
Recombination Hot Spots and Human Disease
Recombination between homologous DNA sequences occurs in all organisms, and the resultant exchange of information is critical for the survival of species. Recombination is an essential cellular process catalyzed by proteins explicitly expressed for this purpose. It provides an effective means of generating genetic diversity that is important for evolution. The proteins involved in recombination allow cells to retrieve sequences lost when DNA is damaged by radiation or chemicals, by replacing the damaged section with an undamaged strand from a homologous chromosome. The process of homologous recombination has also been used to study gene function by way of gene knockouts. However, recombination and factors involved in recombination may also be a source of harmful mutations and disease.
Specific DNA sequences are known to mediate or enhance the rate of recombination in the genomes of many organisms. Attempts to identify and decipher recombination hot spots have focused on determining the influence of various DNA sequences on the rate and type of DNA rearrangements. The human nuclear genome contains a large number of highly repeated DNA sequence families (Jelinek and Schmid 1982; Hardman 1986; Vogt 1990), broadly classified as tandemly repeated DNA or interspersed repetitive DNA. Because of their role in mediating disease-causing recombination errors, a brief overview of the various repeats is presented.
Tandemly repeated DNA is characterized by blocks or arrays of tandemly repeated DNA sequences. They are subclassified based on the size of the blocks or arrays of tandem repeats into satellite (0.1 to >2 Mb), minisatellite (0.1–2.0 kb), and microsatellite (∼150 bp) DNA. Satellite DNA is further sub-classified based on the size of the repeat unit within these blocks into types 1 (25–48 bp), 2 and 3 (5 bp), α (alphoid DNA; 171 bp), and β (Sau3A family; 68 bp). Minisatellite DNA consists of telomeric DNA that has a 6-bp repeat unit, and the polymorphic VNTRs (variablenumber tandem repeats) or hypervariable minisatellite DNA, where the size of the repeat unit ranges from 9 to 24 bp. VNTRs have been shown to be hot spots for homologous recombination in human cells (Wahls et al. 1990). Microsatellite DNA consists of small arrays of tandem repeats (usually 1–4 bp units) that are interspersed throughout the genome, in blocks consisting of <150 bp.
In contrast to tandemly repeated DNA, interspersed repetitive DNA consists of repeat units dispersed throughout the genome. Based on the repeat unit length, two major classes are recognized: short interspersed nuclear elements [(SINES) e.g., the Alu repeat family] and long interspersed nuclear elements [(LINES) e.g., the LINE-1 or L1 element (Singer 1982)]. The Alu repeat containing a 280-bp repeat unit occurs approximately once every 4 kb in the human genome. Mispairing between such repeats has been shown to be a frequent cause of deletions and duplications. Breakpoints of disease-causing deletions have been clustered within Alusequences in genes for the low density lipoprotein receptor (LDLR) (Lehrman et al. 1985, 1987), and the complement component 1 inhibitor (C1I) (Stoppa-Lyonnet et al. 1991). Such observations have suggested a general role for Alu sequences in promoting recombination and recombination-like events. Alurepeats or other dispersed repetitive elements are also thought to have played a role in the evolution of clustered multigene families by mediating unequal crossover events that lead to gene duplications. The average length of the L1 element repeat unit is 1.4 kb. In addition, there are smaller repeat sequence families belonging to this class, including the THE-1 (transposable humanelements), MER (mediumreiteration), HERV (humanendogenous retroviruses), and RTLV (retrovirus-like elements) repeats. Members of many of the interspersed repeat families are considered retrotransposable elements, that is, unstable DNA elements that can migrate to different regions of the genome by transposition via an RNA intermediate. These endogenous retroposons are thought to have played an important role in shaping the genomes of vertebrates by intracellular transposition events and by generating hot spots of recombination (Leib-Mosch and Seifarth 1995).
The various families of repetitive elements are highly relevant to a number of different mechanisms of mutagenesis in human genes. As discussed in the following sections, recombination between such sequences can lead to rearrangements, including deletions, duplications, inversions, and fusion genes.
Deletions and Duplications Caused by Homologous Recombination
Hot spots of homologous recombination between misaligned repetitive elements have been observed at the duplication and deletion breakpoints in a number of human genetic diseases. In Escherichia coli, direct repeats in close proximity can mediate efficient RecA-independent intramolecular recombination. A replicational model for DNA recombination between direct repeats was suggested by Bi and Liu (1996). They proposed that misalignment of repeats at the replication fork creates a recombinogenic intermediate that can be differentially processed and that the proposed sister-strand recombination mediated by direct repeats might be a general mechanism of deletion or duplication of repeated sequences in prokaryotic and eukaryotic genomes.
Large-scale deletions and duplications may be generated by the pairing of nonallelic interspersed or tandem repeats, followed by breakage and rejoining of chromatid fragments. Repeat DNA sequences may predispose to abnormal chromosome pairing and unequal crossing-over, with deletions and duplications representing the reciprocal products of such events. Large deletions within duplicated regions may occur either interchromosomally because of misalignment of non-sister chromatids during meiosis (Pentao et al. 1992; Chance et al. 1994) or intrachromosomally because of either sister chromatid exchange during mitosis or DNA slippage during replication (Krawczak and Cooper 1991).
Charcot-Marie-Tooth disease type 1A (CMT1A) and hereditary neuropathy with liability to pressure palsies (HNPP) are two autosomal dominant peripheral neuropathies resulting from DNA rearrangements that are reciprocal products of an unequal crossing-over event between misaligned flanking CMT1A–REP (repeat) elements on chromosome 17p (Chance et al. 1994; Patel and Lupski 1994) (Fig.1). The proximal and distal CMT1A–REP elements are ∼30 kb in length, extremely AT rich (64% A + T), and display 98% sequence identity (Reiter et al. 1996). Given the high degree of homology between the proximal and distal repeats, a recombination event can potentially occur anywhere within the 30-kb region. However, through the detection of novel junction fragments from the recombinant CMT1A–REP elements in both CMT1A and HNPP patients, a 1.7-kb recombination hot spot within the ∼30-kb CMT1A–REP was identified in 75% of CMT1A duplication patients and 84% of HNPP deletion patients examined (Reiter et al. 1996). Sequence analysis showed there was no particular increase in the degree of sequence identity over this 1.7-kb region, and, interestingly, a mariner transposon-like element (MITE) was identified in the vicinity of the hot spot. Three exons were identified in the repeat, one of which showed homology at the amino acid level to the conserved region of several insect transposases.Kiyosawa and Chance (1996) further investigated the MITE and found that it is probably nonfunctional, as several stop codons were found in its open reading frame. However, it is possible that this nonfunctional mariner sequence may be a target for a functional form of the transposase protein transcribed from a gene located elsewhere.
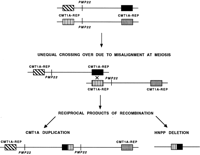
Generation of the CMT1A duplication and HNPP deletion attributable to unequal crossing-over. The proximal and distal CMT1A–REP elements (boxes) flanking the 1.5-Mb region containing the PMP22 gene (vertical line) on two different chromosomes are depicted. Unequal crossing-over because of misalignment at meiosis between the distal and proximal CMT1A–REP elements results in two reciprocal recombination products: the CMT1A duplication (3.0 Mb) chromosome with twoPMP22 genes and the HNPP deletion (1.5 Mb) with noPMP22 gene.
Northern blot analysis with the distal CMT1A–REP, which encompasses the putative transposase, identified a low-abundance transcript expressed in testes but not in ovaries (Reiter et al. 1996). This finding was interesting, because until recently it was considered that the unequal crossing-over resulting in CMT1A and HNPP occurred solely during male meiosis. Investigation of the origin of de novo duplications revealed a paternal origin for the mutation in 13 sporadic cases (Raeymaekers et al. 1991; Palau et al. 1993; Wise et al. 1993;Hertz et al. 1994). Palau et al. (1993) proposed that male-specific factors may operate during spermatogenesis to help form the duplication and/or stabilize the duplicated chromosome. However, Blair et al. (1996) on analysis of de novo duplications in eight families, found seven to be paternal and one, the first reported, to be maternal in origin, indicating that this was not a phenomenon associated solely with male meiosis. Two loci within the duplication region (D17S122 and D17S61) that were physically mapped within 1.0 Mb of each other were found to span an average genetic distance of 4 cM in males and 14 cM in females (Pentao et al. 1992). This large discrepancy between the genetic and physical distances of these duplicated markers suggests that this region appears to be extremely prone to meiotic recombination. Because recombination fractions for the duplicated region in CMT1A are larger in females than in males, oogenesis may afford greater protection from misalignment during synapsis, and/or there may be lower activity of those factors or mechanisms that lead to unequal crossing-over at the CMT1A locus (Blair et al. 1996).
Allelic variants of the human cytochrome P450 CYP2D6 gene have also been shown to arise from homologous unequal crossing-over involving a 2.8-kb direct repeat (Steen et al. 1995). The cytochrome P450 enzyme debrisoquine 4-hydroxylase metabolizes many different classes of commonly used pharmacological drugs. Among Caucasians, 5%–10% are classified as poor metabolizers (PM) because of autosomal recessive inheritance of two CYP2D6 deletion alleles. In these individuals, administration of average therapeutic doses results in toxic plasma concentrations and adverse drug reactions. In contrast, up to 5% demonstrate ultrarapid metabolism (UM) caused by an inherited duplication of functional CYP2D6 genes, thus requiring higher doses of drugs to maintain desired plasma levels. A 2.8-kb repeated sequence (CYP–REP), containing an Alu element and a tandem 10-bp direct repeat flanks the active CYP2D6 gene in the wild-type allele. It is thought that the CYP2D6 deletion and duplication alleles are reciprocal products, generated by homologous recombination between nonallelic CYP–REP elements.
Large-scale rearrangements are detected in 80% of familial and 65% of sporadic cases of juvenile nephronophthisis (NPH), representing the most frequent inherited cause of chronic renal failure in children (Konrad et al. 1996). Large homozygous deletions (250 kb) involving a 100-kb inverted duplication were found at the NPH1 locus on chromosome 2q13. Further characterization of this region revealed the presence of low-copy repeats.
X-linked icthyosis is a disease characterized by an extremely high frequency of submicroscopic deletions involving the steroid sulfatase (STS) gene (Bonifas et al. 1987; Conary et al. 1987; Ballabio et al. 1989a,b). Eighty-four percent of patients with steroid sulfatase deficiency possess deletions of their STS genes (10 exons spanning 146 kb of genomic DNA). The deletions have breakpoints clustered around or within a number of low-copy repetitive sequences called S232 type repeats, flanking the STS gene (Yen et al. 1990). These repeats resemble VNTRs (Li et al. 1992) and indicate that the high frequency of deletions at this locus may be attributable to recombination involving these repetitive sequences.
Deletion of both growth hormone (GH1) genes causes the autosomal recessive disease familial growth hormone deficiency type 1A. In 9 of 10 patients with GHI gene deletions, the crossovers occur within two 99% homologous, 594-bp regions flanking the GH1gene (Vnencak-Jones et al. 1988; Vnencak-Jones and Phillips 1990). The presence of these highly homologous DNA sequences flanking theGH1 gene predisposes recurrent unequal recombination events.
Fascioscapulohumeral muscular dystrophy (FSHD) is an autosomal dominant neuromuscular disease that has been linked to deletions within a tandem array of 3.2-kb repeats (D4Z4) adjacent to the telomere on chromosome 4q35 (van Deutekom et al. 1993). The majority of sporadic cases of FSHD are associated with de novo deletions at the D4Z4 locus. The D4Z4 sequence contains two homeoboxes and two previously described repetitive sequences, LSau and a GC-rich low-copy repeat designated hhspm3 (Hewitt et al. 1994; Lee et al. 1995). D4Z4 is a member of a dispersed family of homeobox-containing repeats, subsets of which are clustered on the short arms of acrocentric chromosomes (Lyle et al. 1995). Analysis of the evolutionary distribution and structural organization of D4Z4 showed that tandem arrays closely related to D4Z4 were conserved at loci syntenic to human 4q35-qter in apes and lower primates, suggesting a functionally important role for these sequences (Clark et al. 1996; Winokur et al. 1996). However, no single gene has yet been associated with FSHD, and the etiopathogenesis of FSHD is not yet known. It has been proposed that the deletions mediated by these repeat sequences may invoke a position effect on a nearby gene.
The most common molecular defect underlying α thalassemia involves a deletion of one or both of the duplicated α-globin genes (Dozy et al. 1979; Higgs et al. 1979). The mechanism by which the α thalassemia deletions occur is related to the underlying molecular structure of the α-globin complex (Embury et al. 1980; Lauer et al. 1980). Each α gene is located within a region of homology, ∼4 kb long (thought to have resulted from an ancient duplication event), and interrupted by short nonhomologous regions. During evolution, these homologous segments were divided by insertions and deletions to give rise to three homologous subsegments referred to as X, Y, and Z. The duplicated Z boxes are 3.7 kb apart, and the duplicated X boxes are 4.2 kb apart. Misalignment and reciprocal crossover between these segments at meiosis give rise to chromosomes with either single (Embury et al. 1980) or triplicated α-globin genes (Goossens et al. 1980). Recombination between the homologous X or Z boxes (4.2 or 3.7 kb apart) gives rise to chromosomes with a 4.2- or 3.7-kb deletion with one α-globin gene and the reciprocal chromosome with three α-globin genes. Although the breakpoints of these deletions vary within these regions of homology, the mechanism of homologous unequal recombination appears to be the same. These recombination events have been reported in several ethnic groups (Higgs et al. 1989).
Inversions Caused by Homologous Recombination
Occasionally, highly similar inverted repeats may be located within or close to a gene. The high degree of sequence similarity between inverted repeats may predispose to pairing of the repeats by a mechanism that involves a chromatid bending back on itself. Subsequent chromatid breakage at the mispaired repeats and rejoining can result in an inversion, thus disrupting a functional gene. An example of pathogenic inversions caused by such a mechanism is seen in hemophilia A. Forty-five percent of patients with severe hemophilia A have an inversion and disruption of the coagulation factor VIII gene mediated by an unequal crossing-over event (Lakich et al. 1993). Within intron 22 of the factor VIII gene lies another gene, F8A, which has two additional copies situated 500 kb upstream (telomeric) of the factor VIII gene on Xq but in the opposite orientation. Evidence suggests that the tip of Xq flips back on itself, aligning the homologous intragenic and extragenic F8A sequences in meiosis. Unequal crossing-over results in recombination between one of the upstream genes and the intragenic F8A gene, generating an inversion of the intervening factor VIII gene sequence (Fig.2). Such inversions result in disruption of the factor VIII gene, separating exons 1–22 from exons 23–26 by 200–500 kb. As the single X chromosome in males remains largely unpaired in meiosis, the tip of the chromosome is free to flip up on itself—a phenomenon that does not occur in female meiosis, where both X chromosomes pair along their length like autosomes, apparently restricting the movement of Xqter. Rossiter et al. (1994) hypothesized that pairing of Xq with its homolog inhibits the inversion process and, therefore, the event should originate predominantly in male germ cells. They examined 20 informative cases in which the inversion originated in a maternal grandparent, and by analysis of DNA polymorphisms determined that it occurred exclusively in the male germ line. In addition, they showed that all but one of the 50 mothers of sporadic cases resulting from an inversion were carriers. These data supported their hypothesis that factor VIII gene inversions leading to severe hemophilia A occurred exclusively in male germ cells.
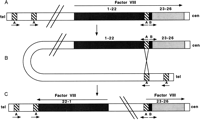
Inversion and disruption of the factor VIII gene mediated by the F8A gene in intron 22. (A) The genomic organization of the factor VIII gene showing the position and orientation of the three copies of the F8A genes (A) (two upstream of factor VIII and one within intron 22) and one copy of the F8B gene (B) within intron 22. Arrows indicate the direction of transcription of the factor VIII, A and B genes. (B) The homologous F8A sequences (A) within and outside the factor VIII gene mediate intrachromosomal recombination with the proximal A gene (presented here) or distal A gene. (C) The recombination product resulting in an inversion and disruption of the factor VIII gene.
Another example of an inversion caused by homologous recombination is that involving the iduronate-2-sulfatase (IDS) gene. Deficiency of the enzyme IDS results in Hunter syndrome, an X-linked recessive disorder also known as mucopolysaccharidosis II. A linkedIDS pseudogene-like sequence (IDS2) was detected ∼90 kb downstream of the IDS gene (Bondeson et al. 1995;Rathmann et al. 1995). This region is involved in a homologous recombination event with the IDS gene in 20% of patients with Hunter syndrome (Bondeson et al. 1995). The intrachromosomal recombination between homologous sequences present in the IDSgene and the IDS2 locus results in disruption of theIDS gene in intron 7 with an inversion of the intervening DNA. No detectable deletions or insertions are observed as a result of this inversion event.
Unequal exchange between large homologous repeats leading to genome variation in the absence of disease-causing mutations was reported recently in the Xq28 region, which harbors the neighboring genes for filamin (FLN1) and emerin (Small et al. 1997). Flanking the 48-kbFLN1/emerin region are two large inverted repeats, 11.3 kb in length, that exhibit 99% sequence identity (Chen et al. 1996). During the characterization of a rare mutation involving a complete deletion of the emerin gene and a partial duplication of the FLN1gene in a patient with Emery–Dreifuss muscular dystrophy (EMD), a common rearrangement resulting from mispairing of these large repeats was identified (Small et al. 1997). Recombination betweeen these inverted repeats leads to a complete inversion of the 48-kbFLN1/emerin region without any sequence alteration in either gene. This inversion was detected in the heterozygous state in 33% of normal females and helps explain the discrepancies between the genetic and physical map distances in this region of the X chromosome (Small et al. 1997). These investigators speculate that regional variation in the genetic map of humans may reflect the influence of similar, as yet uncharacterized, inversions mediated by large inverted repeats.
Fusion Genes Generated by Recombination
Besides deletions, duplications, inversions, or other rearrangements, recombination between homologous sequences also results in the creation of fusion genes. Sequence homology between members of gene families or pseudogenes results in such rearrangements.
Several hemoglobin variants containing fused or hybrid globin chains have been described. Hemoglobin Lepore (Hb Lepore) was the first to be reported and is an example of gene fusion resulting from the deletional removal of intervening DNA sequence (Gerald and Diamond 1958). This hemoglobin, which is synthesized in reduced amounts, is an abnormal molecule with the amino-terminal 50–80 amino acids of δ-globin and the carboxy-terminal 60–90 residues of β-globin. The reduction in globin synthesis is caused by the reduced synthesis of mRNA encoding the fusion product to a level intermediate between those of the δ-globin and β-globin genes. Thus, Hb Lepore contains normal α chains, and its non-α chain is a δ–β fusion chain. Three different varieties of Hb Lepore have been described (Hb Lepore Hollandia, Hb Lepore Baltimore, and Hb Lepore Boston), in which the transition from δ to β occurs at different positions (Baglioni 1962; Barnabus and Muller 1962; Ostertag and Smith 1969). Misalignment of chromosome pairing during meiosis results in pairing of the δ-chain gene with the β-chain gene instead of its homologous partner. The fusion chains arise by nonhomologous crossing-over between part of the δ locus on one chromosome and part of the β locus on the complementary chromosome (Fig. 3). This mechanism gives rise to two abnormal chromosomes: the Lepore chromosome, which has no normal δ or β loci but has a δ–β fusion gene, and the anti-Lepore chromosome, with a β–δ fusion gene (Hb anti-Lepore) and normal δ or β loci. A variety of anti-Lepore hemoglobins have been described, including Hb Miyada, Hb P-Congo, Hb Lincoln Park, and Hb Nilotic (Lehmann and Charlesworth 1970; Ohta et al. 1970; Badr et al. 1973;Honig et al. 1978). Hemoglobin Kenya is analogous to Hb Lepore, except that the abnormal hybrid chain is a γ–δ fusion chain (Huisman et al. 1972). The anti-Kenya chromosome contains intact Aγ-, δ-, and β-globin loci. The Lepore variants result in the clinical phenotype of β or δβ thalassemia. The anti-Lepore variants and Hb Kenya are not associated with any significant hematolgical changes.
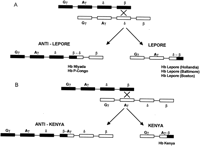
Fusion genes resulting from unequal crossing-over at the β-globin gene cluster. Genes within the β-globin gene cluster are shown. Fusion genes arising from unequal crossing-over between the β and δ-globin genes (Hb Lepore/anti-Lepore) and the Aγ and β genes (Hb Kenya/anti-Kenya) are shown in A andB. (A) The generation of Hb Lepore and Hb anti-Lepore: The different types of Hb anti-Lepore fusion alleles that have been described include Hb Miyada and P-Congo, where the crossover regions occur between residues β12 and δ22 and between residues β22 and δ87, respectively. The different types of Hb Lepore alleles that have been described are Hb Lepore Hollandia, Baltimore, and Boston, where the crossover regions occur between residues δ22 and β50, residues δ50 and β87, and residues δ87 and β116, respectively. (B) The generation of Hb Kenya and Hb anti-Kenya: For Hb Kenya, the crossover region is between residues γ81 and β86. No Hb anti-Kenya variants have been described.
Unequal crossover or unequal sister chromatid exchange between a functional gene and a related pseudogene can result in deletion of the functional gene or formation of fusion genes containing a segment derived from the pseudogene. Alternatively, the pseudogene can act as a donor sequence in gene conversion events and introduce deleterious mutations into the functional gene. In steroid 21-hydroxylase deficiency, virtually all pathological mutations arise as a result of sequence exchanges between the functional 21-hydroxylase gene (CYP21B), and a very closely related pseudogene (CYP21A) (Fig. 4). The two genes occur on tandem 30-kb repeats that show ∼97% sequence identity. The repeated segments also contain other duplicated genes—namely, the complementC4A and C4B genes. About 25% of pathological mutations at the 21-hydroxylase locus are large deletions resulting in removal of 30 kb of DNA resulting from unequal crossover or unequal sister chromatid exchange (Sinnott et al. 1990). The remaining 75% of mutations are point mutations where small-scale gene conversions of the CYP21B gene are thought to occur. A small segment of the CYP21A gene containing deleterious mutations is inserted into the CYP21B gene replacing a short segment of the original sequence. Analysis of one such mutation that arose de novo showed that the conversion tract was 390 bp in length (Collier et al. 1993).
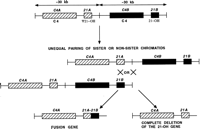
Steroid 21-hydroxylase gene mutations as a result of exchange of sequence between the functional 21-hydroxylase gene (21B) and its closely related pseudogene (21A). The duplicated complement genes (C4A and C4B) and steroid 21-hydroxylase genes (21A and 21B) located on tandem 30-kb repeats are shown. Unequal crossover or unequal sister chromatid exchange results in either formation of a fusion gene or complete deletion of the functional 21-hydroxylase gene.
Glucocorticoid-suppressible hyperaldosteronism (GSH) is an autosomal dominant form of hypertension caused by oversecretion of aldosterone. Gene fusion between the cytochrome P450 genesCYP11B1 and CYP11B2 has been shown to cause GSH (Lifton et al. 1992). CYP11B1 and CYP11B2 are two highly homologous genes closely linked on chromosome 8q22 that encode steroid biosynthetic enzymes catalyzing cortisol production and aldosterone production under the control of corticotrophin (ACTH) and angiotensin, respectively. Production of a hybrid gene due to unequal meiotic crossing-over between CYP11B1 andCYP11B2 results in a new gene that contains the promoter ofCYP11B1 and the coding region of CYP11B2. This fusion gene produces an enzyme that catalyzes the formation of aldosterone but is sensitive to ACTH. As a result, normal levels of ACTH, which normally maintain low levels of cortisol, lead to excessive production of aldosterone with consequent hypertension and hypokalemia.
The genes involved in visual dichromacy or red/green color blindness are those encoding the red and green visual pigments. These are highly homologous (98% sequence identity in exons, introns, and 3′ flanking regions) and are linked in tandem on chromosome Xq28. The red/green gene arrays are composed of a single red pigment gene and one or more green pigment genes located downstream (3′) of the red pigment gene. Gene expression studies indicate that when several green pigment genes are present, only the most proximal is expressed in the retina. Deficiencies in red/green color vision arise from unequal recombination of these normal X-linked genes (Nathans et al. 1986). Such events lead to deletions of the green pigment genes or the formation of full-length hybrid genes consisting of portions of both red and green pigment genes. (Fig. 5). With a few exceptions, deletion of the green pigment genes leaves a single red pigment gene and is associated with deuteranopia (G−R+). Affected individuals are dichromatic, as they completely lack green cones. Individuals with 5′-green–red-3′ fusion genes have deuteranomaly (G′R+), a milder type of color vision defect, with a slightly red-shifted absorption maximum for the green pigment. Individuals with 5′-red–green-3′ fusion genes are associated with protan abnormalities (R− or R′). Those who have a hybrid gene only are always protanopic (R−) and are therefore, dichromats. Those who have normal green genes in addition are either protanopic (R−) or protanomalous (R′) and have a milder defect with slightly green-shifted absorption maximum of the red pigment.
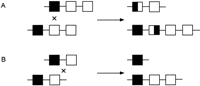
Unequal recombination in the color vision gene cluster. Unequal recombination resulting in creation of fusion genes (A) or loss of genes (B) is shown. (▪) Red cone pigment gene; (□) green cone pigment gene.
Hybrid SMN (survival motorneuron) genes have been identified in patients with autosomal recessive spinal muscular atrophy (SMA) (Hahnen et al. 1996). The SMN gene is a strong candidate for SMA and is present as two highly homologous copies, telomeric SMN and centromeric SMN (TELSMN and CENSMN,respectively) within the SMA region. A large percentage of SMA patients (90%–98%) carry homozygous deletions in TELSMN, affecting either exon 7 or both exons 7 and 8. Hybrid SMN genes were identified in 42 patients with SMA, who showed homozygous deletions of exon 7 but not of exon 8 of the TELSMN copy (Fig.6). Besides the SMN gene, which is present in at least two copies per chromosome, all other genes and markers present in the SMA region are also present in several copies, and these regions are prone to unequal crossing-over resulting in deletions, duplications, and gene conversion events. A putative recombination hot spot represented by recombination-stimulating sequence elements (TGGGG and TGAGGT) was identified in exon 8 of the SMN gene. These sequences are homologous to polymerase arrest sites (Weaver and DePamphilis 1982) and the deletion hot spot consensus sequences in the immunoglobulin switch region (Gritzmacher 1989) and the α-globin gene cluster (Nicholls et al. 1987).
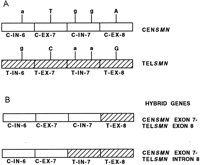
Generation of hybrid SMN genes. (A) The two highly homologous copies of the SMN gene, centromeric SMN(CENSMN) and telomeric SMN (TELSMN) are depicted. The two copies are distinguished by base changes in introns (IN) 6 and 7 and exons (EX) 7 and 8 as shown. The bases within introns and exons are depicted as lower- and uppercase, respectively. (B) Of the 42 patients examined (Hahnen et al. 1996), 40 showed hybrid genes containing CENSMN introns 6 and 7 and exon 7, and TELSMN exon 8; 2 of the 42 patients had a hybrid gene containing CENSMN intron 6 and exon 7, and TELSMNintron 7 and exon 8.
Sex-Specific Meiotic Recombination Hot Spots
Meiotic recombination in the genome does not occur randomly but tends to be concentrated in hot spots, regions with relatively high recombination rates separated by stretches of diminished recombination. There is a striking sexual dimorphism in cytogenetic chromosome length, with human female pachytene chromosomes being 50% longer than those in males (Wallace and Hulten 1985). It seems likely that the higher rate of recombination seen in human females versus males is a reflection of the more condensed state of male chromosomes. Chromatin conformation may influence meiotic recombination in humans. This is suggested by the observation that during spermatogenesis, the X and Y chromosomes are transcriptionally inactive and experience restriction of recombination, whereas the X chromosomes, which are transcriptionally active during oogenesis, participate in unrestricted recombination (Handel and Hunt 1992). Recombination is probably prevented during meiosis in males by specific heterochromatization of the sex chromosomes (McKee and Handel 1993). There is virtual absence of crossing-over in constitutive heterochromatin, which is highly condensed and devoid of transcribed genes; this may be further evidence for an influence of chromatin structure on recombination.
In an effort to determine the pattern of chromatin condensation and recombination at meiosis in an imprinted region, Robinson and Lalande (1995) carried out fine-scale genetic mapping in the 4-Mb Angelman Syndrome/Prader–Willi Syndrome (AS/PWS) region. Their results indicated that the male/female recombination ratio varies significantly over short regions. A male recombination hot spot was localized to a region that is adjacent to, but outside, the putative AS/PWS-imprinted regions. In females, a region of relatively high recombination was observed that spans a domain of paternal allele-specific transcription, implicated in the Prader–Willi syndrome.
The preponderance of CMT1A duplication and factor VIII gene inversion in the male germ line has been discussed in the previous sections.
Proteins Involved in Homologous Recombination and their Role in Cancer
Human cancer is commonly associated with rearrangements of DNA that result in deletions of tumor suppressors and altered expression or amplification of proto-oncogenes. Multiple mutations are present in most human tumors, and these genetic modifications appear to be necessary to produce and select premalignant, malignant, and metastatic cells. Destabilization of genes in cancer could be explained by pre-existing hot spots or by creation of de novo hot spots by the rearrangements themselves. In this section proteins involved in homologous recombination, and “recombination hotspot-binding” proteins and their role in carcinogenesis are discussed.
Homologous recombination is a fundamental biological process, the biochemical understanding of which is most advanced in E. coli. The proteins involved in promoting genetic exchange include RecA, RecBCD (exonuclease V), RecE (exonuclease VIII), RecF, RecG, RecJ, RecN, RecOR, RecQ, RecT, RuvAB, RuvC, SbcCD, SSB proteins, DNA polymerase I, DNA gyrase, DNA topoisomerase I, DNA ligase, and DNA helicases. Collectively, they define biochemical events essential for efficient recombination (Kowalczykowski et al. 1994). In addition to these proteins, a cis-acting recombination hot spot sequence chi (5′-GCTGGTGG-3′), is also known to play an important role (Smith 1989). The central events in homologous recombination are the pairing of homologous molecules and the initiation of strand exchange. The E. coli RecA protein is the prototype of proteins that can catalyze these reactions by promoting interaction between homologous DNA molecules (Kowalczykowski and Eggleston 1994). Evidence is accumulating that all organisms have a protein that shares significant functional homology to this bacterial protein, suggesting that the fundamental mechanisms of recombination are conserved in all species (Heyer 1994). Homologs of RecA are known in yeast (Saccharomyces cerevisiae; ScRad51) (Aboussekhra et al. 1992; Basile et al. 1992;Shinohara et al. 1992); mouse (MmRad51) (Morita et al. 1993); and human (HsRad51) (Shinohara et al. 1993; Yoshimura et al. 1993).
Genes involved in recombination in the yeast S. cerevisiaeinclude those of the RAD52 epistasis group (RAD50–RAD57), involved in meiotic and mitotic recombination (Shinohara et al. 1993; Bishop 1994) and double strand break (DSB) repair (Game 1993). Accurate repair of these genotoxic lesions is essential for the prevention of chromosomal fragmentation, translocations, and deletions, which can lead to carcinogenesis through activation of oncogenes and/or inactivation of tumor suppressor genes. Key genes in the RAD52 DNA repair pathway includeRAD54 and RAD51. The primary sequence of manyRAD52 group genes is conserved from yeast to human. Analysis of the phenotype of mouse MmRad54 −/− cells by Essers et al. (1997) demonstrated that homologous recombination in these cells was reduced compared to wild-type cells, implying that homologous recombination contributes to the repair of DSBs in mammalian cells.
ScRad51 mutants also show defects in genetic recombination and repair of damaged DNA (Game 1993). A role of ScRad51 in meiosis is shown by its presence in meiotic nuclei along with ScDMC1, another homolog of RecA specific to meiosis (Bishop et al. 1992). Evidence for the role of HsRad51 protein in meiosis comes from the findings that antibody to HsRad51 stained murine synaptonemal complexes early in meiosis, and stained numerous foci in nuclei of human cells exposed to DNA-damaging agents (Haaf et al. 1995; Plug et al. 1996). The role of the MmRad51 gene in mitosis and meiosis was shown by the finding of high levels of transcription of homologs of the Rad51 gene in lymphoid and reproductive organs (Shinohara et al. 1993). A homozygous MmRad51 mutation (MmRad51 −/−) was lethal early in murine embryogenesis (Tsuzuki et al. 1996). Lim and Hasty (1996) showed that the embryonic lethal phenotype of MmRad51 −/− is suppressed by a mutation in the p53 oncogene. Sturzbecher et al. (1996) reported that p53 interacts with HsRad51 and RecA. These observations suggest that functional wild-type p53 may select directly the appropriate pathway for DNA repair and control the extent and timing of the production of genetic variation via homologous recombination. Therefore, rearrangements may occur as a direct consequence of a defect in p53-mediated control of homologous recombination processes attributable to mutations in the p53gene.
Individuals with mutations in either the BRCA1 orBRCA2 tumor suppressor genes have a dominant predisposition to breast and ovarian cancer (Smith et al. 1992; Easton et al. 1993;Wooster et al. 1994; Gayther et al. 1997). Colocalization and coimmunoprecipitation experiments have shown that human BRCA1 protein associates with HsRad51 in mitotic and meiotic cells (Scully et al. 1997). Thus, there appears to be a role for BRCA1 in nuclear processes that leads to normal chromosomal recombination and control of genome integrity. Embryonic lethality and radiation hypersensitivity mediated by Rad51 was shown in mice lacking Brca2 (Sharan et al. 1997). Using a yeast two-hybrid assay, Sharan et al. 1997 also identified an interaction between Brca2 and MmRad51. The homozygous mutant phenotypes of Brca1, Brca2, and Mmrad51 are similar, indicating that these genes function in similar pathways (Hakem et al. 1996; Lim and Hasty 1996; Sharan et at 1997). The association of Rad51 with Brca1 and Brca2 and the resultant sensitivity of MmRad51 −/− and Brca2 −/− cells to irradiation may explain the high penetrance of early onset cancer phenotypes exhibited by patients with either BRCA1 or BRCA2 mutations. In mammary epithelial cells that have lost BRCA1 or BRCA2 activity, the HsRad51-mediated DNA repair mechanisms may be compromised, thus destabilizing the genome. Thus, Rad51 probably suppresses tumor formation through its interaction with both Brca1 and Brca2, all three of which may be involved in detecting and repairing DSBs, thereby controlling cell cycle progression (Sharan et al. 1997).
A recombination hotspot-binding protein, translin, which is associated with chromosome translocations and binds to consensus sequences at breakpoint junctions of chromosomal translocations in many lymphoid malignancies, was reported by Aoki et al. (1995). ReHF-1, a recombination hot spot-associated factor specifically recognizes novel target sequences at the sites of interchromosomal rearrangements in T-cell acute lymphoblastic leukemia (T-ALL) (Kasai et al. 1994). Nuclear proteins have been identified that bind to target sequences within the recombination hot spot regions of the Bcl-2oncogene that is involved in rearrangements associated with follicular lymphomas (Aoki et al. 1994). These proteins appear to be similar to ReHF-1. Nuclear proteins were also shown to bind to the recombination hot spot region of the retinoic acid receptor α gene on chromosome 17 (Tashiro et al. 1994, 1995), which along with the PML gene on chromosome 15 is involved in the 15;17 translocations found in acute promyelocytic leukemia. The interaction of these proteins with conserved target sequences at chromosomal breakpoint junctions suggests that they may be involved in enzymatic mechanisms reminiscent of the general features of DNA recombination or replication events in E. coli or S. cerevisiae.
Recombination is of eminent importance in germ cells to generate genetic diversity during meiosis and to safeguard DNA from genotoxic damage in somatic cells. Identification of human homologs of genes encoding components of the recombination and replication pathways of lower organisms will yield insight into mechanisms of disruption of these pathways that lead to human disease. Further characterization of the human genome has allowed identification of previously unknown sequence elements that may be involved in the recombination process. Understanding the complex mutational mechanisms in disease genes will allow us to discover new hot spots and mechanisms of recombination resulting in human disease.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL pragna{at}bcm.tmc.edu; FAX (713) 798-8526.
- Cold Spring Harbor Laboratory Press








