.20,θR − 0.15,...,θR + 0.25}; if R does not have well-defined spacing, take this set to be {θR}.
- 4.
- Find peakR and θR. If the end of the trace has been reached, go to step 5; otherwise go to step 3.
- 5.
- If d is “leftward,” stop. Otherwise set R toS and d to “leftward,” and go to step 3.
Phase 2: Locating Observed Peaks
Observed peaks are found by scanning the four trace arrays for regions that are concave, that is, satisfy 2 × v(i) ⩾ v(i + 1) + v(i − 1) where v(i) is the trace value at point i.For each such region, the trace values are summed to estimate the peak area. If this area exceeds 10% of the average area of the preceding 10 accepted observed peaks and 5% of the area of the immediately preceding peak, it is accepted as an observed peak; otherwise it is ignored. The ratio of the peak area to the ten peak average is stored as the relative area of the peak.
The location of the observed peak is taken to be the position in the trace where the peak area is bisected. Because in some cases, a single peak may later need to be split into two, three, or four virtual peaks, the locations of the positions that trisect, quadrisect, or pentisect the area are also found.
Phase 3: Matching Observed and Predicted Peaks
Peak matching consists of assigning an observed peak to each predicted peak. This is the most complex part of the base-calling procedure and consists of three stages: finding easy matches (called fixed peaks); using a dynamic programming algorithm to align observed and predicted peaks that were not matched in stage one; and matching observed peaks that were not assigned in the first two stages but appear to represent genuine bases.
It is useful to define the shift of an observed peak relative to a particular predicted peak as the distance between their locations, divided by the period of the predicted peak. The shift may be positive, negative, or zero, with, for example, a positive shift value indicating that the observed peak lies to the left of the predicted peak. The shift change is the difference between the shifts calculated for an observed–predicted peak pair and the adjacent observed–predicted peak pair; this is relevant in compressions, where several observed–predicted pairs may have large shifts, but small shift changes. For notational clarity in the following, we also make the following definitions: The observed peak that is assigned as the called peak associated to a predicted peak is denoted theobs_peak; prior to the final choice of the obs_peak, thebest_obs_peakis a working peak associated to the predicted peak; and thebest_uncalled_peakis an observed peak associated to the predicted peak but not called.
For each predicted peak all observed peaks (if any) are found that are closer to that predicted peak than to any other, and of these the one with the largest relative area is designated thebest_obs _peakof the predicted peak. The observed peak relative area values are then recalculated using the running average area of the 10 preceding assignedbest_obs_peaks.
In the first stage of peak matching, for any group of four or more consecutive predicted peaks that havebest_obs_peaks with relative areas >0.2 and shifts between −0.2 and 0.2, the corresponding observed peaks are designated as fixed and assigned to the predicted peaks.
For the second stage, each observed peak not assigned in stage 1 and having relative area greater than 0.1, and each predicted peak to which no observed peak was assigned in stage 1, is considered. Each possible pairing of an unused observed peak with an unused predicted peak is assigned a score calculated as the observed peak area, times a penalty factor <1 that takes into account the direction and magnitude of the shift. Right shifts are penalized more than left shifts, reflecting the fact that fragments may migrate faster but not slower than their idealized rate; for example, a left shift of 0.5 scales the area by 0.95, whereas a right shift of −0.5 scales the area by 0.85. Large shifts are disallowed. A modified dynamic programming algorithm is then used to find the alignment of observed and predicted peaks having the highest total score, subject to the constraints that each shift be in the allowed range and each shift change be <0.7. A single observed peak may be split into as many as four observed peaks assigned to consecutive predicted peaks. Splitting is disallowed when the observed peak relative area falls below 1.6 and when the shift of an observed peak component falls outside the range −0.5 ≤ shift ≤ 2.1.
The third stage has two parts. In the first part, for each observed peak that was not assigned to a predicted peak in the first or second stage, phred checks whether the nearest predicted peak has an assigned observed peak. If so, the observed peak is assigned as thebest_uncalled_peakof the predicted peak (unless a larger peak has already been so assigned). If not, phred assigns it as thebest_obs_peakof the predicted peak, unless the predicted peak already has abest_obs_peakwith larger relative area; in the latter case phred assigns the observed peak as thebest_uncalled_peakunless there is already a larger assignedbest_uncalled_peak.
In the second part, any predicted peak without an assigned observed peak is checked to see whether it has abest_obs_peak not already assigned to any predicted peak. If it does, thisbest_obs_peakis assigned as the obs_peak for the predicted peak. If thebest_obs_peakhas already been assigned, theobs_peak of an adjacent predicted peak is checked to see whether it has a relative area exceeding the minimum value for splitting and has not been split the maximum number of times; if so, it is split and assigned to the predicted peak.
If no suitable observed peak can be assigned to a predicted peak, the corresponding base-call is defined to be N. This occurs very rarely.
Phase 4: Finding Missed Peaks
Occasionally, following completion of the above three phases there remain one or more well-resolved observed peaks that clearly represent bases but have not been assigned to predicted peaks. This can occur when a severe compression, extensive noise, or a lane processing aberration interferes with peak prediction, resulting in underestimation of the number of peaks in a region so that some observed peaks have no free predicted peak to which they can be assigned. To recover such peaks, following the matching phase each remaining unmatched observed peak is checked to see whether it (1) has the largest of the four signals at its time point, (2) meets a minimum size criterion, (3) is unsplit, (4) is flanked by resolved peaks, and (5) is such that adding it results in improved peak spacing. If all conditions are met, an additional predicted peak is created and the observed peak is assigned to it.
Accuracy Assessment Methods
In principle, the accuracy of a basecalling program is easily measured by aligning read sequences produced by that program to the correct sequence and tabulating discrepancies. There are a number of subtleties in such an analysis, however, and it is easy to get misleading results. The details of the alignment algorithm can significantly affect how errors are classified. Moreover, it is important to allow for variability of error rates within and between reads, since otherwise the most error prone parts of the reads, or a small subset of reads with a very high error rate, can unduly influence the results. It is also worth recognizing that not all regions of a read are equally important; the higher quality part tends to be the most useful in practice, and the error rates in that part are therefore of most relevance.
We assessed the accuracy of the phred and ABI base-callers in several large sets of reads from cosmid clones sequenced in three laboratories (Table 1). These included (set 1) 9 mammalian (human and mouse) cosmids, sequenced by L. Rowen in L. Hood’s laboratory (University of Washington); (set 2) 9 C. elegans cosmids from the Washington University Genome Sequencing Center (R. Waterston); and (set 3) 36 human chromosome 7 cosmids, from the University of Washington Genome Sequencing Center (M. Olson). In each case the sequencing strategy consisted of a moderate to high depth (6× − 10×) shotgun phase in which dye primer reads were obtained from M13 subclones, followed by a finishing phase in which additional reads (mostly dye terminator) were used to close gaps and resolve low quality regions. Trace processing was performed using ABI analysis software.
Cosmid Set Descriptions
The sets differed somewhat in the machine running conditions that were used and in the sequencing polymerase (see footnote to Table 1); in particular we separate the analyses of the cosmid sets one and two from the analyses of cosmid set three because set three had average read lengths several hundred bases longer than the other two sets, which would complicate interpretation of combined results. Dye primer reads were analyzed separately from dye terminator reads.
For each cosmid we created two FASTA formatted files, one containing the ABI base-called reads, and the other containing the phredbase-called reads (as generated from the ABI-processed trace files). Each of these was screened to mask out vector sequence, and the screened reads were then aligned to the finished cosmid sequence and discrepancies tabulated.
Alignment of the reads to the finished sequence was performed by use of a restricted Smith–Waterman (Smith and Waterman 1981) algorithm as implemented in the program cross_match(P. Green, in prep.), which searches bands in the Smith–Waterman matrix surrounding word matches between the query and target sequences. It is important to recognize that the details of the alignment (its precise extent and the positions and types of the discrepancies) depend to some degree on the parameters used in the Smith–Waterman algorithm, which in our case were +1 for a match, −2 for a mismatch, −4 for a gap-initiation penalty, and −3 for a gap-extension penalty. The error rate in sequence reads tends to increase progressively toward the end of the read, and the local alignment stops when the error rate starts becoming high enough to produce a negative score. With our parameter values, this occurs where the error rate starts to exceed roughly 30% (depending on the distribution and type of the discrepancies). In regions containing multiple errors, there may be several possible ways of aligning the read against the cosmid sequence. For example, with compressions (where typically a set of peaks are shifted leftward), frequently the base-caller will get the number of bases correct, but it will omit one base (the first shifted peak in the group) and then compensate by inserting an additional base (orN) following the last shifted peak. Such a miscalled region may be aligned against the true sequence in either of two ways: by having a single deletion and a single insertion flanking several correctly matching bases; or by having no indels but multiple substitution discrepancies. Which of these has the higher score will depend on the relative sizes of the indel and mismatch penalties, as well as on the precise sequence of the shifted bases. Our parameter values tend to modestly favor alignments with substitution discrepancies rather than indels.
The relative performances of the ABI and phred base-callers were assessed by use of a computer program written to compare thecross_match outputs for the two sets of reads for each cosmid. The number of aligned bases for each method (i.e., the number of bases in the Smith–Waterman alignment, summed over all reads) was first determined. For meaningful comparison of the error rates of two different base-calling methods it is important to restrict attention to the part of each trace for which errors can be counted for both methods simultaneously (if one instead counts errors for each method in the entire part of the read alignable for that method, the more accurate method may actually appear less accurate, because in general it will have a longer alignable read length and the additional aligned bases will be in a lower quality part of the trace that has an intrinsically higher error rate). We did this by considering for each trace the segment of the cosmid sequence that is aligned to the ABI-called read bycross_match, the segment of the cosmid that is aligned to the phred-called read, and then taking the intersection of these two segments (this intersection is called the set of jointly alignable bases for the trace). Discrepancies inside this segment were tabulated; those outside it were ignored. Discrepancies were classified as substitutions (not counting Ns),Ns, insertions, and deletions, and tabulated in 100-base intervals with respect to position in the read sequence. The error rate for a particular error type in a particular 100-base interval in a data set is computed as the total number of errors of that type in that interval, divided by the total number of jointly alignable bases in that interval in the data set.
Table 2 shows the number of jointly alignable bases in each interval of 100 read bases in the cosmid sets. From positions 100 to 199, the number of aligned bases is nearly equal to the number of aligned reads multiplied by 100, reflecting the fact that most reads have alignable lengths of 200 or more. The number of aligned bases in positions 1 to 99 is lower, in part because the first few bases of the read are from the sequencing vector and in part because lower trace quality for the first 50 or so peaks sometimes results in enough basecalling errors to prematurely truncate the alignment. The number of aligned bases decreases slowly from position 200 up to about position 400 in cosmid sets one and two, and up to about position 800 in the data sets of cosmid set three, reflecting the fact that most reads have relatively high data quality in these regions. Thereafter, the numbers of aligned bases drop rapidly owing to degradation of trace quality toward the end of the read.
The Total Number of Jointly (phred and ABI) Aligned Bases in Each Data Set by Read Position and Chemistry
RESULTS AND DISCUSSION
We compared the accuracy of the ABI and phredbase-callers in three sets of cosmids (see Table 1, and Accuracy Assessment Methods). Although there is some variability within each set, on average cosmid sets 1 and 2 may be considered to be of average quality (Taq polymerase, short gels), whereas cosmid set 3 is of high quality (TaqFS polymerase, long gels). Sets 1 and 2 had very similar error profiles and were combined for the analyses described below. Dye primer and dye terminator reads from each set were analyzed separately.
Figures1 F2 F3 F4summarize the error rates by error type and read position in thephred and ABI reads for each data set. (To permit meaningful comparison, we count errors only in the part of each trace for which both the ABI and phred base-calls are accurate enough to be jointly alignable; see Accuracy Assessment Methods.) Consistent with our quality designation above, set three has fewer errors at corresponding read positions than do sets 1 and 2, and the dye primer reads in each set have fewer errors at corresponding positions than the dye terminator reads. In general the error rates for both ABI andphred are quite low in the middle (high quality) parts of the reads, rising sharply later in the reads. There is an inflection point (reduction in the rate of increase) in many of the overall error rate plots later in the reads that corresponds closely with the point at which the number of alignable bases drops (Table 2). This appears to be attributable to a subpopulation of reads that have significantly higher quality, and thus longer alignable lengths and lower error rates, than the bulk of the reads.
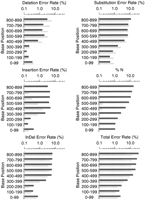
Phred (gray bars) and ABI (black bars) error rates for dye primer data in cosmid sets 1 and 2.
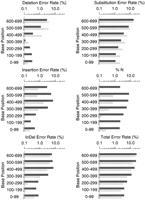
Phred (gray bars) and ABI (black bars) error rates for dye terminator data in cosmid sets 1 and 2.
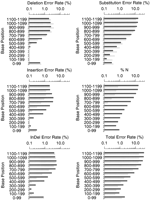
Phred (gray bars) and ABI (black bars) error rates for dye primer data in cosmid set 3.
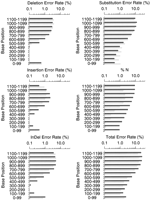
Phred (gray bars) and ABI (black bars) error rates for dye terminator data in cosmid set 3.
Phred’s total error rates (lower right panel in Figs. 1 F2 F3 F4) are lower than ABI’s in all parts of the read, in each data set. Overall, for the dye primer data, phred has ∼41% fewer errors than ABI in sets 1 and 2, and 52% fewer errors in set 3; for the dye terminator data, phred has 39% fewer errors in all three sets. It is interesting that the relative improvement is greatest in the highest quality data set (set 3 dye–primer reads). Table 1shows that the phred-called reads on average had about 5% longer alignable read length than the ABI reads, which is consistent with phred’s greater accuracy throughout the read. To ensure that these results are not attributable to a small subset of reads with high error rates, we repeated the analyses after excluding the 5% of ABI reads and the 5% of phred reads having the highest error rates. The same relative differential (40%–50% fewer errors withphred) was seen on the pruned data sets.
It is instructive to consider the different error types. Insertion or deletion errors (indels) are more serious than substitution errors for many purposes [e.g., in expressed sequence tag (EST) data, where they may change the reading frame thus making homology detection more difficult]. The combined indel rate for phred is lower throughout the read length in each data set, except past base 1000 in set 3 where ABI does slightly better. In the regions of higher trace quality (i.e., before the sharp dropoff in numbers of aligned bases)phred’s insertion and deletion rates are both lower than ABI’s. After this point, the rates for both ABI and phredincrease substantially but with different tendencies. Thephred deletion rates increase somewhat more quickly than ABI’s and are substantially higher than ABI’s towards the end of the trace; conversely the ABI insertion rates increase more rapidly thanphred’s. It appears that the ABI base-caller is more likely to add a peak than to remove one, while phred avoids high insertion rates with its tightly constrained peak prediction algorithm. The elevated phred deletion rates later in the trace are probably attributable to the fact that phred freezes the predicted peak spacing value where it can no longer reliably determine it, which tends to underestimate the number of predicted peaks when the true spacing decreases.
Phred makes roughly the same numbers of substitution errors as ABI, except for the terminator reads of cosmid sets one and two where the phred substitution rate is about twice the ABI rate in the high quality part of the trace. However, ABI calls substantially moreN’s than phred throughout, in part becausephred only calls an N when it cannot find an observed peak to associate to a predicted peak location, while the ABI software assigns an N when either it cannot find a peak or the call is uncertain because of noise peaks. The combined substitution +N rate is substantially higher for ABI than forphred in all data sets.
Because we consider any failure to predict the correct base as an error, N’s are errors. This is the only sensible policy, as otherwise a base-caller could inflate its accuracy simply by callingN’s in any low-quality region; for example, phredcould attain an error rate of <1 in 10,000 by designating every base with quality value <40 as an N (Ewing and Green 1998). Assigning an N to an ambiguous peak has some merit in the absence of quality values because it helps to indicate low trace quality, but is not useful when quality values are available. Nonetheless in fairness to the ABI software one must acknowledge that its performance under our criteria could be improved by modifying it to always guess a base. If we assume (optimistically) that by doing so the ABI software could convert 75% of its N’s to correct base-calls, with the remaining 25% becoming substitution errors, then the ABI substitution error rates would approximately equal thephred rates in the high quality regions of the traces, but would exceed the phred rates later in the traces in all cases except the terminator data for cosmid sets one and two. Total error rates would still be less for phred than for ABI.
Sensitivity to Lane-Processing Method
We believe that phred’s observed peak detector and its methods for aligning observed peaks to predicted peaks will work well regardless of the trace source. This is not true of the peak prediction algorithm, however, which was tuned on ABI-processed traces and works well only when the trace processing includes a step to render the peak spacing relatively uniform; without such peak spacing normalization,phred may predict peaks poorly in some regions of the trace, resulting in increased numbers of indels. Other specifics of the lane processing may also affect phred performance. The ABI mobility correction appears to be less accurate toward the beginning and end of the trace, affecting basecalling accuracy there. In addition, it appears that the ABI trace processing broadens peaks excessively, which has the effect of reducing resolution and increasing the numbers of deletion errors.
We have recently developed a trace-processing program, plan,that reads raw trace data and processes the trace arrays. Preliminary tests indicate that phred’s performance on traces processed using plan is comparable to, or slightly better than, that using ABI processed traces.
Further Improvements
What further accuracy improvements are possible? The high-quality part of the trace presents different issues from the later, lower quality part. For most applications where an accurate finished sequence is required, the high-quality part of the trace is by far the most important, since it is the only part usable in deriving the final sequence. Raw data quality in the later parts of the trace is such that, although improved basecalling accuracy is certainly possible, it would be extremely difficult to reduce the error rate to the level necessary to infer highly accurate sequence.
In the high-quality part of the trace the major sources of error at present are compressions in dye primer data, and highly variable peak heights or missing peaks in dye terminator data. Both of these are sequence-context dependent, and classifying the specific sequence motifs that are most error prone is a promising approach to improving accuracy. It is particularly important to improve phred’s ability to identify and resolve CC and GG compressions, which are often difficult even for skilled human finishers to detect by eye, and we have recently made some progress in this regard using rules for hairpin-prone sequences.
Improved accuracy in the lower quality part of the trace would be useful in single read applications (e.g., EST sequencing), and to improve repeat discrimination and assist in making joins in the early phases of shotgun sequencing projects (which helps make collection of additional data more efficient by delineating the sizes of regions where more data are needed). The major gains to be made here are in improved peak spacing estimation and in processing to improve peak resolution.
Program Availability
C source code is available at no charge to academic researchers for research purposes, and by commercial license from the University of Washington to other users; contact Brent Ewing atbge{at}u.washington.edu.
Acknowledgments
This work was partly supported by a grant from the National Human Genome Research Institute. We thank several people for helpful suggestions and/or data sets, in particular David Ficenec, Bob Waterston, Darren Platt, Asif Chinwalla, Shawn Iadonato, and Lee Rowen.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL phg{at}u.washington.edu; FAX (206) 685-7344.
- Received December 5, 1997.
- Accepted February 3, 1998.
- Cold Spring Harbor Laboratory Press








