
Pathological Consequences of Sequence Duplications in the Human Genome
Abstract
As large-scale sequencing accumulates momentum, an increasing number of instances are being revealed in which genes or other relatively rare sequences are duplicated, either in tandem or at nearby locations. Such duplications are a source of considerable polymorphism in populations, and also increase the evolutionary possibilities for the coregulation of juxtaposed sequences. As a further consequence, they promote inversions and deletions that are responsible for significant inherited pathology. Here we review known examples of genomic duplications present on the human X chromosome and autosomes.
Gene duplication is an important mechanism in the evolutionary process. As analyzed by Ohno in his classic monograph(1970), these duplication events liberate copies of the gene to diverge and take up new functional roles in the organism, while the master gene is constrained to preserve its original role. Families of genes, often including those in clustered repeats, have been encountered since the beginning of molecular biological analysis. The notable early examples included the HOX genes (Krumlauf 1994); members of the immunoglobulin superfamily, such as the immunoglobulins, the T-cell receptors, and the major histocompatibility complex (MHC) genes (Hood et al. 1985); the globin genes (Orkin and Kazazian 1984); and the small and large rRNAs (Srivastava and Schlessinger 1991). Evolution has played with the regulatory possibilities, resulting in mechanisms as varied as the globin switch, immunoglobulin diversity, and the succession of expression of tandem HOX genes during development. Recently we reviewed some features of the resulting distribution of repetitive elements and genes in the human genome (Mazzarella and Schlessinger 1997). In this discussion we expand on some of the consequences of sequence duplications as they relate to human physiology and disease.
The ability to detect and analyze duplication in genomes has been expanded enormously by the explosive progress of long-range sequencing analyses. On a statistical and comparative level, the inference of repetitive elements and motifs has shown that a variety of sequences of unknown function, as well as functional segments of genes, are spread through the genome (for an interesting recent discussion, see Babbitt and Gerlt 1997). Duplication and divergence are central to the generation of diversity and new genes. Duplication can involve rare noncoding or coding sequences, and can occur with or without associated clustering. Thus, members of the actin and tubulin families are scattered in the genome, but globin genes show a more complex pattern involving clustering. As part of the pattern, for example, restriction mapping of the short arm of chromosome 16 has revealed that three different alleles of the α-globin gene lie, respectively, 170, 350, and 430 kb from the telomere (Wilkie et al. 1991). Polymorphic length variation at this locus is postulated to have arisen by nonhomologous exchanges between the subtelomeric repeats on different chromosomes. Furthermore, heterozygosity for the telomere polymorphism may have an effect on meiotic segregation. Because most nonhomologous pairing resulting in nondisjunction occurs at telomeres, trisomy of chromosome 16 may be more frequent in heterozygotes for the subtelomeric region (Speed 1988). Interestingly, trisomy of chromosome 16 is the most common trisomy seen in early natural abortuses (Bond and Chandley 1983).
A variety of specialized selective pressures could promote the development of sequence clusters. They include
- 1.
- An increase in expression. This can occur by simple expansion of tandem repeats, like the rRNA and 5S genes, or by the duplication of sequences at nearby but noncontiguous positions, as exemplified in the discussion below.
- 2.
- Preservation of function of genes on the Y chromosome, which must retain activity in the face of accumulating mutations with no available cognate chromosome to rescue defects by recombination (Muller 1964;Rice 1987); recently a number of genes on the Y chromosome have been shown to be duplicated many times in tandem (Lahn and Page 1997).
X-linked Clusters and Pathology
Repeated sequences can mediate local deletions, duplications, and inversions, with a number of consequences for genome diversity and genetic pathology; the range and seriousness of such events is increased when repeats occur near one another but not directly juxtaposed. Perhaps because it is one of the first regions of the genome to be analyzed in detail, the telomeric cytogenetic band q28 of the X chromosome shows a number of clusters. It also affords corresponding instances in which significant human pathology results from the interaction of the duplicated sequences (Table1). Figure 1 provides a sketch of current information about some clustered sequences on the X chromosome. Figure 2 illustrates recombinational events that have been detected in Xq28. More than 10% of 2.5 Mb of sequence determined to date for Xq28 is duplicated at least once. The duplications are usually nearby in the genome, but also include a 26.5-kb segment found on both chromosome 16p11.1 and Xq28 (Eichler et al. 1996).
Sequence Duplications and Consequences
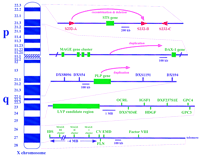
X chromosome regions with duplicated sequences involved in pathology. The regions are (from top to bottom) the X-linked ichthyosis region in Xp22.3, the dosage-sensitive sex reversal candidate region in Xp21.3, the Pelizaeus-Merzbacher region in Xq22, the lymphoproliferative syndrome in Xq25, and regions in Xq28 (further diagrammed in Fig. 2). The genes and probes indicated are all described in the Genome Database and include (STS) steroid sulfatase; (S232-A,B,D) three repetitive sequences that cross-hybridize with genomic clone CRI-S232; (MAGE) melanoma antigen gene family; (DAX-1), critical region for adrenal hypoplasia congenita; (PLP) proteolipid protein; (DXS8096, DXS54, DXS1191, and DXS94) polymorphic markers; (LYP) lymphoproliferative syndrome locus; (OCRL) oculocerbrorenal (Lowe) syndrome gene; (IGSF1) immunoglobulin superfamily gene 1; (HDGF) hepatoma-derived growth factor gene; (GPC3) glypican 3; (DXS7034E and DXF237S1E) expressed sequence tags; (GPC4) glypican 4; (IDS) iduronate-2-sulfatase gene; (CV) color vision genes; (FLN) filamin gene; (EMD) Emery-Dreifuss muscular dystrophy (emerin) gene; andfactor VIII gene.
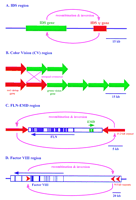
Proposed recombination events between homologous sequences affecting genes in Xq28. Recombinations lead to, respectively (A) Hunter syndrome; (B) color blindness, (C) polymorphism (inversion) and occasional further crossover/deletion to give EMD muscular dystrophy, and (D) hemophilia A.
The classic locus for tandem repeats is color vision. Extensive polymorphism is associated with color blindness in ∼1 in 12 white men and 1 in 200 women [McKusick 1994; OMIM no. 303800 (green pigment) and OMIM no. 303900 (red pigment); The Online Mendelian Inheritance in Man (OMIM), edited by Dr. Victor A. McKusick and colleagues, is found at URL http://www.ncbi.nlm.nih.gov/omim/]. The vast majority of color blindness results from variations in a tandem set of one to four red and one to seven green pigment genes (Nathans et al. 1992; Neitz and Neitz 1995), and phenotypic differences in color vision between individuals are the direct result of the ratio of expressed red genes to green genes. The six exons of the red and green pigment genes and their intragenic regions are 98% identical, suggesting that they arose recently in evolution by duplication events (Vollrath et al. 1988). The spectral differences between the two pigments are attributable to base changes in exon 5. In addition, the two genes are distinguished by a major polymorphism, in which the red gene has a 1.8-kb insertion in intron 1, with a resultant increase from 13.4 to 15.2 kb in genomic span compared to the green gene. Green pigment genes are proposed to be duplicated by homologous recombination in intergenic crossover events, whereas red genes are duplicated in intragenic events (Neitz and Neitz 1995).
Emery-Dreifuss muscular dystrophy (EMD; Emery 1989; OMIM no. 310300) results from lesions in the emerin gene, which is comprised of 6 exons spanning ∼2 kb in a GC-rich region of Xq28 (Bione et al. 1994). This 220-kb region of the genome is gene-dense, containing at least 14 known genes (Chen et al. 1996). Located adjacent toEMD and transcribed in the opposite direction is the filamin gene (FLN). This gene encodes an actin binding protein, with 48 exons spanning ∼26 kb. Flanking a 38-kb segment containing the two genes are two 11.3-kb inverted repeats that are 99.2% identical (Chen et al. 1996). It has been shown that these repeats can lead to a complete deletion of the emerin gene as well as a partial duplication of the adjacent FLN gene, apparently resulting from mispairing of the inverted repeats followed by a double recombination event (Small et al. 1997).
Recombination between the inverted repeats also apparently contributes to the persistence of both the homogeneity of the 11.3-kb sequences and the inversion of the intervening 38-kb region containing theFLN and EMD genes. The inversion is frequent enough to make 33% of females heterozygous for the region (i.e., having one X chromosome with the region in one orientation and the other X with the opposite orientation; Small et al. 1997). The data also suggest an explanation for reported discrepancies between genetic and physical map distances in this region of Xq28 (Small et al. 1997).
The Xq28 region also contains at least two other regions in which nearby but nontandem duplications are involved in inherited disease. Hunter syndrome (mucopolysaccharidosis type II) is an X-linked lysosomal storage disorder caused by a deficiency in the activity of the enzyme iduronate-2-sulfatase (IDS) (Young and Harper 1982; OMIM no. 309900). About 3 kb of the IDS gene is duplicated 20 kb distal to the active gene (Bondeson et al. 1995a,b; Timms et al. 1995, 1997), and a significant fraction of Hunter syndrome cases (15%) are caused by recombination between the gene and its pseudogene, with the consequent deletion of the intervening material (Bondeson et al. 1995b). In addition to localizing the duplicated segment, genomic sequencing found several nearby genes that are affected by more extensive deletions in severe Hunter syndrome cases with additional phenotypes.
Hemophilia A (coagulant factor VIII deficiency; OMIM no. 306700) is another of the paradigmatic X-linked recessive disorders. The 26 exons of the factor VIII gene are scattered in 180 kb of genomic DNA and are transcribed in the telomeric to centromeric direction. A CpG island is located ∼10 kb downstream from exon 22, in the largest 32-kb intron of the gene. The CpG island appears to function as a bidirectional promoter encoding two different transcripts, referred to as factor VIII-associated genes A and B (Levinson et al. 1990, 1992). The part of intron 22 that contains the CpG island is repeated in extragenic copies situated ∼300 kb and 400 kb telomeric to the 5′ end of the factor VIII gene (Naylor et al. 1995). DNA sequencing and chemical mismatch analysis have demonstrated that these three repeat units are 9.5 kb long and 99.9% identical. About 45% of the cases of the severe form of hemophilia A arise by recombinational inversion occurring between the intragenic copy and one of the extragenic copies of the sequence (Lakich et al. 1993; Naylor et al. 1993; Tuddenham et al. 1994). This results from homologous mispairing and a single crossover event. As a consequence, the factor VIII gene becomes disrupted, with exons 1–22 dissociated from and flipped to an orientation opposite that of exons 23–26.
The melanoma antigen gene (MAGE) family is comprised of 12 genes found in three clusters of four genes, all in Xq28 (Rogner et al. 1995), and an additional cluster of 4 genes located in Xp21.3 (Lurquin et al. 1997). The coding region of each MAGE gene is a single exon. Those in Xq28 are 69–98% identical, and those in Xp21.3 are 66–81% identical; there is 45–63% identity between the genes at the two locations. These genes encode a melanoma antigen, with products detected from six of the genes in Xq28 (MAGE A1, A2,A3, A4, A6, and A12) in lung cancers, sarcomas, leukemias, colon cancers, and breast carcinomas (van der Bruggen et al. 1991; De Plaen et al. 1994; De Smet et al. 1994). Similarly, two of the genes from the Xp21.3 region (MAGE B1and B2) are expressed in a significant fraction of tumors from different histological origins (Lurquin et al. 1997). The identification of the tumor-specific antigen genes within these clusters is significant, because they might be candidates for immunotherapeutic intervention. In addition, the MAGE genes could be involved in hereditary disease as they could again provoke gene dosage changes in Xq28, or in Xp21.3, where the cluster maps within the critical region for the dosage-sensitive sex (DSS) reversal [locus duplication of that region results in a male-to-female sex reversal phenotype (Bardoni et al. 1994; OMIM no. 300018)].
X-linked ichthyosis (OMIM no. 308100) provides a well-characterized example of pathology caused by duplications at some distance from one another. This disease was mapped to Xp22.32, and ∼90% of ichthyosis patients were found to be deleted for the entire steroid sulfatase gene (Ballabio et al. 1989; Shapiro et al. 1989). Molecular analysis of the region revealed four homologous sequence elements, one distal and three proximal to steroid sulfatase (STS), distributed over 2.5 Mb. Subsequent studies showed that the majority of deletion patient breakpoints occurred within these homologous sequences, indicating recombination between these noncoding duplicated elements (Yen et al. 1990).
Pelizaeus-Merzbacher disease (PMD) is located in Xq22, and in many individuals is caused by a duplication of the proteolipid protein (PLP) gene (Woodward et al. 1998; OMIM no. 312080). Analysis of patient DNAs has shown that the duplication can vary from 500 kb to 1.65 Mb in length, although the patients all share the same distal end, differing in the proximal end of the duplicated region. Because affected males are homozygous for a variety of polymorphic markers in the region, it appears that the duplicated alleles are derived from the same chromosome. Therefore, the duplication may arise by intrachromosomal rearrangement.
Another disease that may be caused by a recombination between homologous but relatively distant repeats is the X-linked lymphoproliferative disorder (OMIM no. 308240). Several patients have similarly sized deletions in Xq25, and mapping of the region in the vicinity of the breakpoints with PCR-based markers shows that some sequences are repeated in the areas that border the deletions (Porta et al. 1997). In addition, a gene of the immunoglobulin superfamily occurs outside of the deletion borders but near the disease locus, and exhibits striking homology to natural killer (NK) receptors (Mazzarella et al. 1998). This observation may be a coincidence, but lymphoproliferative patients are deficient in NK cell activity (Sullivan et al. 1980), and there may be functional clustering of genes in the region.
Autosomal Clusters and Pathophysiology
Comparable events documenting the relationship between sequence duplication and disease have been observed on autosomes (Table 1). Figure 3 characterizes 12 instances, categorized roughly as arising from unequal crossover between clusters of related gene sequences (A–F), changes involving an intrachomosomal recombination step (G–K), and an example of a putative unequal crossover followed by gene conversion (L). Here we outline these cases further.
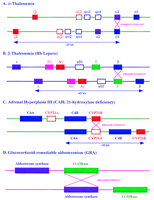
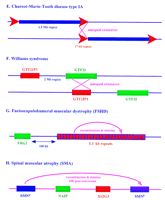
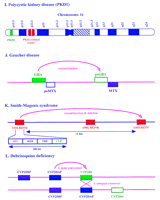
Probable recombination mechanisms between duplicated sequences in well-characterized autosomal diseases. These recombinations resultin in (A) α-thalassemia; (B) β-thalessemia; (C) 21-hydroxylase deficiency; (D) glucocorticoid-remediable aldosteronism; (E) Charcot-Marie-tooth disease type IA; (F) Williams syndrome; (G) facioscapulohumeral muscular dystrophy; (H) spinal muscular atrophy; (J) Gaucher disease; (K) Smith-Magenis syndrome, and (L) debrisoquine deficiency. Also shown is the physical relationship of duplicated sequences homologous to the polycystic kidney disease region (I).
The hemoglobinopathies (Fig. 3A,B) are classic autosomal examples of sequence duplication leading to human pathology (Maniatis et al. 1980;Collins and Weissman 1984; Orkin and Kazazian 1984; Antonarakis et al. 1985; Higgs et al. 1989). The hemoglobin tetramer is composed of two α (or α-like) subunits and two β (or β-like) subunits. The α gene cluster is found in chromosome 16p13.3 and is comprised of two active α genes (α 1 andα 2), two pseudogenes (ψα 1 andψα 2), and the embryonically expressed α-like ζ gene (ζ 2) and its pseudogene (ψζ 1). The majority of lesions of the α-globin gene cluster are the products of deletion, resulting in α-thalassemia (OMIM no. 141850; Fig. 3A). The α 1 andα 2 genes are nearly identical at the nucleotide level and encode identical proteins. The level of homology between the two genes extends ∼1 kb into the 5′ flanking region, and overall the two genes are highly homologous over 4 kb. Homologous exchanges appear to promote unequal crossover events, resulting in one chromosome with added globin genes and the other chromosome with less or no globin genes. As a result of such unequal genetic exchanges, individuals may have as many as 6 α genes, although the excess production of α-globin appears to have no negative consequences.
The β gene cluster is located at 11p15.5 and is composed of the embryonic ε gene, two fetalγ genes (Gγ andAγ), the δ gene, and the β gene and its pseudogene (ψβ 1). Deletions of the β cluster resulting in β-thalassemia (OMIM #141900; Fig. 3B) are rare as the homologous regions between the cluster members are limited to portions of the exons. One type of cataloged defect is of particular interest here: a deletion of ∼7 kb observed in patients with Hb Lepore. Analysis of these individuals reveals a hybrid gene produced by fusion of the 5′ portion of the δ globingene with the 3′ portion of the β globingene. This observation suggests that an unequal crossover has occurred between the adjacent genes. A similar recombination mechanism has been postulated for the fusion of Aγ andβ in patients with Hb Kenya.
Pathology based on numbers of repeats certainly can extend to dispersed gene families. The P-450 superfamily (involved in Fig. 3C,D,L) consists of >10 gene families and 100 genes that are localized to a least 6 different chromosomes, including 6, 7, 10, 15, 19, and 22 (Nebert et al. 1991). Four of these cytochrome P-450 enzyme families are responsible for the metabolism of numerous substrates including steroids and drugs (Nebert and Gonzalez 1987).
About 95% of the cases of congenital adrenal hyperplasia (CAH; adrenal hyperplasia III) are caused by deficiency of the enzyme 21-hydroxylase (21-OHase; OMIM no. 201910; Fig. 3C), which is one member of the cytochrome P-450 superfamily. This autosomal recessive disorder is based on events occurring within the MHC locus in 6p21.3 (Werkmeister et al. 1986). Molecular analysis of the region in normal individuals reveals two 21-OHase genes alternating with two complement four genes (C4A and C4B; Donohoue et al. 1986; Werkmeister et al. 1986). The 21-OHase genes include one inactive pseudogene (CYP21A) and the other (CYP21B) encoding the active gene product. Homology between the gene and the pseudogene is 98% in the coding regions and 96% in the intronic regions (White et al. 1985). The disease state arises from several different mechanisms including point mutation, deletion, duplication, and gene conversion. The latter three lesions probably result from recombinations at meiosis between the pseudogene and the active gene. One study has demonstrated that an unequal crossover between CYP21A and CYP21Bgenes results in deletion of the active gene, and that such crossovers occur at specific regions with the homologous genes (Donohoue et al. 1989).
Glucocorticoid-remediable aldosteronism (GRA) is a rare autosomal dominant disorder in 8q21 (also known as glucocorticoid-suppressible hyperaldosteronism; GSH; OMIM no. 103900; Fig. 3D; Lifton et al. 1992). Present in this region of the genome are another two members of the P-450 family, aldosterone synthase (CYP11B2) and steroid 11-β-hydroxylase (CYP11B1), which are 95% identical and arranged in a head-to-tail configuration. The GRA disorder arises from an unequal crossover between the two genes, which produces a chimeric gene containing the 5′ regulatory region of the11-β-hydroxylase gene fused to the coding sequence of aldosterone synthase. Aberrant expression of aldosterone synthase activity in the adrenal fasciculata results because its transcription is now controlled by adrenocorticotropic hormone (ACTH) because of the 11-β-hydroxylase regulatory sequences.
Pathology based on gene dosage and unequal crossing-over is seen in Charcot-Marie-Tooth disease type IA (CMT1A), a dominant peripheral neuropathy mapped to chromosome 17p11.2-p12 (OMIM no. 118220; Fig. 3E). A large 17-kb repeat is involved in the etiology of this disease. Two copies flank a 1.5-Mb region in unaffected individuals; but in patients, physical mapping of the region detected a tandem duplication of a 1.5-Mb segment (Pentao et al. 1992). Further analysis of chromosomes from patients showed that they contain three copies of the 17-kb repeat rather than the usual two. Duplication was suggested to have arisen by misalignment of the 17-kb repeat sequences followed by unequal crossover during meiosis. Thus, the duplication in most patients has been termed a kind of segmental trisomy (Matise et al. 1994; Schiavon et al. 1994). In striking support of this notion, Huxley et al. (1996) created a mouse model sharing many of the features of CMT1A by pronuclear injection of a yeast artificial chromosome (YAC) containing the locus.
Deletion rather than additional copies is involved in Williams syndrome, an autosomal dominant syndrome based on 7q11.23 (OMIM no. 194050; Fig. 3F). A similar deletion of ∼2 Mb, apparently arising independently many times, has been characterized in many affected individuals (Nickerson et al. 1995; Osborne et al. 1996). About 90% of Williams patients are hemizygous for the elastin gene, having deleted the copy from one chromosome (Nickerson et al. 1995). The mechanism of deletion is unknown, but is again likely to involve the pairing of homologous sequences and a crossover that loses the intervening DNA. Recent evidence indicates that the homologous sequences may involve theGTF2I gene and its pseudogene. The GTF2I gene encodes the transcription initiator binding protein TFII-I, a phosphorylation substrate for the Bruton’s tyrosine kinase, and maps near the telomeric breakpoint of the 2-Mb deletion; its pseudogene GTF2IP maps close to the centromeric breakpoint (Perez-Jurado et al. 1998).
Complex recombination and deletion events are thought to underlie facioscapulohumeral muscular dystrophy (FSHD), an autosomal dominant myopathy in 4q35 (Wijmenga et al. 1990; Lunt and Harper 1991; OMIM no. 158900; Fig. 3G). Analysis of a polymorphic EcoRI fragment tightly linked to the FSHD disease region revealed rearrangements in FSHD patients (Upadhyaya et al. 1991). Further analysis of this polymorphic marker showed that it can vary in size from 10 kb in affected individuals to 300 kb in normal individuals (Lee et al. 1995). The disease state has been correlated with the deletion of an integral number of 3.3-kb tandemly repeated units contained within the EcoRI fragment (van Deutekom et al. 1993). These repeats contain two known repetitive elements and two homeodomain motifs, although no corresponding transcripts have been detected (Hewitt et al. 1994). FISH experiments suggest that the repeated units are members of a 3.3-kb repeat family found in the heterochromatic regions of the genome (Lyle et al. 1995). This suggested that the deletion of an integral number of the repeats may lead to position effect variegation, repressing transcription of a nearby gene and thus leading to FSHD. A candidate gene (FRG1) has been identified ∼100 kb centromeric to the repeat. It appears to belong to a multigene family with related sequences on multiple chromosomes, although there is thus far no evidence for the postulated repression of its transcription in patients (van Deutekom et al. 1996).
Recombination/deletion or gene conversion can be invoked in spinal muscular atrophy (SMA), an autosomal recessive disorder that is classified into three forms [Pearn 1980; Melki et al. 1990, 1994; OMIM no. 253300 (type I); OMIM no. 253550 (type II); OMIM no. 253400 (type III); Fig. 3H]. All three types of SMA map to 5q11.2–q13.3, a region of the genome containing multiple copies of different markers and genes (Thompson et al. 1995; Wirth et al. 1995). Three cDNAs found in the region have been used as probes to detect deletions in various SMA patients. Both copies of the neuronal apoptosis inhibitory protein (NAIP) gene and the XS2G3 gene are deleted in ∼50% of the patients with the most severe form of the disease (type I) and may contribute to the severity of the disease (Lefebvre et al. 1995; Roy et al. 1995). The third gene, the survival motor neuron (SMN) gene, is present in two nearly identical copies, referred to as the centromeric SMN gene (SMNc ) and the telomeric SMN gene (SMNt ) (van der Steege et al. 1995; McAndrew et al. 1997). The SMNt gene is absent in 95% of SMA patients, as a result either of sequence conversion (SMNt conversion to SMNc , giving rise to two SMNc copies) or SMNt gene deletion (DiDonato et al. 1997a). Sequence conversion is in fact known to be a common event in the milder forms of the disease (types II and III) (DiDonato et al. 1997b).
Comparably complex interactions of multiple copies of a long (50 kb) region are involved in autosomal dominant polycystic kidney disease (ADPKD), one of the most common genetic diseases, with a reported incidence of 1 in 1000 individuals (Gabow 1991; Fig. 3I). There is considerable variability in the age of onset and severity of the disease. Some of the variability can be explained by linkage to different genetic loci, with polycystic disease 1 (PKD1) occurring on 16p13.3 (OMIM no. 601313), PKD2 on chromosome 4 (OMIM no. 173910), and PKD3 (OMIM no. 600666) as yet unmapped (Brook-Carter et al. 1994;Bogdanova et al. 1995; Daoust et al. 1995). In general, PKD1 appears to be a less severe form. The analysis of PKD1 has been complicated by the occurrence of at least three additional copies of a 50-kb region, containing the entire PKD1 gene with the exception of 3.5 kb at the 3′ end, on 16p13.1 (European Polycystic Kidney Disease Consortium 1994). The duplicated genomic regions are >95% identical. Interestingly, all of these copies produce polyadenylated transcripts but it is not known whether they encode proteins.
Gaucher disease (OMIM no. 230800; Fig. 3J) presents a relatively simple case of recombination-based deletion between repeated segments. The disease results from glucocerebrosidase deficiency and is the most common inherited lysosomal enzyme disorder. The glucocerebrosidase (GBA) gene is encoded by 11 exons (Choudary et al. 1985;Horowitz et al. 1989) and is located on chromosome 1q21 (Ginns et al. 1985). A pseudogene (psGBA) significantly contributes to the disease condition (Tsuji et al. 1987) and is located ∼16 kb telomeric to GBA (Winfield et al. 1997). A number of mutations occurring in the pseudogene are detected in the encoded products of patients affected by the disease, apparently resulting from recombination between the two homologous sequences (Eyal et al. 1990;Latham et al. 1990; Zimran et al. 1990). In this case, the extent and evolutionary history of the duplication can be discerned partially. The duplication includes a second gene sequence for metaxin (MTX). One copy is adjacent and on the DNA strand opposite the psGBAgene; a corresponding pseudogene (psMTX) is nearby on the same strand. Analysis of sequence from the region indicates that the overall duplication extends ∼14 kb, and occurred at an evolutionary time before the insertion of a 6.1-kb segment and several Alu sequences (Winfield et al. 1997).
A “common deletion” spanning 5 Mb is also seen in more than 90% of the patients affected with Smith-Magenis syndrome (SMS) in chromosome 17p11.2, ∼500 kb proximal to the CMT1A disease region (Chen et al. 1997; OMIM no. 182290; Fig. 3K). Analysis of the region revealed three 200-kb low-copy repeats, two flanking the deletion and one in the middle of the deleted region. Further characterization of the repeats has shown that each repeat represents a gene cluster containing significant homologies to four different genes: coactosin-like protein (CLP), signal recognition particle (SRP), type-I keratin (KER), and the TREoncogene (TRE). It is unclear whether these genes are functional copies or pseudogenes. Examination of patient DNA showed that recombination almost always occurred between the proximal and distal repeats, presumably by intrachromosomal rearrangement, although other mechanisms are possible.
Still another instance involving the P-450 superfamily (Fig. 3, cf. L with C and D) involves a gene cluster of four members (theCYP2D subfamily cluster) localized at 22q13.1. It contains the functional CYP2D6 gene and two highly homologous pseudogenes, and is important in the metabolism of ∼20% of commonly prescribed drugs (Gonzalez et al. 1988; Kimura et al. 1989; OMIM no. 124030; Fig.3L). Five to 10% of white populations are poor metabolizers of the antihypertensive debrisoquine and other plant alkaloids because of a genetic deficiency at the P-450 CYP2D6 locus (Meyer et al. 1990). Several haplotypes of this gene cluster have been identified by restriction fragment length polymorphisms (Skoda et al. 1988). One of the haplotypes was found to contain four CYP2D-related genes, instead of the three found in most individuals (Heim and Meyer 1992). Comparison of the genes suggests that an early point mutation was followed by a crossover and gene conversion event. This would result in a net yield of three pseudogenes and a mutant CYP2D6 gene, resulting in the deficient metabolism of debrisoquine and other drugs (Heim and Meyer 1992).
Homologous recombination between repeated sequences has also been implicated as the mechanism by which a “common deletion” is produced in Prader-Willi syndrome (PWS; OMIM no. 176270) and Angelman sydrome (AS; OMIM no. 105830) patients in chromosome 15q11–q13 (Christian et al. 1995; Huang et al. 1997). Seventy percent of the individuals afflicted with PWS and AS result from a ∼4-Mb deletion in the parental and maternal genomes, respectively. In addition, this region is subject to duplications and supernumerary marker formation. The recent construction of a detailed YAC map encompassing the region should aid in the resolution of which elements are responsible for this chromosomal rearrangement (Christian et al. 1998).
Low-copy repeats that lower the dosage of critical genes may also be involved in the deletion events seen in DiGeorge syndrome (DGS; OMIM no. 188400) and Velocardiofacial syndrome (VCFS; OMIM no. 192430). These syndromes are caused by haplo insufficiency of genes in chromosome 22q11. DGS is the more severe of the two disorders, including the VCFS phenotype as well as additional abnormalities. About 80%–85% of the DGS/VCFS patients have been shown to have deletions of more than 1 Mb. Using FISH it has been shown that several low-copy repeat families flank the DGS/VCFS locus (Halford et al. 1993). Recently, a novel transmembrane protein has been identified as deleted in >80% of VCFS patients (Sirotkin et al. 1997). Further molecular studies of the region should specify or discount the role of the low-copy repeats in the deletion mechanism.
Although we have concentrated on nuclear events, we note that comparable sequence duplications and comparable consequences are also observed in the human mitochondrial genome. For example, one-third of the patients with Kearns-Sayre syndrome (KSS) have a “common deletion” of their mitochondrial mtDNA, sometimes associated with a tandem duplication (Holt et al. 1988: Zeviani et al. 1988; Moraes et al. 1989; OMIM no. 530000). This common deletion was found to be mediated, presumably through homologous recombination, by a 13-bp repeated sequence present in normal mitochondrial DNA (Schon et al. 1989; Mita et al. 1990). Furthermore, it appears that duplications of the region are more prevalent in heart tissue, an observation possibly correlated with the extremely high numbers of mitochondria in cardiomyocytes (Fromenty et al. 1997).
The incidence and variety of such pathological changes in DNA would be still further sharply increased if deletions or additions that involve highly repetitive elements distributed throughout the genome were also included. These range from the expansion of microsatellite repeats to crossovers between copies of Alu or other repetitive elements, all of which have been excluded here to focus on locally duplicated longer sequence tracts.
Summary: Consequences of Clustering
Detailed structural analyses of the genome are increasingly revealing clusters of sequence that provide a snapshot of evolution generating new genetic possibilities. In simple cases, nearby repeated sequences can lead to deletions, inversions, and the production of considerable diversity. A second source of clustering arises when a sequence moves from one genomic site to another. This creates the possibility for coregulation of the juxtaposed sequences, even when they are quite dissimilar.
The significance of duplications is thus dependent on their frequency, dosage effects, and location, as well as the time at which they occurred in human evolution. For example, classic examples show that newly arising extra copies of a gene or a chromosome (as in the extreme case of trisomy 21) can be as detrimental as deletions. The repertoire of possible pathology then naturally increases for gene duplications that have had time to diverge. In an extreme example, two neighboring transporter genes on chromosome 7 that have diverged considerably cause two different diseases when mutated [Pendred syndrome (Everett et al. 1997) and congenital chloride diarrhea (Hoglund et al. 1996)].
The relative contributions of deletions/additions and point mutations to genetic pathology will depend on factors like the size of the gene, incidence and severity of the effects of lesions, and selective factors. In this context, nearby repeats increase qualitatively the frequency of dynamic changes in DNA composition; in instances such as Williams syndrome, color blindness, and hemophilia A, those changes are involved in a very large fraction of analyzed cases. The highest incidence can be quantitated in well-studied examples like hemophilia A, where the overall incidence is ∼1 in 10,000 to 20,000, and ∼40% of cases arise from repeat-catalyzed inversion events (Fig. 2; Table 1). This frequency is thus comparable to the combined incidence of all other deleterious changes in a gene that spans 180 kb of genomic DNA. In other cases, like Williams syndrome, rates of 1 in 100,000 are not uncommon.
To estimate the potential impact of the range of such effects, we can ask just how frequent are duplications that are evolutionarily deep-seated potential sources of pathology? Long-range sequencing on the X chromosome has progressed far enough to suggest that levels of 5–10% of the genome are duplicated at least once (as in Figs. 1 and2). On autosomes, sequencing is only now beginning to accumulate rapidly (and regions with duplications are generally harder to sequence). Nevertheless, it is notable that in situ studies with cosmid probes on chromosome 7 found more than one site for the order of 10% of the cosmids (Green et al. 1994), and Korenberg et al. (quoted inPennisi 1998) have found similar levels of cross-hybridizing loci for bacterial artificial chromosomes, particularly in gene-rich pericentromeric and subtelomeric regions.
Therefore, if we speculate that duplications comparable to those we discuss here are likely to be spread through the genome, then every individual will have an appreciable chance of having undergone such an event (that is, an inversion, addition/deletion, or deletion occurring at a rate of 1 in 10,000 to 100,000 per gene, and taking place in any of 10,000 susceptible genes). Thus, the limited number of examples shown here are a very small tip of a very large iceberg. The precise determination of the extent of duplications is coming from the sequencing of the human genome that has already provided some of the examples, and will continue to provide the probe reagents to assess the range of incidence of variation in copy number, inversions, and deletions.
What Are the Practical Consequences for Genetic Investigations?
First, workers investigating a genetic locus are well-advised to ask what are the neighboring genes. The number of instances in which the next gene is potentially relevant to functional analysis is going up very rapidly. For example, in two instances in which we have been recently involved in some of the studies, the X-linked anhidrotic ectodermal dysplasia (EDA) gene is juxtaposed with two other genes that show high levels of expression in skin (Kere et al. 1996) and the gene responsible for the Simpson-Golabi-Behmel syndrome, encoding glypican 3 (Pilia et al. 1996), turns out to be next to the glypican 4 (Watanabe et al. 1995) gene (work in progress; see GenBank accession no. AC00240).
Second, the processes of nearby duplications and interactions give rise to very appreciable diversity between individuals and populations.
Third, inversions, deletions, and other changes in DNA are favored by these clusters. They occur at frequencies on the order of 10−4–10−6, sufficient to result in very significant contributions to the comparable rates of incidence of genetic disease.
Acknowledgments
We thank our colleagues, including Lucio Luzzatto, Dan Longo, Reid Huber, and Giuseppe Pilia, for careful reading and suggestions.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL schlessingerd{at}grc.nia.nih.gov; FAX (401) 558-8331.
- Cold Spring Harbor Laboratory Press








