
Small Open Reading Frames: Beautiful Needles in the Haystack
… and a time for all things; a time for great things, and a time for small things.
Miguel de Cervantes (1547–1616)
The completion of genome sequences from model organisms creates new opportunities and resources for both basic and applied research. The genome sequence of several bacterial genomes as well asSaccharomyces cerevisiae represent landmark achievements (Goffeau et al. 1996, 1997). The total genome sequence era offers many opportunities to explore the wealth of information contained within a genome, but it is also one of the most challenging phases for researchers and emphasizes a need for global approaches to study biological problems. One of these challenges is identifying and defining very small protein-coding genes, which can easily escape detection because they are “buried” in an enormous pile of meaningless short ORFs. Yet the subset of small, functional ORFs (here abbreviated smORFs) probably encode very interesting proteins in all organisms, including humans.
The Difficulties of Defining Meaningful smORFs
All long DNA sequences, including random ones, contain many open reading frames (ORFs)1 of 1–99 codons in length; biological sequences also contain many ORFs >99 codons long that correspond to real protein-coding genes. The “gray area” surrounding the ad hoc 100-codon boundary presents two special problems for biologists: (1) ORFs of 100–150 codons include numerous artifactual ORFs (Fickett 1995; Das et al. 1997); and (2) the set of ORFs of 1–99 codons, among which the probability of being biologically meaningless is exceedingly high, nevertheless contains numerous interesting genes, which are easily missed because of the sheer number of small ORFs. To illustrate the magnitude of this problem, we plotted the total number of ORFs in the yeast genome of all lengths between 2 and 1000 codons (Fig. 1)1 of 1–99 codons in length; biological sequences also contain many ORFs >99 codons long that correspond to real protein-coding genes. The “gray area” surrounding the ad hoc 100-codon boundary presents two special problems for biologists: (1) ORFs of 100–150 codons include numerous artifactual ORFs (Fickett 1995; Das et al. 1997); and (2) the set of ORFs of 1–99 codons, among which the probability of being biologically meaningless is exceedingly high, nevertheless contains numerous interesting genes, which are easily missed because of the sheer number of small ORFs. To illustrate the magnitude of this problem, we plotted the total number of ORFs in the yeast genome of all lengths between 2 and 1000 codons (Fig. 1); there are ∼260,000 ORFs from 2 to 99 codons long.
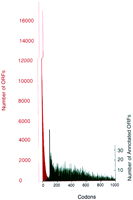
The total number of ORFs of the indicated length encoded in the S. cerevisiae genome are shown in red. The total number of annotated ORFs in SGD are plotted in green. Note that the scale for the total number of ORFs is 100-fold compressed relative to the number of annotated ORFs. Therefore, the difference in magnitude of these curves is actually under-represented by 100-fold. A curve shaped similarly to the red curve but of smaller amplitude is obtained if only the interfeature regions are searched for total ORFs (not shown). The black vertical line at 100 amino acids indicates the cutoff chosen for annotating the genes. (We thank M. Cherry of SGD for kindly providing the data for this graph.)
Because of these problems, ORF length was the key criterion for deciding which ORFs to annotate in the yeast genome. On the basis of simulations with random sequences, all ORFs of at least 100 contiguous codons (including the first ATG) and not entirely contained within a longer ORF on either strand were automatically designated for annotation (Dujon 1994). Using this criterion for S. cerevisiae, the sequence of 12,068 Mb of DNA encompassing 16 chromosomes defined a total of ∼6275 ORFs in addition to genes specifying RNAs (Goffeau et al. 1996).
Both computational and experimental techniques can be used to evaluate the coding potential of a putative ORF. A codon adaptation index (CAI), based on similarity to the preferred codon usage for highly expressed genes in that organism (Sharp and Li 1987), can be used to help predict the likelihood that an ORF represents a highly expressed gene and has been used to help define coding sequences. The average CAI for the entire set of 331 ORFs on chromosome XI is 0.170 (Dujon et al. 1994). ORFs of 100–150 codons with a low CAI (<0.1) were annotated on many yeast chromosomes as questionable ORFs because they may not represent real genes. However, most yeast transcripts are not highly expressed but, rather, are present at one to two copies per cell (Velculescu et al. 1997). Moreover, the smaller an ORF becomes, the less robust the CAI measurement becomes as the contribution made by each individual codon becomes heavier and skews the overall value. Thus, CAI values will become progressively less useful as ORF length decreases.
Termier and Kalogeropoulos (1996) examined the probability of functionality of short ORFs and described computational techniques based on a combination of codon usage, amino acid composition, and dipeptide frequencies in the encoded protein to estimate the likelihood of gene function. Again, these features will fluctuate most dramatically as ORF length decreases. Thus, these computational methods must be combined with some sort of functional analyses to help find the needles in the haystack. We note that in organisms with many spliced genes the problem is somewhat different than in yeast and bacteria because exon definition occurs before ORF definition and in fact may well help with defining the latter.
smORFs
Small proteins include a number of important classes, such as mating pheromones, proteins involved in energy metabolism, proteolipids, chaperonins, stress proteins, transporters, transcriptional regulators, nucleases, ribosomal proteins, thioredoxins, and metal ion chelators. (See Table 1 for a set of 32S. cerevisiae proteins of <7.5 kD encoded by smORFs.) In multicellular organisms, there is already a rich diversity of short peptides including many hormones, antibacterial defensins, cecropins, and magainins. There are also small ORFs encoding transporter proteins, homeobox proteins, transcription factors, and kinase regulatory subunits reported fromCaenorhabditis elegans(http://www.sanger.ac.uk/Projects/C_elegans). How many more interesting smORFs lie buried and undiscovered in fully sequenced yeast and bacterial (nematode and human...) genomes?
A Sample of Some Proteins Encoded by smORFS
Despite the accepted practical lower limit of 100 codons, at least 100S. cerevisiae proteins <100 amino acids long have already been identified by genetic or biochemical techniques. But the total number of such proteins may be much higher. In Eschericia colithere are 381 proteins of <100 amino acids in length represented among a total of 4288 annotated ORFs (8.9%;http://www.genetics.wisc.edu/). Analysis of the yeast mitochondrial genome results in 32 ORFs, of which 4 are smaller than 100 amino acids (http://speedy.mips.biochem.mpg.de/), suggesting that 12.5% of encoded proteins are encoded by ORFs of <100 codons. An independent estimate of this ratio was obtained by examining the set of proteins identified by amino acid sequencing of randomly selected two-dimensional gel spots of total proteins from the fully sequenced cyanobacterium Synechocystis. Of these proteins, 11.8% were encoded by ORFs of <100 codons (http://www.kazusa.or.jp/tech/sazuka/cyano/proteome.html). The latter two calculations are probably somewhat biased toward small proteins but provide at least an upper limit for the number of smORFs. Extrapolating these ratios of smORFs to long ORFs to the entire yeast genome, there might be as many as 800 smORFs in the nuclear genome.
Identifying and Characterizing smORFs in S. cerevisiae
A genome-wide project to disrupt all known yeast ORFs is currently under way (http://sequence-www.stanford.edu/group/yeast_deletion_project/deletion.html). However, this project will not discover new smORFs but, rather, depends on sequence databases like SGD (http://genome-www.stanford.edu/Saccharomyces/) and MIPS (http://speedy.mips.biochem.mpg.de/) to identify genes and bases its decision to disrupt ORFs on this basis. This type of project urgently requires input from the yeast community both on removal of questionable ORFs >99 codons long and annotation and inclusion of smORFs.
Defining smORFs is not a trivial task; several approaches used in parallel should help to identify these genes and help elucidate their biological role. However, all of the methods have severe limitations, and we invite suggestions on additional tools that might help to solve this problem. We describe some approaches currently in use in S. cerevisiae that will assist in identifying smORFs and the limitations of these methods.
Conventional Genetic Techniques
The ease of classical and recombinant genetic approaches has made it possible to define many genes in S. cerevisiae. Standard procedures for mutagenesis and genetic screens have been extremely useful in defining gene functions in S. cerevisiae. However, the small target size of smORFs makes them difficult targets for mutagenesis.
Computational Biology Approaches
Probably the most powerful computational tool available is homology searching. A six-frame translation from each intergenic region of the genome could be individually used in database searches against expressed sequence tag (EST) and protein databases to identify smORFs corresponding to evolutionarily conserved proteins (Koonin et al. 1994). A second approach would be to generate a database of all smORFs and search their 5′ and 3′ noncoding regions for conserved motifs. It may be that there are special problems associated with expressing short ORFs and that there are special consensus sequences involved with overcoming these problems. Such nucleotide sequence signals could then be used as probes to identify additional candidates for smORFs.
Serial Analysis of Gene Expression
The serial analysis of gene expression (SAGE) technique (Velculescu et al. 1995, 1997) has been used to identify, quantitate, and compare global gene expression patterns in S. cerevisiaeand is based on two principles: (1) a 9- to 10-bp sequence tag derived from a defined region in any poly(A)+ transcript uniquely identifies that transcript; and (2) multiple tag sequences concatenated within a clone are obtained in a single sequencing lane. SAGE identified 4665 genes (corresponding to 76% of all annotated ORFs) with transcript levels ranging from 0.3 to 200 copies per cell. In addition to identifying genes predicted by the genome sequencing efforts, SAGE also identified ∼160 transcripts (varying from 1 to 94 copies/cell) corresponding to ORFs of 60–98 codons. The 30 most abundant of these transcripts were observed at least nine times. Several of the corresponding genes are evolutionarily conserved, as at least 7 of 20 smORFs examined have homologs in human, mouse, or C. elegans.Northern blot analysis for three of these has confirmed high level expression. Studies in progress will determine the expression, translation, and possible functions of these smORFs (M.A. Basrai, R.K. Kitagawa, D.E Bassett, Jr., V.E. Velculescu, B. Vogelstein, K. Kinzler, and P. Hieter, in prep.). These results suggest that SAGE can be used on a genome-wide level as a primary screen for identifying genes encoding small proteins not predicted by the genome sequence. The number of smORFs identified will be limited by the number of tags analyzed, the physiological state from which they are isolated, and the restriction enzyme used to define the 9-bp tag (currently the 4-bp cutter, NlaIII). If this enzyme does not cut the cDNA of interest, this transcript will be missed. A possible source of false positives with this method may be that fortuitous ORFs in the 3′-untranslated region (UTR) of another transcript could show up as potential smORFs.
Transposon Methods
The Yale Genome Analysis Center is undertaking a large-scale functional analysis of the S. cerevisiae genome. Insertional mutagenesis based on a bacterial Tn3 derivative has been used to create a collection of strains each with lacZ inserted at a random genomic location along with an in-frame hemagglutinin (HA) tag. The multifunctional transposons identify genes expressed at different times in the life cycle and determine the subcellular locations of the encoded gene products as well as the phenotype of the disrupted strains (Burns et al. 1995; Ross-MacDonald et al. 1997). Fusions have been detected in both known and unknown genes. This technique has also identified fusions in numerous smORFs not annotated by the genome sequencing efforts (http://ycmi.med.yale.edu/YGAC/home.html). These results will allow researchers who identify a yeast gene to determine immediately whether that gene is expressed at a specific time during the life cycle and whether its gene product localizes to a specific subcellular compartment. The success of this strategy, like other expression-based strategies, will be limited by the number of insertions analyzed and the physiological state of the cells from which they are isolated. A number of examples of what appear to be false positives (i.e., fusions to ribosomal DNA) have been reported so far, but others appear to represent novel small genes.
Smith et al. (1996) have described a genetic footprinting method based on the endogenous yeast transposon Ty1; ORFs are evaluated for function by subjecting pools of cells with random Ty1 insertions to various selections and comparing the Ty1 insertion pattern before and after selection. The Ty1 insertions are detected by a PCR approach that requires the use of predetermined target primers corresponding to regions of interest. This method could be useful for identifying smORFs if primers against interfeature regions (regions lying between known ORFs, tRNA genes, or other sequence “features”) were included in the analysis.
Chip Methods
The chip-based methods for analysis of gene expression represent a powerful tool for identifying transcripts (Schena et al. 1995; Shoemaker et al. 1996), including small transcripts corresponding to smORFs. Currently available chips are based on previously defined sequences of interest. Although it would be possible to create arrays of interfeature genomic regions, a confounding issue is that the 3′ ends of yeast transcripts are not systematically defined—some transcripts contain long 3′ UTRs and thus defining the appropriate boundaries of these interfeature regions could not be done in an automated manner. These overlapping transcript ends would create a high background on the chip hybridizations. However, S. cerevisiaeEST data and the results from experimental approaches for known genes could be examined thoroughly to define one or more predictive 3′-end formation consensus sequences. This information might allow an explicitly designed chip to identify small novel transcripts, whether they correspond to ORFs or not.
Integrated Protein Identification and Analysis Approaches
Two-dimensional gels combined with tandem mass spectrometry can be used to identify proteins in relatively complex mixtures. When this type of data is combined with a complete genome sequence, hits are virtually guaranteed. A systematic project of this type is under way at a biotechnology resource center at the University of Washington (http://cellworks.washington.edu/). Both very complex mixtures of yeast proteins and various purified multiprotein complexes are being analyzed by these methods, and it is anticipated that many new small proteins will be identified by this type of procedure. This approach of reverse genetics will aid in the identification of the smORFs.
Conclusion
The approaches described above should be complemented by additional in vivo experimental data to establish the identity of a cloned gene. It is clear that emerging new technologies applied globally to any given model organism will further our understanding of fundamental biological problems. We used several different computer resources to try to determine the number of known smORFs. The output of data obtained by these methods varied widely, depending on the database and search engine that was used. As these are essentially a black box to most end users, it will be extremely useful to have a database specifically designed to catalog and evaluate the possible functionality of smORFs.
Acknowledgments
We thank A. Bairoch, Doug Bassett, Mike Cherry, Mark Johnston, Guy Plunkett III, and John Spieth for helpful discussions and information.
Footnotes
-
↵1 We define an ORF as a segment of DNA capable of encoding a protein beginning with an ATG and ending at a termination (stop) codon (including nested ORFs). We ignore ORFs initiating with other codons.
- Cold Spring Harbor Laboratory Press








