
A High-Resolution Physical and Transcript Map of the Cri du Chat Region of Human Chromosome 5p
Abstract
A high-resolution physical and transcription map has been generated of a 3.5-Mb region of 5p15.2 that is associated with the Cri du chat (CDC) syndrome. Utilizing a variety of resources including a natural deletion panel, a chromosome specific radiation hybrid panel, and YAC, PAC, and BAC genomic clones we have ordered >60 STSs within this region. Approximately 45% of these STSs were obtained from publicly available whole genome maps, thus allowing for integration of this map with currently available resources. Thirteen of these markers were ESTs. In addition, >70 exon trapped products have been mapped on the natural deletion panel and bacterial clone resource. The combination of these resources has allowed for the identification of 17 transcripts within this region, all of which represent candidate genes for CDC. Further characterization of the genomic contig also revealed that this region of 5p15 contains a large number of repetitive elements.
[The sequence data described in this paper have been submitted to GenBank under accession nos. G31374–G31412,B07604–B07657. On-line supplementary material concerning markers used, primers, PCR product sizes, and annealing conditions is available athttp://www.cshl.org/gr.]
Cri du chat (CDC) syndrome is associated with deletions of 5p15 and is one of the most common contiguous gene disorders with an incidence of 1 in 50,000 live births (Niebuhr 1978). Hallmarks of this syndrome include severe mental retardation, speech delay, prenatal and postnatal growth delay, hypotonia, microcephaly, a round face with down-slanting palpebral fissures, hypertelorism, epicanthal folds, low-set and/or poorly formed pinnae, broad nasal bridge with prominent nasal root, microretrognathia, and a plaintive, high-pitched cry similar to the mewing of a cat (Lejeune et al. 1963;Niebuhr 1978; Baccichetti et al. 1988; Church et al. 1995). There have been no reports of CDC without a cytogenetically visible chromosome rearrangement, suggesting that several megabases of DNA must be deleted to produce the classical CDC phenotype. Previous cytogenetic studies indicate that there is a CDC critical region in 5p15.2–15.3 (Niebuhr 1978). A few patients have been described who present with only a subset of characteristics (Baccichetti et al. 1988; Smith et al. 1990;Overhauser et al. 1994; Church et al. 1995). Analyses of the abnormal chromosomes from these individuals have resulted in the correlation of individual phenotypic characteristics with deletions of specific portions of 5p (Overhauser et al. 1994; Church et al. 1995).
Recently, attempts have been made to identify the smallest region of DNA commonly deleted in individuals with classical CDC (Overhauser et al. 1994; Church et al. 1995). Reports for the localization of speech delay and the catlike cry have been consistent, with speech delay localizing to the distal part of the terminal cytogenetic band 5p15.3 and the catlike cry localizing to the proximal portion of this cytogenetic band (Baccichetti et al. 1988; Overhauser et al. 1994;Church et al. 1995). However, there is some disparity over the region involved in producing the characteristic facial features and severe mental retardation associated with the syndrome (Overhauser et al. 1994; Church et al. 1995). Two nonoverlapping, but adjacent regions on 5p have been described as producing the typical facial features and severe mental retardation associated with CDC (this region will be abbreviated CDCMR) (Overhauser et al. 1994; Church et al. 1995). The inconsistency revolves around the diagnosis of patient HHW962, who was described by Overhauser et al. (1994) as having typical CDC and byChurch et al. (1995) as not having typical CDC. We have recently identified a new patient (lymphoblasts HHW1961; hybrid HHW1985) who has few features of CDC. This individual reduces the region of interest, which is defined by the markers L28349 and D5S1623E, and lies in bins II–IV (Fig. 1). This supports the more proximal localization of the CDCMR region.

Natural deletion panel defining the CDCMR region. HHW1985 represents the distal breakpoint for the region, and HHW1249 represents the proximal breakpoint. The thick, black vertical lines represent the portions of the chromosome that remain. The thin horizontal lines demarcate the individual bins. Roman numerals at the rightindicate the individual bin names.
In an effort to elucidate the molecular etiology of CDC, we have constructed a high-resolution physical map of the CDC critical region. Prior to this mapping effort, the only maps of this region were from whole genome mapping efforts (Hudson et al. 1995; Dib et al. 1996). No genes had been localized and a total of 20 monomorphic and polymorphic sequence-tagged sites (STSs) had been localized to this region (Overhauser et al. 1987; Hudson et al. 1995; Dib et al. 1996; Grady et al. 1996). Recently, whole genome radiation hybrid (RH) maps have provided 13 additional STSs that are associated with expressed sequence tags (ESTs) (Schuler et al. 1996). These resources provided the basis for the construction of this map.
We have constructed an STS-based, 100-kb resolution, sequence-ready physical map. This map is bounded by the markers L28349 and D5S1623E and encompasses ∼3.5 Mb of DNA. Approximately 45% of the STSs needed to generate this map were derived from public databases, which allow it to be integrated with available whole genome and chromosome-specific maps (Hudson et al. 1995; Dib et al. 1996; Grady et el. 1996; Schuler et al. 1996). This resource will be invaluable for understanding the molecular basis of CDC.
RESULTS
Patient Identification
An individual with a de novo 5p deletion was recently identified (lymphoblast HHW1961; hybrid HHW1985). He was born at term by cesarean section for fetal distress. Birth weight and length were at the tenth centile and head circumference was at the fifteenth centile. Abnormalities noted at birth, including imperforate anus, hydronephrosis, and defects of several lumbosacral vertebrae led to karyotype analysis, which revealed 46, XY, del(5p15.2 → 5pter)de novo. There was an inconsistent history of a soft cry, possibly catlike. Evaluation at 8 weeks age revealed normal features except for a low nasal bridge, anteverted nasal tip, slightly small chin, mildly incurved fifth fingers, and hypotonia. The cry was soft and monotonic but not typically catlike. He rolled over at 4 months, crawled at 9 months, was able to sit alone at 10 months, and walked with support at 15 months. First meaningful words were spoken at 13 months. Developmental assessment at 33 months revealed that he is delayed by 9–12 months in several areas, primarily in speech and language. He has grown along the fifth centile in height and the tenth centile in weight, but at 16 months of age; his head circumference was 44 cm (less than third centile, average for 6 months). We will continue to follow this patient’s progress, but preliminary evaluation suggests that he does not have typical features of CDC and thus narrows the CDC critical region to bins II–IV (Fig. 1).
Physical Map
Natural Deletion Panel
The entry point to high-resolution physical mapping in this region consists of a natural deletion panel that divides the CDCMR region into seven bins (Fig. 1). These natural deletions are maintained in a rodent background and are amenable to positive and negative scoring of markers using either PCR or Southern blot analysis (Dana and Wasmuth 1982). In this manner, 10 STSs have been mapped to bin I, 1 STS to bin Ia, 7 STSs to bin II, 9 STSs to bin III, 1 STS to bin IIIa, 35 STSs to bin IV, and 5 STSs to bin V. One STS could not be localized to a discrete natural deletion bin (D5Z11).
RH Mapping
Although placement of markers on the deletion map provides localization of these STSs, there is no marker order within the bins. Estimated bin size ranges from <100 kb to ∼2 Mb. Therefore, it was desirable to obtain marker order within these bins to facilitate genomic clone orientation. The analysis of a chromosome 5-specific RH panel has allowed us to obtain order for 13 of 16 markers (Fig.2). The markers D5S2769 and D5S817 could not be ordered relative to each other, nor could the markers D5S2895, L28276, and D5S2768 be distinguished. Three sets of markers, D5S478/D5S667, L28289/D5S2905, and L28311/D5S2081 were ordered with odds of inversion >200:1 but <1000:1. All other markers could be ordered with odds >1000:1. The order obtained agrees with that determined using the bacterial clones with one exception (Figs. 2 and3). The markers D5S17 and D5S2905 are inverted on the RH map relative to the physical map (Figs. 2 and 3).

RH map of the CDCMR region. Markers are placed along the vertical line in the order derived by RH mapping. The horizontal lines indicate natural deletion breakpoints. The numbers to the left of the markers show the distance in centiRays while the numbers to the right give the odds for local inversion between pairs of markers. Asterisks indicated odds of local inversion which are not statistically significant.
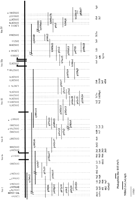

Physical map for the CDCMR region. The thick horizontal line at the top of the page represents the chromosome; thick black lines intersecting this one correspond to the natural deletions shown in Fig. 1. Bin designations are noted above the chromosome. Markers are shown above this line and are arranged in the order derived from STS content mapping. Markers with asterisks indicate that they were also placed on the RH map; names in boldface type indicate that the STSs were derived from an EST. The brackets above the markers indicate unknown marker order. The horizontal lines just below the chromosome represent YAC clones; the horizontal thin lines below that represent bacterial-based clones. Designation of clone names: (cy) CEPH YAC; (FS5Y) flow-sorted chromosome 5-specific YAC library; (p) PAC clone; (b) BAC clone. Slanted lines at the ends of genomic clones indicate that they are chimeric; slanted lines within a clone indicate an internal deletion. Coordinates given below the genomic clones represent exon-trapped products derived from the CDCMR region. Exon clones in bold are present only once within the contig. Exon-trapped products that identify nonoverlapping clones are not in bold. If the sequence is predicted to be an almost exact copy, the same clone name has been used; if the sequence is predicted to be a related sequence, the designation rs has been added. The relative positions of cDNAs identified by exon amplification are indicated by horizontal lines below the exon clones. The exons used to detect the transcript are noted in parenthesis.
The correlation of kilobase pairs to centiRays for this RH map was determined using two markers present in end clones of yeast artificial chromosome (YAC) cy881c11, which is estimated to be 800 kb. This YAC is not chimeric and appears to be intact (Fig. 3). These markers (D5S2769 and D5S2768) are 24 cRays apart, giving an estimate of 33.3 Kb per cRay. This is consistent with the estimate of 29 Kb per cRay determined for the entire chromosome 5 RH map (J.D. McPherson, B. Apostol, C.B. Wagner-McPherson, S. Hakim, R.G. DelMastro, N. Aziz, E. Baer, G. Gonzales, M.C. Krane, R. Markovitch et al., in prep.). The distance between D5S432 (the most distal marker in bin I) and D5S737 (the most proximal marker in bin V) is 38 cRays by two-point analysis of these two markers or 142 cRays when the distance is added between all of the markers along the entire RH map. These markers encompass the entire CDCMR region and have an estimated physical distance of 1.2–4.6 Mb.
YAC Clone Analysis
Selected STSs from each bin were used to identify YAC clones for this region. Five YACs were isolated from the Centre d’Etude du Polymorphisme Humain (CEPH) I YAC library using PCR screening (Dausset et al. 1992). One YAC was isolated from a chromosome 5-specific YAC library (L. Deaven, pers. comm.); three YACs were identified by inspection of a whole-genome YAC map (Hudson et al. 1995). Isolation of YAC end clones indicated that five of nine YACs are chimeric (Fig. 3). One YAC (cy63e7) appears to contain an internal deletion, as a result of the absence of marker D5S2867. Using estimates based on distances obtained by RH mapping, >90% of the entire region is represented in YAC clones. YAC coverage between the markers D5S2768 and D5S2866 is not complete. All of the clones in this region are <400 kb and all are chimeric. Within this region the markers D5S2872, D5S810, D5S2877, D5S1537, and D5S2081 are not contained in any YAC clones. This is also a region in which there are three natural deletions within 200 kb of each other. The 1.6 Mb of the CDCMR region distal to the gap is contained within a contiguous set of YAC clones. The CDCMR region proximal to the gap is contained within a single YAC, although this YAC is chimeric. The markers WI-12799, WI-15727, WI-18203, D5S1623E, D5S1665E, and SGC35145 lie proximal to this YAC and are not contained in any YACs. The markers WI-12799 and WI-15727 are in bin IV but are known to be proximal to cy160d6, based on bacterial clone data (discussed below). The other STSs are known to be proximal to cy160d6, as they were placed in bin V by natural deletion mapping.
Bacterial Clone Identification and Analysis
In an initial attempt to identify bacterial clones in the CDCMR region, colony filters containing a chromosome 5-specific cosmid library were screened with radiolabeled complex probes derived from YACs from the region (Munroe et al. 1994). Fluorescence in situ hybridization (FISH) of these cosmids to metaphase chromosomes from normal lymphoblast cells indicated that the vast majority did not map to the region of interest (U. Bengtsson, D.M. Church, R. Shiang, and J.J. Wasmuth, unpubl.). This was determined to be attributable to the presence of low- and intermediate-copy repetitive sequences associated with 5p15.2 (see Discussion) (Simmons et al. 1995, 1997).
To circumvent this problem, an STS-based approach was adopted. A total human P1-derived artificial chromosome (PAC) library was obtained (P. de Jong, pers. comm.), and clones were assessed for STS content as described in Methods (markers used are available as an on-line supplement at http://www.cshl.org/gr). Initially, STSs were obtained from whole genome maps, from bacteria phage clones that had been mapped previously to this region, and YAC end clones (Overhauser et al. 1987;Hudson et al. 1995; Dib et al. 1996; Grady et al. 1996). The PAC library was initially screened with 24 such STS markers. In the initial screen of this library, four STSs identified no clones (L28289, L28432, D5S2905, and D5S2906). These markers were radiolabeled, as were three exon-trapped products (1d6, 1e3, 1f2) and an EST clone (NIBA2) from the region. These probes were hybridized to high-density grids of a PAC library containing an additional sevenfold coverage of the human genome (P. de Jong, pers. comm.). Approximately 50% of the PAC clones identified in this screen localized to the CDCMR region by subsequent STS screening, representing a marked increase in efficiency of clone detection compared with complex YAC clone probes. Overlap of all bacterial based clones was established by STS content, using either the previously described markers or STSs generated from the ends of these genomic clones. STSs that were not positive for any of the previously isolated bacterial clones, but were localized on the natural deletion panel, were used to screen the PAC library. STSs that identified no new clones in the PAC library were used to screen a total human bacterial artificial chromosome (BAC) library (Research Genetics).
A complete, STS-linked contig was established in the distal part of the CDCMR region as a result of this study. This contig is defined by the markers L28349 and D5S2864 and spans ∼1.6 Mb. Another complete contig in the proximal region, spanning 1.3 Mb, has been constructed between the markers D5S2899 and WI-15727. There is one virtual gap in the STS linkage of the proximal contig (between markers L28432 and D5S2866), as evidenced by fingerprinting, which suggests that these clones overlap (data not shown). In addition to STS content mapping, clone overlap was confirmed by fingerprint analysis (data not shown). The sizes of individual clones were determined by NotI digestion and pulse field gel electrophoresis analysis (data not shown). There was one discrepancy between the natural deletion binning and bacterial clone STS content, marker WI-11436. Natural deletion binning suggested that this marker mapped to a novel bin, whereas genomic clone STS content suggested placement within bin IV, where the marker has been placed. A small island of clones containing the markers L28311–D5S2081 does not connect to the surrounding contigs of bacterial clones by STS analysis or fingerprinting analysis. The two gaps in this region are predicted to be <100 kb based on RH mapping. This is also the region for which no intact YAC clones could be obtained (Fig. 3).
Although STS content does not confirm that the proximal contig crosses the HHW1249 breakpoint, data from exon-trapped products suggest that these clones may cross the breakpoint (Fig. 3). Exon-trapped product 10a1 maps to bin V and to the clones p207f15, p4m11, and p29o12 (Fig.3). However, given the repetitive nature of this region, the connection of the proximal contig to a clone in bin V (p143i2) will be necessary to ensure that this breakpoint is covered within this contig.
Transcript Map
Exon Amplification
To begin transcript identification, bacterial clones from this region were analyzed for potential coding content using exon amplification (Buckler et al. 1991; Church et al. 1994). The resulting exon-trapped products were initially characterized by sequence analysis. One hundred twenty-one unique clones were obtained and compared to the public databases using the BLASTN and BLASTX programs (Altschul et al. 1990). Approximately 12% of the exon-trapped products were known repetitive elements of various classes (mer22, Line,KpnI, υ repeats).
Table 1 shows the results obtained from BLAST analysis of the exon-trapped products. Twenty-three exon-trapped products showed significant homologies (P < 10−3) to entries in the database. All others had weak or no homologies.
Results of BLAST Searches with the Exon-Trapped Products from the CDCMR Region
Ninety-eight nonredundant clones were subjected to Southern blot analysis using the CDC natural deletion panel (Figs. 1 and4). In this manner, map location and copy number of the sequence could be assessed. Seventeen percent of these clones were shown to be repetitive elements (Fig. 4A). In addition, a number of exons identified related sequences within the CDCMR region (Figs. 3 and4C). Three exons (4h2, 9a4, and 9b6) identified related sequences on other chromosomes (data not shown).
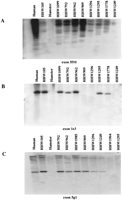
Representative Southern blots of various classes of exon-trapped products from the CDCMR region. (A) A repetitive exon; (B) a single copy exon; (C) an exon that identifies one major band, and other related sequences, two that map to the CDCMR region, and one that maps to another chromosome. Exon 5g1 is the same as exon 1f1.
Exon-trapped products that were single copy or appeared to have few related sequences within the CDCMR region were radiolabeled and hybridized to colony filters of the bacterial-based genomic clones. In this manner, 76 exon-trapped products were mapped to the CDCMR region contig. As expected from the data obtained by hybridization of these clones to genomic Southern blots of the CDC deletion panel, several exon-trapped products identified clones determined to be nonoverlapping by STS content mapping (Fig. 5). In some cases, the signal intensity was the same for all clones (Figs. 3 and 5B), and in others the PAC/BAC from which the exon clone was derived gave a stronger signal than the signal seen in the nonoverlapping clones (Figs. 3 and 5C). Clones in which a weaker hybridization signal was seen were assumed to have a related sequence rather than an exact copy of the exon-trapped product. Exon 4a8 was derived from cosmid clone ccI5-120, which lies in bin I but is not connected to the CDCMR contig (Table 1).
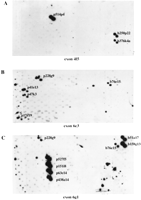
Colony hybridization of exon-trapped products from the CDCMR region to colony filters (stamped in duplicate) of genomic clones from the CDCMR bacterial clones. (A) An exon that identifies overlapping clones; (B) an exon that identifies nonoverlapping clones. (Note all clones appear to have near equal hybridization intensity.) (C) Exons that hybridize to nonoverlapping clones but with differing hybridization intensities.
EST Mapping
In an effort to increase the density of the transcript map in this region, ESTs from the publicly available RH consensus map were integrated with our map using all of the above physical reagents (Schuler et al. 1996). There are 43 ESTs listed within the genetic interval between D5S630 (distal to the CDCMR region) and D5S1954 (proximal to the CDCMR region) (Fig. 3). This number was reduced to 29 nonredundant ESTs by BLASTN analysis (Altschul et al. 1990). Three of these were part of known genes (NIBA2, Dap-1, and Trio) (Feinstein et al. 1995; Debant et al. 1996; Simmons et al. 1997). Initially, STSs derived from 25 of these ESTs (including the three known genes) were mapped to 5p using the natural deletion panel (Fig. 1). Data were not obtained for four STSs, either because of inability to establish primer conditions or inconsistent results within the deletion panel. Three ESTs mapped to bin I, seven mapped to bin IV, four mapped to bin V, five were distal to bin I region, and two were proximal to bin V region. Once a bin location was established, the genomic clones from that bin were assayed for the presence of the EST. Two of the ESTs in bin I were contained within the bacterial clone contig (WI-13430 and NIBA2); the other (Dap I) is distal to the bacterial clone contig, but contained within the YAC contig. Twelve of the 13 EST clones have identified bacterial genomic clones. Only one, WI-18203, was not contained in either a YAC or bacterial clone.
DISCUSSION
Elucidating the molecular etiology of aneuploidy syndromes is a daunting task. These are disorders of gene dosage and most likely involve many genes. One molecular approach to analyzing these disorders is to define the smallest region commonly deleted in patients sharing common manifestations of the disorder. Relatively small regions have been defined for some disorders, such as Wolf–Hirschorn syndrome (165 kb) (Wright et al. 1997). In other cases, the paucity of patients with informative deletions can make this task more difficult, as is the case with CDC. We have currently defined the CDCMR critical region as bounded with the markers L28349 and D51623E, which spans ∼3 Mb of DNA.
We have generated a high-resolution physical and transcription map of a 3.5-Mb region of 5p15.2 that is associated with the severe mental retardation and characteristic facial features of the CDC syndrome. The inclusion of markers from six whole genome maps [Genethon and the Cooperative Human Linkage Center (CHLC) genetic maps, the Whitehead, Stanford, and Consensus RH maps and the Whitehead YAC map] allows for the integration of this map with those already available. Prior to this effort, 12 markers were present on at least two of the maps, and only one marker (D5S478) was present on five of the whole genome maps. The high-resolution physical and transcription map presented here provides a significant resource for examining the molecular etiology of the CDC syndrome.
The contigs contained in this map are presently being sequenced at the Human Genome Center at the Lawrence Berkeley National Laboratory (J.-F. Cheng, pers. comm.). As these clones cover six natural deletion breakpoints, they will provide a useful starting point for identifying the junction fragments in patients and characterizing the molecular mechanisms of these deletions. These genomic clones provide a useful diagnostic tool to assess new individuals with deletions of 5p using FISH. In addition, this provides a resource for other disorders that have been localized to this region. The cartilage disorder, chondrocalcinosis (CC), was mapped to 5p15.2 in a large English kindred (Hughes et al. 1995). The flanking markers for this disorder are D5S810 (in the CDCMR region) and D5S416 (proximal to the CDC region), which means the regions for CDC and CC are overlapping. Thus, this resource provides information for CC as well as CDC. A recent report suggests that an asthma susceptibility locus in African Americans also maps to 5p15 (The Collaborative Study on the Genetics of Asthma 1997).
RH mapping proved to be useful in the initial stages of this project when there were only 16 markers and no genomic clones. The inability to order some of the markers using this method suggests that the limit of resolution of this technique had been reached. All markers that could not be ordered with statistical significance were shown to be within 100–150 kb of each other on the bacterial clone map. The order obtained by RH mapping was consistent with STS content of the genomic clone map with one exception, the order of D5S17 and D5S2905 (Figs. 2and 3). Given the repetitive nature of this region, it is possible that this region is more sensitive to radiation or more prone to deletion during the generation of the hybrid cell lines. One other explanation is that there is a difference in the donor DNA used to construct the RH panel as compared to the donor DNA used to construct the genomic libraries.
A minimal tiling path consisting of 30 PAC/BAC clones with three gaps has been generated between the markers L28349 and D5S1623E. There is a minimal tiling path of seven YACs, with four gaps for this same region (Fig. 3). Sequencing of end clones in the region between D5S2872 and D5S2081 identified many repetitive elements, such as Line, mer andKpnI repeats (data not shown). Other sequences derived from clones around this region did not identify known repetitive elements but were suspected to be repetitive as they identified genomic sequences from throughout the genome (data not shown). It is possible that the many repetitive elements within this region underlie its instability.
Exon amplification was used to scan the genomic clones for potential coding content (Buckler et al. 1991; Church et al. 1994). This method of gene identification is expression independent and has been used successfully in the identification of numerous disease genes (The Huntington’s Disease Collaborative Group 1993; Mercer et al. 1993;Zhang et al. 1994). cDNA selection was not used because of the numerous repetitive elements within this region (Lovett et al. 1991). Other attempts to apply cDNA selection to 5p15.2, distal to the CDCMR region, resulted in ∼50% of the clones containing repetitive sequences, despite extensive prehybridization with Cot1 DNA (Simmons et al. 1995,1997). It would be useful, however, to perform cDNA selection on this region, as reports suggest that these two techniques identify a diverse population of clones (Yaspo et al. 1995). As this contig becomes increasingly well characterized, it may be possible to develop the proper molecular tools to allow sufficient suppression of the numerous repetitive elements in this region so that hybridization based approaches could be used for gene identification.
Current estimates of gene density in the human genome suggest that there could be from 50 to 150 genes within this 3.5-Mb region. It is difficult to speculate on the gene coverage that the combined exon/EST resource provides for this region until full-length cDNAs corresponding to these clones are analyzed. There are a total of 17 candidate transcripts within the CDCMR to date. In addition, there are 64 unique, low- or single-copy exon-trapped products. Of the 17 transcripts, 11 were identified only by a random mapping approach, 4 were identified solely by exon-trapped products, and 2 transcripts were identified by both methods (NIBA2 and Trio). The relatively small degree of overlap between the two gene resources indicates the power of a combined approach for gene identification.
The majority of the transcripts and exon-trapped products identified in the CDCMR region are of unknown function. One exon (9a7) shares homology with the plakophilin family of molecules. Plakophilin is a component of the desmosome and is involved in mediating cell–cell contact (Alberts et al. 1994). BLAST analysis of the NIBA2 gene also reveals homology to plakophilin family members. Based on map location, it seems likely that these two genes are part of the same transcript. Another gene identified in the CDCMR region is Trio. This gene was identified as an interactor with the leukocyte-associated transmembrane tyrosine phosphatase receptor (Debant et al. 1996). Trio is a multidomain gene containing two functional guanine nucleotide exchange factors and a protein serine/threonine kinase domain (Debant et al. 1996). Although the biological significance of this interaction is unclear, it is suspected to be involved in cell remodeling associated with cell movement. In addition, a novel member of the dynein family of genes has been identified. There are three distinct families of cytoplasmic dyneins (DHCs 1, 2, and 3), all of which have different roles in cell motility (Vaisberg et al. 1996). It is interesting to note that the three genes associated with functions are thought to be involved in cell–cell interaction and cell motility. Experiments are under way to assess the transcription level of these genes in patients with 5p deletions. As syndromes ofaneuploidy are the result of aberrations in gene dosage, it is anticipated that genes involved in the molecular etiology of CDC can be elucidated in this manner.
METHODS
STS Derivation
Twenty-nine STSs were obtained by inspection of publicly available maps (Overhauser et al. 1994; Hudson et al. 1995; Dib et al. 1996; Grady et al. 1996; Schuler et al. 1996). The remaining STSs were developed using sequence derived from phage, PAC, and YAC clones (this study; Overhauser et al. 1987). Sequences that were determined not to contain repetitive elements were used to develop oligonucleotide primers for PCR amplification (http://www.cshl.org/gr).
Construction of RH Map
A chromosome 5-specific RH panel was constructed using a hamster fibroblast cell line with human chromosome 5 (HHW105) as its only human DNA (J.D. McPherson, B. Apostol, C.B. Wagner-McPherson, S. Hakim, R.G. DelMastro, N. Aziz, E. Baer, G. Gonzales, M.C. Krane, R. Markovitch et al., in prep.). One hundred seventy-seven clones were screened by PCR analysis using 15 STSs (Fig. 2). Amplification was performed a minimum of two times for each marker placed on the map, with the exception of the markers D5S2769 and D5S2081. PCR analyses were performed using the conditions as specified (http://www.cshl.org/gr). Amplification products were electrophoresed through 2% agarose gels and visualized with ethidium bromide using an ultraviolet light source.
All of the STSs placed on the RH map had initially been mapped using a well-characterized natural deletion panel of somatic cell hybrids (Church et al. 1995 and unpubl.; Fig. 1). Marker order was determined by analyzing STSs within the same natural deletion bin using the program fourpoint (Cox et al. 1990). CentiRay distance and linkage of all markers were analyzed using the program twopoint (Cox et al. 1990).
Identification of YAC Clones
The CEPH I YAC library was prepared for screening by PCR analysis as described previously (Dausset et al. 1992). A chromosome 5-specific YAC library (L. Deaven, pers. comm.) was obtained in seventeen 96-well microtiter plates. These plates were arranged in a two-dimensional array from which clones were pooled as plate pools, row pools, and column pools (McCormick et al. 1993). DNAs from these pools were prepared in agarose blocks using the protocol of Schwartz and Cantor (1984). After extensive washing in ddH2O, the agarose was digested using Gelase according to the manufacturer’s instructions (Epicentre Technologies). This DNA was diluted 50-fold for PCR analysis. Both libraries were screened using a subset of the STSs. Three additional YACs were identified from a whole genome YAC-based map (Hudson et al. 1995).
YAC ends from CEPH libraries were cloned using vectorette PCR with modified primers (Riley et al. 1990; Munroe et al. 1994). Ends from the chromosome 5-specific YAC were obtained as described previously (Shero et al. 1991).
Identification of PAC Clones
A total human PAC library containing a threefold representation of the human genome, stored in three hundred twenty-one 384-well microtiter plates, was obtained from P. de Jong (pers. comm.) (Ioannou et al. 1994). These plates were arranged in a three-dimensional array similar to the one used by Aburatani et al. (1996). Briefly, the plates were placed in 9 blocks consisting of 36 plates each in a 3 × 3 × 4 array. Bacterial colonies from each plate were collected. In addition, super row and super column pools from each block were collected using multipettors. DNA was obtained from these pools via standard alkaline lysis procedures. DNAs from each set of 36 plates were pooled to construct nine block pools. This DNA was extracted using phenol/chloroform and precipitated a second time. DNA pools of BAC clones were obtained from Research Genetics and screened for STS content (Kim et al. 1996).
A subset of the markers that are available on-line were utilized in PCR amplification of the above pools. STSs that did not identify any clones in either of the above libraries were then radiolabeled and used as probes to screen nylon filters of an extended total human PAC library (P. de Jong, pers. comm.).
The ends of the PACs and BACs were identified by restricting the clones with an enzyme that does not cut within the vector sequence (NdeI and XbaI for PACs; NheI for BACs), thus releasing the majority of the insert. The restriction fragments were diluted to promote intramolecular association, ligated using T4 DNA ligase and transformed into DH10B cells, plated onto LB plates with the appropriate selection, and grown at 37°C overnight. Two colonies were selected from each transformation and grown in liquid media. DNA was isolated by alkaline lysis, followed by extraction with phenol/chloroform and isopropanol precipitation. Sequencing was performed with the T7 and SP6 primers using the U.S. Biochemical chain termination sequencing kit (Sanger et al. 1977). The sequence was analyzed using the BLASTN and BLASTX programs (Altschul et al. 1990). STSs were developed to ends that did not have homology to known repetitive elements.
Exon Amplification
Exon amplification was performed as described previously (Church et al. 1994; Trofatter et al. 1995). Briefly, DNA pools containing one to five PAC or BAC clones were pooled and restricted using eitherPstI or a BamHI–BglII double digest. The resulting fragments were subcloned into pSPL3 that had been cut with the appropriate enzyme. Individual transformants were pooled, and episomal DNAs extracted from these pools were electroporated into Cos-7 cells. Cytoplasmic RNA was extracted and used as a template for reverse transcription followed by PCR using vector-specific primers. Exons were sequenced on both strands using the U.S. Biochemical PCR sequencing kit following the manufacturer’s instructions. Sequences were analyzed using the BLAST programs as described above (Altschul et al. 1990).
Southern Blot Analysis
Genomic DNA from various cell lines was prepared according to standard techniques (Sambrook et al. 1989). DNA was digested with either EcoRI or HindIII in the presence of spermidine. Restriction fragments were then fractionated through a 0.9% agarose gel and transferred to Nytran (Schleicher & Schuell) or Hybond N+ (Amersham) under alkaline conditions. Before hybridization, blots were prewashed in 0.1× SSC, 0.05% SDS, at 65°C for 30 min. Hybridizations were performed using the method ofChurch and Gilbert (1984). Probes were labeled by random priming using the method of Feinburg and Vogelstein (1983). The blots were washed to a stringency of either 0.5× or 0.1× SSC at 65°C.
Colony Filter Preparation
Bacterial colonies were stamped onto nylon filters (Genescreen-NEN or Hybond N+, Amersham) using a manual stamping device (WashU Instrument Shop). Colonies were spotted in duplicate and grown overnight at 37°C on LB/agar supplemented with the appropriate antibiotic. The membranes were then denatured in 0.25 n NaOH, neutralized in 1 m Tris, and washed in 2× SSC. The filters were scraped to remove any bacterial debris. Filters were then hybridized, as described above.
Acknowledgments
This paper is dedicated to the memory of John J. Wasmuth. D.M.C. was a trainee on National Institutes of Health (NIH) training grant GM07134. R.S. was supported by the Arthritis Foundation, grant A21320. J.J.W. was supported by Human Genome grant NIH HG00834. We thank all of the families with del(5p), as this study would have been impossible without them. We also thank the expert clinical evaluations provided by Dr. Erik Niebuhr. We are grateful to Rachelle Markovich and Ulla Bengtsson for excellent technical assistance. We also thank Drs. Alan Buckler and Alisoun Carey of Sequana Therapeutics for many inspiring discussions regarding this work and for access to the Research Genetics BAC library. We especially thank Drs. Mary Kay McCormick and Sara Winokur for critical reading of this manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author. Present address: Medical College of Virginia, Department of Human Genetics, Richmond, Virginia 23298-0033.
-
E-MAIL rshiang{at}hsc.vcu.edu; FAX (804) 828-3760.
-
- Received April 21, 1997.
- Accepted June 27, 1997.
- Cold Spring Harbor Laboratory Press








