
Novel Families of Putative Protein Kinases in Bacteria and Archaea: Evolution of the “Eukaryotic” Protein Kinase Superfamily
Abstract
The central role of serine/threonine and tyrosine protein kinases in signal transduction and cellular regulation in eukaryotes is well established and widely documented. Considerably less is known about the prevalence and role of these protein kinases in bacteria and archaea. In order to examine the evolutionary origins of the eukaryotic-type protein kinase (ePK) superfamily, we conducted an extensive analysis of the proteins encoded by the completely sequenced bacterial and archaeal genomes. We detected five distinct families of known and predicted putative protein kinases with representatives in bacteria and archaea that share a common ancestry with the eukaryotic protein kinases. Four of these protein families have not been identified previously as protein kinases. From the phylogenetic distribution of these families, we infer the existence of an ancestral protein kinase(s) prior to the divergence of eukaryotes, bacteria, and archaea.
For many years after the discovery of protein phosphorylation, the prevailing view was that phosphorylation of proteins on serine, threonine, and tyrosine residues was a phenomenon restricted to the eukaryotes, in which these modifications perform important regulatory functions (Hanks et al. 1988; Hunter 1995). In bacteria, an analogous regulatory role was attributed to the two-component sensor kinases and the phosphoenolpyruvate-dependent phosphotransferase systems, which typically catalyze the phosphorylation of proteins on histidine and aspartate residues, respectively (Saier et al. 1990). Further experimental work has brought to light exceptions to these paradigms. Evidence of bacterial (Wang and Koshland 1978, 1981) and archaeal (Skorko 1984; Smith et al. 1997) proteins containing phosphoserine, phosphothreonine, or phosphotyrosine residues has accumulated over the past two decades, and elements of the two-component sensor kinase system have been detected in archaea and eukaryotes (Loomis et al. 1997).
Several unusual mechanisms of Ser/Thr or Tyr protein phosphorylation have been recognized in prokaryotes, but they do not appear universally in bacteria or archaebacteria. There are examples of histidine kinase-related proteins in Bacillus subtilis (Yang et al. 1996) and the eukaryotic mitochondrion (Popov et al. 1992) that mediate phosphoserine formation. In addition, there have been reports of Ser/Thr or Tyr autophosphorylating proteins in bacteria (Ostrovsky and Maloy 1995; Grangeasse et al. 1997).
Recently, genes encoding proteins homologous to eukaryotic Ser/Thr protein kinases have been identified in several bacteria (Kennelly and Potts 1996) and archaea (Smith and King 1995). Myxococcus xanthus encodes numerous paralogous Ser/Thr kinases that show highly significant sequence similarity to one another (Munoz-Dorado et al. 1993). The best characterized of these proteins, Pkn2, has been shown to play a regulatory role in secretion (Udo et al. 1995). Protein kinases of the Pkn2 type have also been recognized in a number of other bacteria (Zhang 1996).
To evaluate the entire repertoire of potential protein kinases in bacteria and archaea and to gain insight into the early stages of their evolution, we conducted an extensive search for proteins with similarity to protein kinases in the sequences of the currently available complete genomes. We report here the identification of four previously unknown families of predicted protein kinases (in addition to the known and newly detected members of the Pkn2 family) with representatives in the bacteria and archaea. Sequence analysis indicates that these protein kinase families are distant members of the eukaryotic protein kinase superfamily (Hanks et al. 1988). The evidence presented here suggests strongly the existence of an ancestral protein kinase(s) prior to the divergence of the eukaryotes, bacteria, and archaea.
RESULTS AND DISCUSSION
Identification of Previously Unknown Kinase Families
In PSI-BLAST searches, members of the new protein kinase families from bacteria and archaea were detected typically with random expectation (e) values <10−4 in the first iteration. After several more iterations, additional members are detected, usually with e < 10−6. Examination of the aligned regions reveals considerable conservation of important sequence motifs shared with the ePK superfamily. Occasionally, after five or more iterations, known members of the choline-kinase or antibiotic kinase families were detected withe ∼ 10−2. The antibiotic kinases are known to be similar structurally to the protein kinases (Hon et al. 1997). The candidate protein kinases in the bacterial and archaeal sequences were distinguished from putative choline or antibiotic kinases by reciprocal PSI-BLAST searches into the GenBank nonredundant protein sequence database. The statistical significance of alignments of the candidate protein kinases amongst themselves and to known protein kinases was routinely several orders of magnitude greater than in those few instances in which they detected choline or antibiotic kinases.
Although these bacterial and archaeal proteins contain the unmistakable fingerprints of enzymes that transfer a phosphate group from ATP to a hydroxyl group on the substrate molecule, it may not be possible to deduce unequivocally the substrate specificity from sequence analysis alone. Nevertheless, the consistently greater sequence similarity to eukaryotic-type protein kinases and the indications of structural similarity (see below) suggest strongly that they catalyze the ATP-dependent phosphorylation of serine, threonine, or tyrosine residues in proteins. This hypothesis is supported by biochemical data in the case of Saccharomyces cerevisiae piD261 (YGR262C), which is a member of one of the identified protein families (see below) and has been shown to possess Ser/Thr-specific protein kinase activity (Stocchetto et al. 1997).
The candidate protein kinases discovered in the complete genomes are listed in Table 1. These proteins were classified into families by single-linkage clustering on the basis of highly significant scores in reciprocal BLAST searches. In addition to the previously identified Pkn2 family of bacterial protein kinases, we identified four families of predicted protein kinases present in bacterial and archaeal genomes; these families are designated the ABC1, RIO1, piD261, and AQ578 families, after their respective prototype members.
Phylogenetic Distribution of Eukaryotic-Type Protein Kinase Homologs
With the exception of the spirochaetes, Borrellia burgdorferiand Treponema pallidum, all of the complete genomes encode some type of protein kinase. Mycoplasma genitalium,Mycoplasma pneumoniae, Haemophilus influenzae andHelicobacter pylori each have a single protein kinase gene.Chlamydia trachomatis contains three members of the Pkn2 family. B. subtilis encodes two members of the Pkn2 family, and two unclassified predicted protein kinases. The other genomes each contain one or more members of the previously undetected families of candidate protein kinases (Table 1). Escherichia coli contains three candidate protein kinases that do not fall into any of the families. The rfaY and rfaP genes are part of a locus involved in the synthesis of the lipopolysaccharide biosynthesis (Roncero and Casadaban 1992); and the inaA gene, which is regulated by the multiple antibiotic resistance and the superoxide stress response systems (White et al. 1992; Rosner and Slonczewski 1994), is closely related to rfaP.
Structural Conservation
Multiple alignments were constructed for the identified families of putative protein kinases (Figs. 1A–D). Where BLAST searches revealed members of these protein families in Arabidopsis thaliana, S. cerevisiae, Caenorhabditis elegans, or Homo sapiens, the eukaryotic proteins were included in the alignment. All detected families contain the most important conserved elements found in the ePK superfamily. The alignments in Figure 1, A–D, show motifs corresponding to the highly conserved residues found in conserved regions I, II, VIb, and VII (Hanks et al. 1988; Smith et al. 1997). Region I of the protein kinases is involved in binding ATP and is characterized by a β-strand leading to a glycine-rich loop that contains the GXGXXGXV motif. Structurally, this motif forms a turn that is followed by another β-strand. Allowing for substitution of the small side chains of Ala and Ser in place of Gly, there is excellent conservation of this structural element, including the aliphatic residues preceding and ending the motif, in the identified protein families. In the ePK superfamily, these residues (typically Leu and Val,respectively) form a hydrophobic pocket that sequesters the adenine ring of ATP. The Lys residue in conserved region II is invariant in the identified candidate protein kinases, as is its presence in a region of predicted β-strand conformation.

Multiple alignments of the identified protein-kinase families. Arrows indicate β-strands in the mouse MAPK structure (PDB accession no.1P38) (Wang et al. 1997). Broken arrows indicate that the respective strand includes gaps introduced by alignment to the protein kinase families described here. All sequences are listed with their GenBank identifiers followed by their systematic ORF names or a short mnemonic name. Numbers preceding, within, or following the aligned regions refer to the number of amino acid residues elided. Question marks indicate unknown peptide lengths in ORFs conceptually translated from EST sequences. Consensus key: Uppercase letters indicate conserved residues by the single-letter amino acid code, lowercase letters indicate conserved classes of amino acids that are highlighted as follows: [yellow (h)] hydrophobic residues (A,C,F,I,L,M,V,W,Y); [blue (p)] polar residues (C,D,E,H,K,N,Q,R,S,T); [red (c)] charged residues (D,E,H,K,R); [green (u)] tiny residues (A,C,G,S); (magenta) other conserved residues, as annotated in the consensus sequence [(s) small residues (A,C,D,N,G,P,S,T,V); (b) bulky residues (E,F,I,K,L,M,Q,R,W,Y); (a) aromatic residues (F,H,W,Y); (l) aliphatic residues (I,L,V), (+) positively charged residues (H,K,R), (−) negatively charged residues (D,E)]. The est_CElegans protein sequence in B is an artificial composite of ORFs from two EST sequences (gi2378893 and gi1121991). The MJ1211 sequence shown here actually differs from gi1591840 by the selection of an upstream start codon, adding 95 amino-terminal residues, including counterparts to the conserved regions I and II of the ePKs. The GenBank identifier of est_human inC (gi2357825) refers to a nucleotide sequence; a conceptual translation of one ORF is shown here.
As in the ePK catalytic loop, the interacting Asp and Asn residues in region VIb are highly conserved and the variation in the other residues of the HRDLKXXN signature is within the range seen in known members of the ePK superfamily. The DFG triplet in region VII, with its interacting Asp and Gly residues, is well conserved in the ABC1 and piD261 families (Fig. 1A,C), whereas it is somewhat divergent in RIO1 and AQ578 families (Fig. 1B,D). Nevertheless, the Asp residue of this motif (with a single exception discussed below) is invariant in these putative protein kinases.
There is no obvious counterpart of the conserved region VIII of the ePKs, with its characteristic APE consensus sequence. This region is often phosphorylated in the ePKs, resulting in activation of the kinase. However, in light of the fact that the S. cerevisiaepiD261 (YGR262C) protein (which does not contain the APE motif) has been shown to possess Ser/Thr-specific protein kinase activity (Stocchetto et al. 1997), this element is apparently not strictly essential for protein kinase catalytic activity. There is some family-specific sequence conservation carboxy-terminal to the conserved region VII DFG triplet (not shown), in certain cases containing conserved negatively charged residues; but there is, as yet, no evidence that these regions play a similar role to ePK conserved region VIII in the regulation of protein kinase activity.
Secondary structure prediction based on multiple alignments (Rost and Sander 1994) confirms that these protein families have a globally similar structure to each other and to the known protein kinases in terms of the presence and spacing of predicted regions of β-strand and α-helical conformation (Fig. 1; data not shown). However, in the absence of detectable sequence similarity, it would be purely speculative to suggest (for instance) that a given predicted α-helical region including a conserved negatively charged residue was the counterpart of ePK conserved region III.
The ABC1 Family
There is a unifying theme seen in certain members of the ABC1 family that have been investigated biochemically. Mutants in the aarFgene of Providencia stuarti and the yigR gene fromE. coli are both deficient in the synthesis of ubiquinone (cofactor Q) (Macinga et al. 1998). The prototypic family member,S. cerevisiae ABC1 (YGL119W), is defective in aerobic respiration because of a reduction in the activity of the mitochondrial bc1 complex that can be overcome by exogenously supplied quinones (Bousquet et al. 1991; Brasseur et al. 1997). One possible explanation for the expansion of this family in the cyanobacteriumSynechocystis could be the role of the Q-cycle in capturing energy from photosynthesis. The precise mechanisms by which mutants in the ABC1 family manifest their phenotypes is unknown. It has been proposed that aarF and yigR may regulate the activity of transcription factors (Macinga et al. 1998) and that yeast ABC1 may regulate the structure/interactions of multimeric protein complexes (Brasseur et al. 1997). There is, of course, ample precedent for protein kinases performing either of these roles in the literature on ePKs.
Several individual members of the ABC1 family deserve further comment. The absence of the critical Asp residue (involved in chelating magnesium ions) in the unusual KYG triplet that replaces the DFG signature in YPL109 suggests that this protein may be a catalytically inactive. There are a number of human ESTs with >90% identity to the sequence of the murine member of the ABC1 family (gi1172105), suggesting that at least one member of the ABC1 family will be identified in humans.
The RIO1 Family
Most of the eukaryotes and archaea listed in Table 1 contain two paralogs of the RIO1 family. These proteins form two (or possibly three) distinct subfamilies of paralogs (Fig. 1B). Phylogenetic tree analysis of the RIO1 family reveals significant bootstrap support for the clustering of subfamily 1, containing representatives from both archaea and eukaryotes (Fig. 2). The eukaryotic members of subfamily 2 are also grouped together with significant bootstrap support; however the statistical support for grouping the archaeal members of subfamily 2 with each other or with the eukaryotic proteins is weaker. The eukaryotic and archaeal members of subfamily 2 are grouped together in Figure 1B on the basis of similarities apparent from a close examination of the sequence alignments.
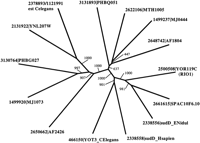
Phylogenetic tree analysis of the RIO1 protein kinase family. Phylogenetic tree analysis of the RIO1 protein kinase family was performed using the neighbor-joining algorithm as implemented in the CLUSTALW program (Higgins et al. 1996). The sequences are labeled as in Fig. 1. Bootstrap values from 1000 replicates are shown.
The consequences of the divergence of these subfamilies on the activity of these proteins are unknown, but the conservation of both paralogs in such a broad phylogenetic range argues that each of them has a unique function. Relatively little is known about the biology of the RIO1 family proteins. The sudD gene was identified in a screen for extragenic suppressors of a mutation in the bimD gene, which is involved in DNA repair and cell-cycle control in Emericella nidulans (Holt and May 1996).
The piD261 Family
A most unusual feature of this family is that two of its archaeal members (MJ1130 and MTH1425) occur as amino-terminal fusions with anO-sialoglycoprotein endopeptidase (OSGP) domain. The other archaeal members of this family, PHCJ009 and AF0665 (which was mislabeled originally as having an OSGP domain); however, do not contain a counterpart to the OSGP catalytic domain and are independent occurrences of the kinase domain found in MJ1130 and MTH1425.
The product of the S. cerevisiae YGR262C gene (piD261) has been recognized as a likely protein kinase (Clemente et al. 1997), and its similarity to MJ1130 has been noted (Hunter and Plowman 1997). Experimental evidence shows that it is a Ser/Thr-specific kinase required for normal growth of the yeast (Stocchetto et al. 1997). Interestingly, this kinase displays some unique features when compared to conventional ePKs; including resistance to staurosporine and a requirement for Mn2+ instead of Mg2+ (Stocchetto et al. 1997).
The AQ578 Family
The AQ578 family displays a distinct relationship to the piD261 family, although there is considerable variability in the region VIb consensus (HRDLKXXN) and the region VII consensus (DFG), suggesting the possibility that the function of these protein families may have diverged after an earlier gene duplication in the archaeal lineage. MTH915 is a highly divergent member of this family, with rather poor conservation in region I. One particular feature of the AQ578 family, namely the conserved negative charge in the third position of the DFG triplet, seems to be unique among protein kinases.
Evolutionary Scenarios
The ABC1 family is found in all three domains of life (bacteria, archaea, and eukaryotes), but there is only a single ABC1 member in the archaeal (Methanobacterium thermoautotrophicum). There are multiple paralogs of the ABC1 family encoded in several of the complete genomes (Table 1), but given the absence in the majority of the archaea, there is no compelling evidence that the expansion of this family occurred prior to the divergence of the bacteria and the eukaryotes. We propose that the ABC1 family evolved in bacteria and entered the early eukaryotes by horizontal transfer, possibly from mitochondria, with subsequent duplication and divergence (Fig.3). One of the ABC1 paralogs in A. thalianais most closely related to the ABC1 proteins found in the cyanobacterium Synechocystis, suggesting an independent chloroplast origin (we label this protein as chABC1, to distinguish it from the other paralog in A. thaliana). The established role of S. cerevisiae ABC1 (YGL119W) in the function of the mitochondrion (Bousquet et al. 1991; Brasseur et al. 1997) lends additional support to an ancient bacterial origin for this protein family. The only archaeal ABC1 family member (found in M. thermoautotrophicum) is most closely related to the bacterial ABC1 proteins and may represent a more recent horizontal transfer.
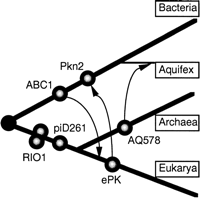
A tentative evolutionary scenario for the protein kinases. (Heavy lines) Major branches of the rRNA-based phylogenetic tree (Pace 1997). (Circles) Points at which individual protein-kinase families are supposed to have emerged; two paralogous subfamilies of the RIO1 family are shown. (Arrows) Postulated horizontal transfer of a protein kinase gene from one lineage to another. The postulated horizontal transfer of an ABC1 family member from bacteria to M. thermoautotrophicumis not shown.
The RIO1 and piD261 families are found in archaea and eukaryotes. From the distribution of the two subfamilies of paralogs in the RIO1 family, we tentatively conclude that an ancestral RIO1 gene existed in the common ancestor of the eukaryotes and archaea and that it had already undergone a gene duplication event before the divergence of these two domains (Fig. 3). With a single exception, the AQ578 family is found only in archaea. It seems likely that AQ578 was introduced into Aquifex aeolicus as one of numerous archaeal genes acquired by horizontal transfer into this hyperthermophilic bacterium (Aravind et al. 1998).
The Pkn2 family clusters most closely with the ePKs; but, as such, appears only in bacteria. We reason that there was an early horizontal transfer of this family from eukarya into bacteria with subsequent expansion, particularly in those bacteria with complex developmental cycles. There have apparently been losses of this gene family in certain bacterial lineages, notably in proteobacteria.
It is interesting to note that all of the bacterial genomes in Table 1that contain a Pkn2-type protein kinase also contain at least one paralog of the PP2C-class protein phosphatases and that several of them contain FHA domain proteins. The PP2C-class phosphatases catalyze the removal of phosphates from phosphoproteins (Bork et al. 1996) and the FHA domain mediates protein–protein binding to targets containing phosphoserine or phosphothreonine residues (Hofmann and Bucher 1995;Sun et al. 1998). The presence of PP2C-class phosphatases and FHA-domain-containing proteins suggests the possibility that Pkn2 family proteins act as protein kinases in a protein-phosphorylation signal-tranduction cascade. Another interesting observation is that the genes encoding certain Pkn2 family members in B. subtilis,Mycobacterium tuberculosis, Synechocystis, andMycoplasma are adjacent to phosphatase-encoding genes, suggesting the existence of a coordinately regulated operon.
Among the genomes that contain a PP2C paralog but do not contain Pkn2 family members (Table 1), H. pylori contains a protein related apparently to the eukaryotic PKC family, but the appearance of FHA domain proteins in T. pallidum cannot be adequately explained as part of an endogenous signal transduction mechanism. The origins of the protein kinase genes in H. influenzae and H. pylori are unclear, but they may derive from horizontal transfer events from their eukaryotic hosts into these pathogens.
Whereas there seems to be no direct evidence for an obvious progenitor of the ePK superfamily in the prokaryotic world, we speculate that it emerged early in eukaryotic evolution from one of the ancient protein kinase families, perhaps the RIO1 or pID261 family, and evolved over time to attain its current diversity.
The phylogenetic distribution of the protein-kinase families identified in the complete genomes points to a complex evolutionary history (Table1; Fig. 3). The ABC1 family is found primarily in bacteria and eukaryotes, whereas the RIO1 and piD261 families are found in archaea and eukaryotes. The AQ578 family is found primarily in archaea whereas the Pkn2 family, although showing a distinct relationship to the ePK superfamily, occurs only in bacteria. It is apparent from the widespread distribution of these genes that the ancestry of the catalytic domain of eukaryotic-type protein kinases predates the divergence of the three domains of life.
METHODS
Databases
We analyzed complete genome sequences from bacteria—A. aeolicus, B. subtilis, B. burgdorferi, T. pallidum, C. trachomatis, E. coli, H. influenzae, H. pylori, M. genitalium, M. pneumoniae, Synechocystis sp., and Mycobacterium tuberculosis—and archaea: Archaeoglobus fulgidus, M. thermoautotrophicum, Methanococcus jannaschii, andPyrococcus horikoshii. T. pallidum data are available for FTP from TIGR at (ftp://ftp.tigr.org/pub/data). M. tuberculosisdata were produced by the M. tuberculosis Sequencing Group at the Sanger Centre and are available for FTP at (ftp://ftp.sanger.ac.uk/pub/tb). C. elegans data was from Wormpep13 (http://www.sanger.ac.uk/Projects/C_elegans/wormpep). Sequence data for all other genomes were obtained from the GenBank database via the Entrez WWW interface at (http://www.ncbi.nlm.nih.gov/Entrez/Genome/org.html).
Sequence Analysis
Searches of the nonredundant (NR) protein sequence database at the National Center for Biotechnology Information (NCBI, NIH, Bethesda) were performed using the gapped BLAST program and the PSI-BLAST program (Altschul et al. 1997); data handling and analysis was managed by the SEALS software package (Walker and Koonin 1997). Single-linkage clustering of proteins by sequence similarity was performed using the GROUPER program of the SEALS package. Multiple sequence alignments were constructed and phylogenetic tree analysis was performed using the CLUSTALW program (Higgins et al. 1996). Consensus sequences were calculated using a script made available by Nigel Brown and Jianmei Lai (http://www.bork.embl-heidelberg.de/Alignment/consensus.html). Secondary structure prediction for proteins was performed using the PHD programs (Rost and Sander 1994).
For the identification of candidate protein kinases, a BLAST database was created with the protein sequences from each of the completely sequenced bacterial and archaeal genomes, including plasmids and extrachromosomal elements. Searches into these databases were performed with a variety of eukaryotic protein kinase sequences using the checkpoint feature of PSI-BLAST. The PSI-BLAST algorithm enhances the sensitivity of BLAST searches by using the information contained in a multiple alignment of sequences similar to the original query to modify the substitution matrix in a position-specific fashion—in essence, developing a profile of the query sequence and the sequences it detects. This profile is then used to search the database again, retrieving sequences that were not detected by the original query sequence at statistically significant levels; such iterations can be run until no new similar sequences are retrieved from the database. The checkpoint feature allows the user to save the profile developed by an iterative PSI-BLAST search on one database and use it to increase the sensitivity of searches into a different database. In this study this feature was used to search the database of complete genomes using profiles developed by searching the GenBank NR protein sequence database with eukaryotic protein kinase sequences as queries.
A complementary approach was taken to ensure the identification of all protein kinase paralogs in the complete genomes. With the SEALS package, each of the bacterial and archaeal protein sequences were used as queries for PSI-BLAST searches into the NR database. The outputs of these searches were analyzed by tallying the number of hits that were members of a large set of known eukaryotic protein kinases; those proteins with a large number of hits against the known protein kinases (even if not highly statistically significant) were targeted for further analysis.
Acknowledgments
We thank D. Roland Walker for valuable help with automated data extraction and database searches, and Kenn Rudd for useful discussions at the initial stages of this project.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL koonin{at}ncbi.nlm.nih.gov; FAX (301) 480-9241.
-
- Received June 2, 1998.
- Accepted August 28, 1998.
- Cold Spring Harbor Laboratory Press








