
High-Precision Genotyping by Denaturing Capillary Electrophoresis
Abstract
Genotyping, as applied to linkage mapping, human identification, or mapping of genetic traits, mandates electrophoretic separation systems that enable a user to identify alleles with high precision to obtain a correct genotype. For 2-bp microsatellites or short tandem repeats (STRs), standard deviations of ±0.3 nucleotide are required to ensure with 99.7% probability the identity or dissimilarity of tested alleles. A complete system, consisting of commercially available laser-induced fluorescence capillary electrophoresis (ABI PRISM 310) and performance optimized polymer 4 (POP-4), was evaluated for microsatellite separations. POP-4 is a low viscosity polymer for use in uncoated fused microbore silica capillaries. It separates DNA fragments that differ in size by 1 nucleotide up to 250 nucleotides and that differ in size by 2 nucleotides for fragments up to at least 350 nucleotides in length in about 30 min. The presence of denaturants and, more importantly, operation at 60°C was mandatory for high-precision and high-resolution sizing operation. Reproducible separation performance was achieved in excess of 100 injections per capillary with resulting standard deviations in the range of 0.04 to 0.17 nucleotide. Comparative sizing of known CEPH (Centre d’Etudes du Polymorphisme Humaine) samples performed at 22 independent test sites showed the usefulness of the system for genotyping with standard deviations of 0.24 nucleotide, or better.
Precise and reproducible sizing of DNA fragments generated by PCR amplification has become a fundamental technology in genetic analysis (Dracopoli et al. 1995). The routine implementation of high-resolution slab gel electrophoresis is responsible for significant advances in human disease genetics (Hazan et al. 1992;Hearne et al. 1992; Schuster et al. 1996), agriculture genetics (Ron 1994; Georges et al. 1995), and forensics (Fregau et al. 1993; Kimpton et al. 1993; Robertson et al. 1995). In recent years, analysis of fluorescent-labeled PCR amplified short tandem repeats (STRs) (Weber 1990; Edwards et al. 1991a,b; Beckman and Weber 1992) resulted in significant gains in throughput and automation. STR analysis requires the size comparison of DNA fragments that may differ in length of as little as 1 nucleotide. Indeed, the routine genotyping that uses microsatellite markers containing dinucleotide repeats presents the greatest challenge as precision and resolution must be simultaneously optimized. Although production-style high-throughput laboratories based on slab gel electrophoresis have been established, gel preparation and sample loading have been the areas most refractory to full automation. Alternatives are continually sought to fully automate high-resolution electrophoresis. Since the first descriptions of electrophoresis in small diameter tubes (Mikkers et al. 1979; Jorgenson and Lukacs 1981), capillary electrophoresis (CE) has been identified as a possible replacement for slab gel-based electrophoresis of nucleic acids (Stellwagen 1987; Landers et al. 1993; Karger et al. 1995). Because of the large surface area of a capillary, heat generated during electrophoresis is efficiently dissipated, allowing for high-voltage electrophoresis runs resulting in rapid analysis. Capillary electrophoresis has the additional benefits of automated filling of the capillary with separation medium and automated sample loading. Thus, CE represents a viable alternative to slab gel-based electrophoresis systems, as the entire process is amenable to full automation.
Although enormous interest has been generated recently in CE for high-resolution DNA fragment analysis, no coherent criteria have been established to gauge the performance of a CE system. The electrophoretic properties governing the separation of DNA fragments differ between CE- and slab-based systems. Those schooled in the art of slab gel electrophoresis cannot solely rely on this collective experience to adequately assess CE. In this report, we identified the critical criteria that influence the performance of automated CE and present experimental data to test these parameters.
For capillary electrophoresis to be useful and accepted in high-resolution DNA fragment analysis, the following criteria must be addressed:
- 1.
- Single-nucleotide separation: Genotyping requires the separation of (mostly rare) length variant alleles from common alleles that can differ in size by one nucleotide. PCR amplification often results in the nontemplated addition of a nucleotide to the 3′ end of the amplification product (Smith et al. 1995; Brownstein et al. 1996). This activity of Taq polymerase can produce a ladder of DNA fragments that differ by 1 nucleotide. In the case of STR markers, regardless of the size of the repeat unit (2, 3, or 4 nucleotides), single-base separation is desired to precisely score alleles. This required high resolution should ideally encompass the size range between 70 and 350 nucleotides that is typically encountered during microsatellite amplification.
- 2.
- Standard deviation (s.d.) in sizing of ±0.15 nucleotide: In the automated genotyping of individuals, allele designations are determined by placing a sized DNA fragment in a size bin. For example, binning of alleles would place fragments of 129.79, 129.93, 130.10, and 130.19 nucleotides all in the 130-nucleotide bin. To achieve a 99.7% confidence in binning with single nucleotide separation, nonoverlapping bins should be established with ±0.45-nucleotide range. This allows for a series of bins of 0.90 nucleotide to be established. For the example given above, the bin would be 130 nucleotides ± 0.45 and would not overlap with the next possible bin at 131 nucleotides ± 0.45. Because 99.7% confidence is derived from 3 standard deviations of sizing precision, the ±0.45-nucleotide range is achieved with 0.15 nucleotide (or 0.45/3) (s.d.) in sizing.
- 3.
- Flowable, low viscosity polymer: The sieving medium should be flowable and of low viscosity to allow the refilling of the capillary after each analysis. A replenishable polymer system extends the life time of a capillary, prevents polymer contamination, and avoids sample carry over.
- 4.
- Polymer compatible with additives: To achieve the high precision required, the polymer should be compatible with denaturants. This allows for increased sizing precision by suppressing secondary structures of DNA strands that could affect their mobility and, therefore, their calculated size.
- 5.
- Polymer provides dynamic coating of capillary: Under high electric fields and elevated pH, uncoated fused silica capillaries impose a force on the separation medium, resulting in a process known as the endosmotic flow (EOF). This unwanted flow is opposed to the direction of migration of nucleic acids. Although coated capillaries can be used to suppress EOF, they are difficult to reliably manufacture and quality control. A sieving polymer used in an automated CE system should apply a dynamic coating that is renewed with each new filling, and thereby efficiently minimizing EOF.
- 6.
- High sensitivity: Post-PCR processing of samples to reduce volume and increase DNA concentration would add both time and costs to a CE system, while at the same time adding an additional step that would be difficult to automate. The system must provide high-detection sensitivity to allow loading of the capillary directly from the PCR reaction and unambiguous detection of signal by automated software.
- 7.
- Unattended operation: To keep up with throughput constraints, the CE system should refill the capillary with fresh polymer after each analysis and load samples sequentially without any user intervention.
- 8.
- Low cost: To compete with conventional gel electrophoresis, the cost per analysis by CE should be comparable.
We present here the validation of a capillary electrophoresis-based automated DNA fragment analysis system. Information derived from our internal research and development efforts, as well as evaluation and testing by worldwide test sites, has allowed us to establish these criteria as essential for a successful CE system. As new instrument systems are developed, and as modifications to existing systems are suggested, a consisting methodolgy for evaluation must be used.
Although in the past, reports have described CE systems that are based on different polymeric separation media in regard to sizing accuracy and resolution (Cohen et al. 1988; Chin and Colburn 1989; Zhu 1989;Heiger et al. 1990; Bocek and Chrambach 1991; Sudor et al. 1991;Grossman 1992), we describe a novel linear polyacrylamide-derived commercially available polymer system specifically developed for high-resolution and high-precision fragment analysis on the ABI 310 Genetic Analyzer. An early version of this separation system, GeneScan polymer (GSP), was shown to be useful for identifying mobility abnormalities in double-stranded bent DNA (Wenz 1994). It has been evaluated for the separation of PCR fragments and restriction digests under denaturing and nondenaturing conditions (Williams et al. 1994), and for determining single-stranded conformation polymorphism (SSCP) (Atha et al. 1998; Inazuka et al. 1997), and, thus, was an obvious choice to be part of the 310 Genetic Analyzer. Although this polymer mixed with urea as a denaturant shows sufficient separation at ambient temperatures to achieve reproducible sizing for 3-nucleotide-repeat STRs, insufficient denaturation conditions rendered its use impossible for high precision applications. Therefore, the polymer was developed further into Performance Optimized Polymer 4 (POP-4). This ready-to-use polymer, containing denaturants (Rosenblum et al. 1997), allows for separation at a temperature of 60°C, which proved to be a necessity for highly precise runs, probably by uniformly melting out intrastrand secondary structures.
The improved denaturation conditions, together with an internal standard added to the sample, results in s.d.s in sizing ranging from 0.06 to 0.19 nucleotide in CEPH family DNAs typed with Genethon dinucleotide repeat markers. A world-wide study involving 22 test sites showed the applicability of POP-4 for multi-user genotyping studies. Recently, a study using POP-4 demonstrated with microsatellite analysis the linkage of autism to a chromosomal duplication (Cook et al. 1997).
RESULTS
Automated Instrument System
We have developed a CE instrument capable of simultaneous multicolor detection and high-resolution separation of DNA fragments. This instrument, the ABI PRISM 310 Genetic Analyzer, is highly automated. In developing this instrument system, we focused on the most demanding of applications, that is, the automated fluorescent analysis of PCR-amplified STR loci. On our system, alleles from an STR locus are PCR amplified from human genomic DNA with one unlabeled primer and one primer labeled at the 5′ end with a fluorescent dye. Denatured PCR products are then coelectrophoresed with an internal size standard (DNA fragments of known size labeled in a different dye color). Multiplex STR amplification products (held in a 48- or 96-well tray) are sequentially injected into a single capillary and detected in real time as they electrophorese past a laser-detection window near the end of the capillary. The laser-induced fluorescence is detected on a CCD camera that simultaneously detects all wavelengths from 525–680 nm. The instrument automatically reloads fresh polymer into the capillary between injections. Each capillary has a lifetime of a least 100 injections, so 96 samples in a single tray can be analyzed by the instrument while completely unattended by the user. The collected data is then analyzed by software that automatically determines allele sizes on the basis of a standard curve from the internal size standard.
Polymer Evaluation for Genotyping
We selected two different STR systems to evaluate the performance of polymers for separation and precision: (1) an allelic ladder derived from the human THO1 locus, which is often used to describe the characteristics of a particular separation system (Williams et al. 1994; Butler et al. 1995; Wang et al. 1995; Mansfield et al. 1996; Wang et al. 1996), and (2) DNAs from four different CEPH families, amplified with Genethon dinucleotide repeat markers (Dib et al. 1996).
The CEPH family-derived samples represent the most demanding separation requirements, because use of dinucleotide repeat containing microsatellites require separation of fragments that differ in size by 2 nucleotides. Taq DNA polymerase often nonspecifically adds an additional nucleotide to the 3′ end of the amplification product (Smith et al. 1995; Brownstein et al. 1996). In these cases, 1-nucleotide separation is required. We selected DNAs from four different CEPH families to track the same alleles both within one family and across the four families. Alleles of the same size from the same locus will not necessarily be of identical sequence because of the accumulation of unique mutations within a family (Garza and Freimer 1996). Although these sequence differences are slight across the different families, their calculated sizes can differ when analyzed under conditions of low denaturation.
Initially, to separate both repeat systems, GSP was used under two slightly different conditions (see Methods): GSP1 to separate the fragments of THO1 (Fig. 1A) and GSP2 to separate the fragments within the CEPH samples (Fig. 3A, below). All THO1 ladder fragments (5, 6, 7, 8, 9, 9.3, and 11) could be resolved, covering a size range of ∼154 to 178 nucleotides. Whereas most of the fragments within the ladder are separated by 4 nucleotides, the difference between alleles 9 and 9.3 is only 3 nucleotides, and the difference between alleles 9.3 and 11 is 5 nucleotides. The resolving power of the polymer was assessed with DNA from an individual known to be a 9.3/10 genotype, which requires 1-nucleotide separation (Urquhart et al. 1994). As shown in Figure 1A (inset), both alleles can be resolved, but under these conditions the separation for this particular genotype was not always reliable.
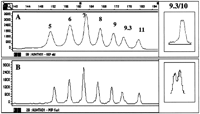
Separation of the allelic ladder HUMTHO1. THO1 was separated by use of condition GSP1 (A) or POP-4a (B). The number of repeat units is indicated. A size scale is aboveA. The separation of the genotype 9.3/10 of THO1, by the described conditions, is shown in the insets.
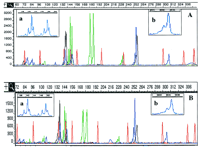
Separation of CEPH sample 1340-033. CEPH sample 1340-033 was separated by use of condition GSP2 (A) or POP-4b(B). The separation of the 12 loci of the sample between 60 and 352 nucleotides is displayed. Insets a and b show a magnification of size ranges, as indicated by the scale above each. Peaks in a are 1 nucleotide apart, and in b, 2 nucleotides.
To ensure with 99.7% confidence a fragments identity, the separation system should reproducibly size DNA fragments of similar length with as.d. of ±0.15, ±0.3, ±0.45, and ±0.63 nucleotide for fragments that differ in size by 1, 2, 3, and 4 nucleotides, respectively. To determine the precision of sizing for all fragments of THO1, the allelic ladder was injected multiple times into the same capillary and the s.d. for the mean size for each allele was determined. As Figure 2 shows, the calculated standard deviations of 0.3 nucleotide or better for all alleles would suggest that precise genotyping for 2-nucleotide alleles with GSP is achievable, but not for samples that contain a genotype ofX/9.3 or X/10, where X represents any allele of THO1.
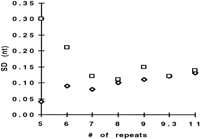
Standard deviation for all alleles in the allelic ladder THO1. The allelic ladder THO1 was separated with either GSP1 (□) or POP-4a (⋄). For the calculation of the standard deviation of sizing for each allele, the sample was injected 15 times in the same capillary.
Although conditions were applied that allowed the precise sizing of standardized allelic ladder fragments, we tested this polymer on samples derived from different CEPH families, which represent a more demanding system in regard to resolution and precision of sizing caused by possible sequence differences between alleles of the same size. As Figure 3 shows, condition GSP2 separates 1-nucleotide differences at least up to 190- and 2-nucleotide differences up to at least 300 nucleotides.
Table 1 shows representative results for sizing alleles in the four different CEPH families for five of the dinucleotide repeat markers by use of GSP. The sizing within each family is very precise, therefore permitting in most instances 1-nucleotide discrimination (A–D). Yet, when averaging the sizing across all four families (E), some loci, like D9S164 and D11S968, show very low s.d.s, whereas others, like D11S934, D10S591, and D11S1320, show larger variations in sizing. Standard deviations for alleles in all 12 loci analyzed varied between 0.04 and 0.50 nucleotide. This variation makes genotyping, as required for multifamily studies impractical. Analysis of the same samples on an automated slab gel sequencer showed for all 12 loci s.d.s that range from 0.05 to 0.12 nucleotide. By use of similar urea concentrations as a denaturant (6.6–8 m), the slab gel system is operated at higher temperatures than the 30°C used for CE. This suggests that inadequate heat denaturation, under GSP conditions, may be responsible for the observed low precision when analyzing data across CEPH family samples. Increasing the CE electrophoresis temperature, with the GSP polymer system, to 42°C did not show precision data comparable with the slab gel system (not shown). Temperatures >42°C resulted in unacceptable loss in resolution as evidenced by broadening of peaks.
Calculation of Standard Deviations for Four Different CEPH Families
Because loss of capillary wall coating of the polymer at higher temperatures was thought to be involved in the loss of resolution, we further developed the polymer to a version (POP-4) having a higher molecular weight than GSP. By replacing GSP with POP-4, the separation distance between peaks of the allelic ladder THO1 remains largely the same. Decreased peak width, however, improves the resolution considerably (Fig. 1B). This enhanced resolution increases the 1-nucleotide separation between genotype 9.3/10 of THO1 (Fig. 1B, inset).
Analyzing the data in the context of sizing precision showed that for POP-4 an improved standard deviation for all alleles in THO1 was achieved. Specifically, s.d.s are below ±0.15 nucleotide, even for alleles 5 and 6 that showed higher s.d. with GSP (Fig. 2). We also found that this increased performance with POP-4 improved the accuracy of sizing for the THO1 alleles. With the GSP protocol, the size difference for alleles of THO1, having as.d. of ±0.15 nucleotide or less, is close to the expected, as a result of the number of repeat units. The difference between alleles 5 and 6, and 6 and 7, however, is considerably larger than would be expected (Table 2, DE vs. DS/GSP). Coincidentally, alleles 5 and 6 show standard deviations that are considerably higher than those of the adjacent alleles (Fig. 2). Separating the allelic ladder THO1, by use of increased POP-4 denaturation conditions, results in a size spacing between all alleles approaching the expected values (Table 2, DE vs. DS/POP-4).
Sizing Accuracy for Allelic Ladders HUMTHO1
The separation of CEPH families samples with POP-4 consistently resolved 1-nucleotide differences up to 250 nucleotides, and 2 nucleotides differences up to at least 350 nucleotides (Fig. 3B) with a cycle time of about 31 min. Reanalysis of the CEPH families samples with POP-4 shows for the representative five loci standard deviations that have improved over the previous analysis with GSP, now allowing the discrimination of 1-nucleotide size differences (Table 1, F). Thes.d.s for alleles in all 12 loci varies from ±0.04 to 0.17 nucleotide and is comparable with data obtained from slab gels.
Temperature Effect on Fragment Sizing
To further show the need of high temperature for denaturation and, therefore, better sizing precision, the following experiment was performed. GeneScan 2500, a size standard having random sequence fragments, labeled with TAMRA (N,N,N′,N′-tetramethyl-6-carboxyfluorescein) fluorophore on both strands of the DNA fragments, was mixed with GS 500, a size standard bearing a TAMRA fluorophore on only one strand of the DNA fragments; the GS 500 size standard was used to size fragments from GS 2500. After denaturation, the samples were separated by use of condition POP-4b at increasing temperatures, ranging from 30°C to 70°C.
All fragments of GS 2500 shown in Figure 4 can be detected as two individual strands at low temperatures. At elevated temperatures, most fragment strands begin to comigrate, or at least, get very close to each other in size. (Fig. 4B, C). One exception is the 94-bp fragment, in which both strands of the fragment migrate evenly separated at all temperatures (Fig. 4 A). With increasing temperature, all fragments of GS 2500 change their mobility relative to the size standard fragments, and, therefore, are sized differentially. These changes in size can be moderate, as low as ∼3 nucleotide (Fig.4A), or more severe 14 nucleotide (Fig. 4C). Whereas the changes in size for each fragment vary, all fragments are sized at higher temperatures at sizes that are close to their actual size on the basis of their known sequence. Although only three examples are shown to demonstrate the different kinds of mobility changes in response to changes in electrophoresis temperature, all fragments examined within the GS 2500 size standard showed varying degrees of mobility changes (H.-M. Wenz, unpubl.).
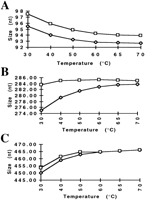
Change in fragment size with increasing temperature. GS 2500 and 500 size standards were mixed and separated by use of condition POP-4b, with increasing temperatures (30°C–70°C). Changes in size for both strands of a fragment from GS 2500 are indicated. The known sequence-based sizes for the three fragments are 94 (A), 286 (B), and 470 nucleotides (C).
Performance Evaluation of POP-4
To ensure consistent performance under the stringent POP-4 denaturation conditions, two additional tests were conducted: First, four CEPH family (1347) samples were analyzed by 22 independant test sites in the United States, Europe, Japan, and Australia. The sizes and standard deviations for alleles, representing six loci, were determined after injecting each sample six times. Table3 shows a summary of the results from the test sites. The sizing and precision data show very reproducible results across 22 different laboratories. For six of the alleles, the standard deviations of ≤0.15 nucleotide allow the identification of alleles that differ by 1 nucleotide, and the remaining three alleles fall within a 2-nucleotide bin.
Test Sites Results
Second, the capillary life span was assessed. The denatured sample, CEPH 1347-02, was injected 100 times into the same capillary over a period of 52 hr, by use of condition POP-4b. After placement of the samples into the autosampler, no additional user intervention was allowed. Figure 5 shows electropherograms of injections 1 and 100 and demonstrates that no considerable change in sample appearance has occurred with a capillary that was in use for 100 sample injections over a period of 52 hr. The sizing range of over 100 injections spans from 0.21 to 0.79 nucleotide for all loci within the sample with corresponding s.d.s of 0.06 to 0.19 nucleotide.
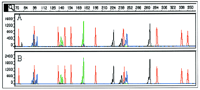
Separation of CEPH sample 1347-02. Electropherograms for injections 1 (A) and 100 (B). Separation conditions are POP-4b. Multiple samples were prepared from sample 1347-02; each sample was injected 5×.
DISCUSSION
The goal of this research was to establish separation conditions for a commercially available CE system that allows the comparative sizing of microsatellites with a precision necessary to allow genotyping for applications such as linkage studies (Hazan et al. 1992;Hearne et al. 1992; Schuster et al. 1996), human identification (Ron 1994; Robertson et al. 1995), or agricultural trait mapping (Ron 1994;Georges et al. 1995).
Although the separation performance with POP-4 is sufficient for most genotyping experiments, the need of 1-nucleotide separation beyond 250 nucleotides might be required in certain instances. A higher concentrated polymer formulation (POP-6), recommended for DNA sequencing, provides 1-nucleotide separation up to at least 400 nucleotides.
The inclusion of an internal size standard with each sample, which is possible when using a multi-color detection system like the ABI PRISM 310, increases the sizing precision by compensating for run-to-run variations (Ziegle et al. 1992). As we have shown, however, it is equally important to provide electrophoretic separation conditions that overcome intra-strand hybrid structures that result in anomalous migration behaviour manifested in poor reproducibility and inaccurate size determinations.
We have presented performance data for two polymers, GSP and POP-4. These formulations were evaluated for the sizing of allelic PCR fragments that contain STRs. As Figure 4 suggests, the application of elevated electrophoresis temperatures, as used with POP-4, improves the accuracy of sizing, presumably by melting secondary intrastrand structures of the single stranded DNA molecules that effect the mobility.
Similar to the data from the multifamily study, in which fragments of similar size from a particular locus have considerable sizing differences under low denaturing conditions (Table 2, A–D) possibly caused by to the accumulation of unique sequence polymorphisms within a family (Garza and Freimer 1996), in SSCP (Orita et al. 1989) single-stranded DNA fragments of similar length, but heterogeneous nucleotide sequence, have differences in mobility, and, therefore, differences in their apparent sizes. Interestingly, the analysis of SSCP can be seen in the presence of denaturants, like 50% urea, or 10% formamide (Glavac and Dean 1993), and Konrad and Pentoney (1993)report that in the presence of 7 m urea, at 35°C, conformations exist in oligomers that affect the electrophoretic mobility.
Whereas the enhanced denaturation conditions clearly improve the sizing accuracy, the calculated sizes still deviate from the real, sequence-based size. This might be attributable to the assumption that the DNA fragments are still not yet completely denatured. Although this is possible for DNA fragments with unusual base compositions, it is more likely that differences in mobility are caused by the different chemistry of the fluorophores used in these studies to label the DNA fragments and their interaction with components of the separation system. The dyes used to label the internal size standard are based on rhodamine (TAMRA or ROX) and differ in their chemical structure from the fluorescein dyes that are used to label the sample DNAs, being either 5-carboxy-fluorescein derived (5-FAM) or 6-carboxy-fluoresceins (FAM, JOE, HEX, and TET). Whereas identical fragments that are labeled with dyes of the same chemical family, when coelectrophoresed, virtually comigrate, the same fragments, labeled with dyes from different chemical families exhibit noticeable mobility differences in the polymer systems used here (Mansfield et al. 1996; H.-M. Wenz, unpubl.). Similar observations have been made in one-lane multi-color DNA sequencing, in which dye-imposed mobility shifts are corrected by applying mobility correction files (Connell et al. 1987). To increase the accuracy in sizing, it might be possible to calculate a normalizing factor (Mansfield et al. 1996) or, alternatively, energy-transfer fluorescent dye-labeled DNA primers (Ju et al. 1995; Wang et al. 1995) that exhibit only very minor size differences could be used.
More important than the absolute sizing accuracy, however, are highly reproducible analyses (Pritchard et al. 1995; Wang et al. 1996). These analyses should provide consistent genotyping for the same allelic STR, when using similar instrumentation and run conditions, independent from the genomic source and from run-to-run. The determination of the standard deviation of DNA fragments that show variability in sizing, like alleles 5 and 6 in the allelic ladder THO1 should provide a strong indicator of the denaturation power delivered by a particular separation system, and, therefore, the achievable sizing precision. The relatively large s.d. in sizing of 0.3 and 0.22 nucleotide for these two fragments with GSP, relative to the other allelic fragments (<0.15 nucleotide) under low denaturing electrophoresis conditions might result from sequence-imposed incomplete denaturation of intrastrand portions that are sensitive to slight variations in the electrophoretic environment. Only in a highly denaturing environment achievable with POP-4, consisting both of added denaturants and high temperature are these structures melted out, behaving similar to the other fragments and result in standard deviations of <0.15 nucleotide for all fragments.
THO1 has been widely used to assess the separation performance of different CE systems. A short review of the literature supports our finding that the sizing precision depends substantially on the applied denaturation conditions: Standard deviations for alleles 6 to 10, that were derived from different amplified samples, were between 0.31 and 0.69 bp when separated at 22°C in nondenaturing 0.8% hydroxyethyl cellulose (HEC), containing 1 μm aminoacridine (Wang et al. 1995). More stringent electrophoresis conditions of 2% HEC, 6m urea and 10% formamide at 22°C showed s.d.for alleles 7–9 of 0.18 nucleotide, but a s.d. of 0.34 nucleotide for allele 6 (Wang et al. 1996). By use of a similar polymer mix at room temperature, Mansfield et al. (1996) report s.d.s for the THO1 allelic ladder fragments ranging from 0.10 to 0.14 nucleotide. The s.d. for alleles 5–11 ranged between 0.10 and 0.37 bp in a capillary filled with cross-linked polyacrylamide and buffer containing 10 μm ethidium bromide (Williams et al. 1994). Interestingly, in this system the spacing between the individual alleles (4 bp) ranged from 4.11 to 2.92 bp, indicating possible conformational effects on the mobility of the fragments, making precise genotyping impossible. In 1% HEC at 25°C, the combineds.d. for all THO1 fragments was 0.14 bp in the presence of the intercalating dye YO-PRO-1. Reducing the dye concentration reduced the overall precision to 0.28 bp (Butler et al. 1995), indicating a relaxation of secondary structures that might interfere with the mobility of fragments.
As this study has shown, the polymer, either as a low-denaturing (GSP) or a high-denaturing formulation (POP-4), is useful for the genotyping of DNA fragments containing short tandem repeats on the ABI PRISM 310. Both formulations have sufficiently low viscosities to allow complete replenishment of the capillary content in 4 min or less. Uncoated capillaries, without any need for additional capillary wall treatment, can be used for at least 100 sample injections without any compromise in precision performance. The automation provided with the instrument allows the operator to realize significant labor savings by avoiding tasks such as gel pouring and sample loading. Also, high detection sensitivity allows the use of unpurified PCR samples, therefore, reducing reagents costs. Whereas GSP showed use in typing of fragments containing repeat units of ⩾3 nucleotides, we recommend POP-4 for all CE-based sizing application because of its superior precision for genotyping experiments.
METHODS
Polymer
GSP polymer (patent application no. PCT/US93/04078) and POP-4 (U.S. patent no. 5,552,028) both are hydrophilic polymers that provide molecular sieving and noncovalent wall coating, when used in uncoated fused silica capillaries (PE Applied Biosystems Division, Foster City, CA). GSP is provided as a 7% stock solution in water that can be diluted and mixed with different additives like urea or glycerol. POP-4, specifically formulated for use in microsatellite analysis, is a 4% polymer solution with 8 m urea and 5% pyrrolidinone. Both polymers have viscosities that allow the filling of a 47-cm-long capillary with an internal diameter of 50 μm in 4 min or less. Both polymers utilize genetic analyzer buffer with 1 mm EDTA (PE Applied Biosystems Division, Foster City, CA).
Instrumentation and Electrophoresis
For all experiments, a laser-based capillary electrophoresis instrument, the ABI PRISM 310 Genetic Analyzer (PE Applied Biosystems Division, Foster City, CA) was used. This instrument uses a multiline argon ion laser, adjustable to 10 mW, which excites multiple fluorophores at 488 and 514 nm. Fluorescence emission is recorded between 525 and 680 nm on a cooled CCD camera. This configuration currently allows the multiplexing and sizing of samples that overlap in size by use of three different fluorophores, plus an additional fluorophore that is attached to an internal size standard. The instrument controls temperature between ambient and 60°C with an accuracy of ±1°C. Electrophoresis voltage is controlled between 100 and 15,000 V. A sample tray holds 48 or 96 samples for unattended operation. Data are collected and automatically analyzed with an instrument specific collection software and GeneScan analysis software (PE Applied Biosystems Division, Foster City, CA). The separation medium is automatically replaced after each sample run. Samples are introduced by electrokinetic injection, typically for 5–10 sec at 7–15 kV. Table 4 provides electrophoresis conditions for the different experiments performed.
Electrophoresis Conditions
Samples
To evaluate the polymer formulations, two different sets of test samples were used. The first was the allelic ladder HUMTHO1 (THO1) with allelic fragments ranging in size from 154 to 178 bp (Table 2). Allele fragments in this ladder are separated by 4 bp, with the exceptions of alleles 9 and 9.3, which are separated by 3 bp, and alleles 9.3 and 11, which are separated by 5 bp. One primer used for the PCR amplification was labeled with the fluorophore 5-FAM (Robertson et al. 1995). The second sample set used DNAs from four different CEPH families (884, 1340, 1341, 1345; Table 1) with 10 different samples per family (Weissenbach et al. 1992). Samples were amplified with panel 15 primers from the Linkage Mapping Set (PE Applied Biosystems Division, Foster City, CA), resulting in the amplification of dinucleotide repeats of 12 different loci ranging in size from 82 to 332 bp. Overlapping loci were labeled with three different fluorescent dye phosphoramidites, allowing their simultaneous detection [(6-FAM)6-carboxyfluorescein; (TET)4,7,4′,7′-tetrachloro-6-carboxyfluorescein; (HEX)4,7,2′,4′,5′,7′-hexachloro-6-carboxyfluorescein]. Typically, 1 μl of a sample without any need for post-PCR sample workup, was diluted in 12 μl of deionized formamide. To each sample, 0.5 μl of an internal size standard (GS 350; PE Applied Biosystems Division, Foster City, CA), labeled with an additional fluorophore (TAMRA) was added for sizing and compensation of run to run variations. The capillary lifetime study was performed with CEPH sample 1347-02 from the Fluorescent Genotyping Demonstration Kit (PE Applied Biosystems Division, Foster City, CA); each sample was injected five times. All samples were denatured at 95°C for 5 min, followed by cooling. Denatured samples could remain on the CE instrument for at least 48 hr, without any loss in signal or change in sizing. For the temperature study, the size standards GS 500 and GS 2500 were used. Both standards are labeled with TAMRA. Whereas GS 500 has a fluorescent label on only one of its strands, GS 2500 is labeled on both strands. GS 500 is a standard containing multiples of monomeric size blocks, GS 2500 is a PstI digest of λ DNA. To determine allelic sizes, GeneScan Analysis 2.1 software (PE Applied Biosystems Division, Foster City, CA) was used. Size standard fragments of GS 350 were used to calculate the size of unknown samples by second order regression. Because of anomalous mobilities (Mansfield et al. 1996; H.-M. Wenz, unpubl.), the 250- and 340-bp fragments were omitted when using GSP, and the 250-bp fragment was omitted when using POP-4.
Acknowledgments
Names of commercial manufacturers are provided for identification only, and inclusion does not imply endorsement by the the FBI. We are grateful to the support unit of the Forensic Science Service, Birmingham, UK, for supplying HUMTHO1. The critical reading and suggestions of Drs. S. Baumhueter, K. Tynan, and K. Lazaruk are acknowledged.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Present address: Forensic Science Research and Training Unit, Federal Bureau of Investigation (FBI) Academy, Quantico, Virginia 22135 USA.
-
↵2 Corresponding author.
-
E-MAIL wenzmh{at}perkin-elmer.com; FAX (650) 638-6666.
-
- Received August 14, 1997.
- Accepted November 18, 1997.
- Cold Spring Harbor Laboratory Press








