
Minisequencing: A Specific Tool for DNA Analysis and Diagnostics on Oligonucleotide Arrays
Abstract
We describe a method for multiplex detection of mutations in which the solid-phase minisequencing principle is applied to an oligonucleotide array format. The mutations are detected by extending immobilized primers that anneal to their template sequences immediately adjacent to the mutant nucleotide positions with single labeled dideoxynucleoside triphosphates using a DNA polymerase. The arrays were prepared by coupling one primer per mutation to be detected on a small glass area. Genomic fragments spanning nine disease mutations, which were selected as targets for the assay, were amplified in multiplex PCR reactions and used as templates for the minisequencing reactions on the primer array. The genotypes of homozygous and heterozygous genomic DNA samples were unequivocally defined at each analyzed nucleotide position by the highly specific primer extension reaction. In a comparison to hybridization with immobilized allele-specific probes in the same assay format, the power of discrimination between homozygous and heterozygous genotypes was one order of magnitude higher using the minisequencing method. Therefore, single-nucleotide primer extension is a promising principle for future high-throughput mutation detection and genotyping using high density DNA-chip technology.
The Human Genome Project will generate a nucleotide sequence of the genome within a few years. Detailed information of all human genes will speed up the identification of mutations that either cause inherited disorders or predispose to multifactorial diseases. Consequently, more efficient methods with the capacity to analyze interindividual sequence variation on a large scale will be required. A promising approach toward cost-efficient DNA diagnostics and comparative sequence analysis is to use arrays of oligonucleotides (DNA chips) as solid support in miniaturized assays (for review, see Southern 1996). The concept of oligonucleotide arrays was originally introduced for de novo nucleotide sequencing by hybridization (Khrapko et al. 1991). Sophisticated technology has been developed for the production of high-density oligonucleotide arrays (Fodor et al. 1991; Pease et al. 1994; McGall et al. 1996), and systems for fluorescence detection and data analysis for this assay format are also under development (Schena et al. 1995).
Despite the impressive technology that is emerging for hybridization to oligonucleotide arrays, a problem originating from the inherent properties of the nucleic acid hybridization reaction itself is a limiting factor in these methods, particularly when single-nucleotide variations are analyzed. The efficiency of hybridization and the thermal stability of hybrids formed between the target nucleic acid and a short oligonucleotide probe depend strongly on the nucleotide sequence of the probe and the stringency of the reaction conditions (Conner et al. 1983). Moreover, the degree of destabilization of the hybrid molecule by a mismatched nucleotide at one position is dependent on the flanking nucleotide sequence. Consequently, it is an impossible task to design a single set of hybridization conditions that would provide optimal signal intensities and discrimination of a large number of sequence variants simultaneously. In the analyses of human genomic DNA this problem is accentuated because a variable nucleotide may be present either in heterozygous or homozygous form. In conventional reverse blot assays for detection of several mutations simultaneously the difficulty of designing universal hybridization conditions can be circumvented by careful adjustment of the length, polarity, and position of the mismatched nucleotide of the oligonucleotide probes (Saiki et al. 1989). The high-density GeneChip (Affymetrix) technology is based on hybridization arrays carrying probes of all possible sequence combinations or “tiled” arrays scanning the sequence flanking the site of the nucleotide variation (Chee et al. 1996; Cronin et al. 1996; Kozal et al. 1996). To facilitate the detection of heterozygous nucleotide variations, a reference target sequence is included in each hybridization reaction and the interpretation of the result is based on the ratios between the hybridization signals from the reference and the test targets with each probe (Hacia et al. 1996). The necessity of using DNA chips composed of tens of oligonucleotide probes per analyzed nucleotide position and reference target sequences has led to a complex setup of assays, and requires mathematical algorithms for interpretation of the data.
We have developed a method, denoted solid-phase minisequencing, for detecting single-nucleotide variations (Syvänen et al. 1990,1993). In this method a detection primer anneals to the target nucleic acid immediately adjacent to a variable nucleotide position. A DNA polymerase is used to specifically extend the 3′ end of the primer with a labeled nucleotide analog complementary to the nucleotide at the variable site. An advantage of minisequencing, compared to hybridization with oligonucleotide probes, is that all variable nucleotides are identified with optimal discrimination at the same reaction conditions. Based on our previous experience of applying the minisequencing principle for the detection of mutations and polymorphisms (Syvänen et al. 1993; Pastinen et al. 1996;Shumaker et al. 1996), we reason that the minisequencing approach is more straight forward and specific for multiplex mutation detection on oligonucleotide arrays than hybridization with allele-specific probes.
Here we describe a solid-phase minisequencing assay for nine disease-causing mutations in an oligonucleotide array format. The mutations were selected as targets because they occur with a high frequency in the Finnish population and population-based screening for them would be clinically relevant. Five of the mutations cause severe recessive disorders [infantile neuronal ceroid lipofuscinosis (INCL;Vesa et al. 1995), aspartylglucosaminuria (AGU; Ikonen et al. 1991), gyrate atrophy (GA; Mitchell et al. 1989) and nonketotic hyperglycinemia (NKH; Kure et al. 1992)] belonging to the “Finnish disease heritage,” (for review, see Peltonen et al. 1995) and occur with a combined population frequency of carriers of ∼0.05. Three mutations in the low-density lipoprotein receptor (LDLR) gene (Aalto-Setälä et al. 1989; Koivisto et al. 1991, 1995) that cause familial hypercholesterolemia (FH) with the estimated prevalence of 1 in 500, both in Finland and worldwide, were also included in the panel. Together, these eight mutations account for 70%–100% of the known cases of these five disorders in Finland. The well-known blood coagulation factor V Leiden mutation (Bertina et al. 1994), which predisposes to venous thrombosis and occurs with an allele frequency of 0.05 in Western populations, was also incorporated into our set of analyzed mutations.
Our results show that single-nucleotide primer extension is an excellent reaction principle for multiplex detection of mutations. Each of the nine mutations constituting both single-nucleotide transitions and transversions, as well as a large (9.5 kb) and a small (7 bp) deletion, were distinguished unequivocally in heterozygous and homozygous patient samples. When compared to hybridization using allele-specific oligonucleotide probes performed in the same array format, the power of discriminating between genotypes was at least one order of magnitude higher.
RESULTS AND DISCUSSION
Design of the Assay
The key steps of the solid-phase minisequencing method for detecting multiple mutations in an oligonucleotide array format (Fig.1) were optimized in preliminary experiments. The protocol, given in Materials and Methods, represents the result of these experiments. One minisequencing primer per mutation to be detected was immobilized in 3 × 3 arrays on a 0.36-cm2area on glass microscope slides. The primers were coupled to the glass surface by mediation of a 5′-amino group (Southern et al. 1992;Lamture et al. 1994) to leave the 3′ end free to be extended in the minisequencing reaction. The coupling efficiency was found to be maximal after incubation for 5–6 hr (∼0.1–0.8 pmole of primer/mm2), and no loss of oligonucleotide from the glass surface was observed after 16 hr incubation periods. To be more accessible for annealing to the template, the primers contained 15 T residues in their 5′ ends (Guo et al. 1994). Nine genomic fragments spanning the disease-causing mutations were amplified in multiplex PCRs. The multiplex PCRs were optimized (Chamberlain and Chamberlain 1994) to yield a similar amount of each product that ranged from 73 to 235 bp in size.

Steps of minisequencing on a primer array.
To choose the optimal DNA polymerase enzyme for the minisequencing on primer array, we compared the AMV and RetroTherm reverse transcriptases and the Tth, T7, ThermoSequenase, TaqEXPRESS and DyNASeq DNA polymerases for their ability to perform sequence-specific incorporation of 33P-labeled dideoxynucleoside triphosphates (ddNTPs). Two DNA polymerases that have been engineered for cyclic Sanger sequencing, the Thermo Sequenase (Tabor and Richardson 1995) and the novel DyNASeq enzyme originating from Thermus brockianus,performed with the highest specificity in our assay. The average ratios between the correctly incorporated nucleotide and misincorporated nucleotides using these two enzymes were as high as 65 and 87, respectively, which was ∼10 times higher than with any of the other enzymes. Based on these results, the DyNASeq enzyme was used in the subsequent experiments.
Comparison of Oligonucleotide Hybridization and Minisequencing
For comparing the specificity to distinguish between different genotypes by minisequencing reaction to that of the hybridization with allele-specific probes, we analyzed PCR products of ∼100 bp in size from the PPT and FV genes using both methods in the same assay format. The samples were either homozygous or heterozygous for the nucleotide at the mutant (INCLFin or FVLeiden) position. For the hybridization we immobilized double sets of allele-specific probes, of 15 and 20 bases in length, on the glass slides. The hybridization probes carried a tail of 15 T residues in their 5′ ends (Guo et al. 1994), and the mutant nucleotide was located in the middle of the probe (Pease et al. 1994) to provide optimal hybridization efficiency and discrimination between the sequence variants. Moreover, to perform a fair comparison, we employed three sets of washing conditions of increasing stringency and analyzed both single-stranded DNA and RNA templates.
In the hybridization experiments the best discrimination between the genotypes at both analyzed nucleotide positions was obtained using RNA as template (Table 1). The results of the hybridization experiments with probes for only two mutations illustrate the difficulty of designing optimal allele-specific probes and universal reaction conditions simultaneously to analyze more than one variable nucleotide. At the site of the PPT mutation, the 15-mer probes performed better than the 20-mer probes. The 20-mer FV probes discriminated better between the genotypes than the 15-mer FV probes when RNA served as template, whereas the 15-mers were better for discriminating between the genotypes in a DNA template. These results are in agreement with the lower G-C content of the FV probes than that of the PPT probes, and with the fact that the FV mutation is a G ′ A transition.
Genotyping the PPT and FV Genes by ASO Hybridization and Minisequencing on Oligonucleotide Arrays
In view of results in the literature (Guo et al. 1994; Lamture et al. 1994; Cronin et al. 1996), the ratios that we obtained between the hybridization signals with the matched and mismatched probes at the homozygous nucleotide positions were acceptable, particularly with the 20-mer FV probes and the 15-mer PPT probes using RNA templates. However, the corresponding ratios between correctly incorporated and misincorporated nucleotides in the minisequencing reactions were significantly higher, as can be seen by comparing the best results obtained by both methods, which are highlighted in Table 1. The ratio between the hybridization signals of the probe for the normal and the mutant allele deviated more from the expected ratio of 1 than the corresponding ratio between the incorporated ddNTPs in the minisequencing reaction. This was attributable to the different thermal stabilities of the hybrids with the allele-specific probes at the heterozygous nucleotide positions (Table 1). Consequently, the power of discriminating between genotypes was as much as 27 times higher at the site of the PPT mutation and nine times higher at the site of the FV mutation using the minisequencing method than with allele-specific hybridization probes (Table 1).
Multiplex Genotyping by Minisequencing
Samples from patients (homozygotes) and disease gene carriers (heterozygotes) at each of the nine sites were analyzed by the multiplex minisequencing method. Figure 2 shows the result obtained in eight samples, as visualized by scanning the primer arrays by the phosphorimaging device. In each of the seven heterozygous samples and in the compound heterozygous GA sample, the signals corresponding to both the normal and mutant nucleotides are clearly visible in the correct positions on the array. A single signal defining a nucleotide in homozygous form is seen in all other positions. At each of the nine mutant sites high signals from correctly incorporated nucleotides, as compared to the average misincorporation, illustrate the excellent specificity of the minisequencing reaction (Fig. 3a). In most cases, the ratio between the signal for the normal nucleotide and that for the mutant nucleotide differed by two orders of magnitude between homozygous and heterozygous samples (Fig. 3b). Thus, the genotype of each sample is unequivocally defined by the minisequencing assay at each analyzed nucleotide position.
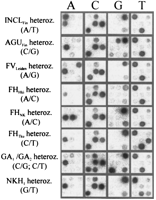
Image obtained from the analysis of eight samples carrying heterozygous mutations by minisequencing on a primer array. The minisequencing primers for detecting nine mutations were immobilized on the arrays in the following order: (Top row, left to right), INCLFin, AGUFin, and FVLeiden; (middle row), FHHki, FHNK,and FHTku; (bottom row) GA1, GA2, and NKH1. The [33P]ddNTP included in the minisequencing reactions is indicated at the top; the genotypes of the samples with the [33P]ddNTP expected to be incorporated at the site of the mutation are given at left.
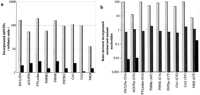
Multiplex detection of nine mutations in samples from heterozygous and homozygous individuals. (a) The signal intensities given for the correctly incorporated nucleotide (lightly shaded bars) at each site are the mean values obtained in 12 samples. The misincorporation signals (dark bars) are the mean value observed in the minisequencing reactions detecting the three other nucleotides at each site in the same 12 samples. (b) The ratio between the signal obtained in the minisequencing reaction corresponding to the normal nucleotide and the signal in the reaction for the mutant nucleotide are plotted on a logarithmic scale for each mutation site. The mutant and normal nucleotides at each position are indicated below. The lightly shaded bars correspond to homozygous normal genotypes. The intermediately shaded bars are heterozygous genotypes, and the dark bars are from individuals homozygous for the mutant allele.
FUTURE PROSPECTS
Already in its present form, the minisequencing primer array can be used as a specific and cost-effective alternative in the screening of relevant mutations in the Finnish population, where typically only one or a few mutations account for the vast majority of the disease alleles. However, to develop high-throughput DNA chips for solid-phase minisequencing assays, some technical issues are to be addressed. Technology for preparing arrays with a high density of primers will be required. The present technology for in situ preparation of high-density oligonucleotide arrays (Fodor et al. 1991; Southern et al. 1992) is not applicable for our method, as the synthesis proceeds in the 3′ → 5′ direction and does not leave the 3′ end free for extension. High-density arrays have also been produced by chemical coupling of NH2-modified cDNAs as miniaturized spots on an activated glass surface using high-capacity printing robotics (Schena et al. 1996). Arrays prepared by this technology could be used for high-throughput genotyping by minisequencing, as our method, in principle, requires only a single primer per variable nucleotide, instead of multiple probes per mutation as in the hybridization methods. The use of a single primer per analyzed nucleotide position requires ddNTPs differentially labeled with four fluorophores. Fluorescent ddNTPs have been shown to be feasible as labels in minisequencing assays (Pastinen et al. 1996; Shumaker et al. 1996;Tully et al. 1996), and custom-built sensitive fluorescence scanners with the ability to detect multiple fluorophores at different wavelengths have been devised recently (Schena et al. 1996; Speicher et al. 1996). Despite the proceeding technical development related to miniaturized arrays for genotyping, a great challenge for the assays still lies in the sample preparation. At present, amplification of DNA templates by PCR limits the number of genomic fragments that can be analyzed efficiently. Methods are required by which a significantly larger number of fragments can be amplified or in which an amplification step is avoided. When the ubiquitous problem of sample preparation has been solved, minisequencing might be the reaction principle of choice for the implementation of high-throughput genotyping on miniaturized arrays in practice.
METHODS
DNA Samples
Blood samples from patients and healthy carriers with previously characterized mutations were collected in accordance with the Helsinki Declaration. The DNA was prepared from the blood samples by a standard method (Bell et al. 1981).
Primers
One PCR primer of each pair contained the T7 RNA polymerase promotor sequence (TTCTAATACGACTCACTATAGGGAGA) in its 5′ end, and the 5′ end of the other PCR primer was modified with a biotin residue (RPN 2012; Amersham, Bucks, UK) during its synthesis (Table2). The minisequencing primers and the allele-specific oligonucleotides (ASOs) contained 15 T residues as a spacer sequence 5′ of the gene-specific sequence and a 5′-amino group added during their synthesis (Aminolink 2, Applied Biosystems, Foster City, CA). Table 2 gives the sequences of the PCR and minisequencing primers. The sequence of the 15-mer ASO probes for detecting the INCLFin mutation was ACTGCCC(T/A)CCTACGG (with the site of the mutation shown in parentheses) and that of the 20-mer probes was CCACTGCCC(T/A)CCTACGGAAT. The sequence of the 15-mer ASO probes for the FVLeiden mutation was GACAGGC(G/A)AGGAATA, and that of the 20-mer probes was CTGGACAGGC(G/A)AGGAATACA. All oligonucleotides were synthesized on an Applied Biosystems 392 DNA synthesizer.
Genes, Primers, and Mutations
Oligonucleotide Arrays
Printed microscope glass slides (Erie Scientific, Portsmouth, NH), containing eight circular wells of 10 mm in diameter lined by a Teflon coating, were used in the minisequencing reactions, and slides with three wells of 14 mm in diameter were used in the ASO hybridizations. The glass slides were washed using a detergent, rinsed with distilled water and ethanol, dried at 80°C for 5 min, and placed in a mixture containing 80 ml of dry xylene, 32 ml of 96% 3-glycidoxypropyltrimethoxy silane (Aldrich, Steinheim, Germany), and 160 μl of 99% N-ethyldiisopropylamin (Aldrich) at 80°C for 16 hr. The slides were then rinsed in ethylacetate and dried at 80°C for 30 min (Lamture et al. 1994; Shumaker et al. 1996). The oligonucleotides carrying a 5′-amino group were applied onto the epoxide-activated glass surface of the wells manually with the aid of a model array below each well. One-half microliter of a 80 μm primer solution in 0.1 m NaOH was applied per spot of the array. The immobilization reaction was allowed to proceed in a humid chamber at 37°C for 16 hr. The slides were subsequently washed in distilled water and stored at 4°C until use.
PCR
The duplex PCRs for the PPT and FV genes were carried out at a dNTP concentration of 0.2 mm, using 2.5 units of Dynazyme II DNA polymerase (Finnzymes, Helsinki, Finland), 150 ng of template DNA, 30 pmoles of the PPT primers and 100 pmoles of the FV primers in 100 μl of 1.5 mm MgCl2, 20 mm Tris-HCl (pH 8.8), 15 mm (NH4)2SO4, 0.1 % Tween 20, and 0.01% gelatin. The PCR reactions for the ASO hybridization experiments with DNA as target were carried out identically, except that the dNTP concentration was 0.07 mmand 3.5 μCi of [32P]dATP (3000 Ci/mmole, Amersham) was included in the reaction mixture. The multiplex amplification of the PPT, AGA, FV, LDLR, OAT, and GCSP genes was carried out at a dNTP concentration of 0.7 mm, using 10 units of Dynazyme II DNA polymerase, 150 ng of template DNA, 35 pmoles of the PPT and LDLR primers; 110 pmoles of the AGA, FV, and OAT primers, and 60 pmoles of GCSP primers in 100 μl of 2.2 mm MgCl2, 20 mm Tris-HCl (pH 8.8), 15 mm(NH4)2SO4, 0.1% Tween 20, and 0.01% gelatin. The PCRs were performed in a Programmable Thermal Controller (MJ Research, Watertown, MA). The cycling parameters for the duplex amplifications were 35 cycles of 1 min at 95°C, 1 min at 56°C, and 1 min at 72°C. The parameters for the multiplex amplifications were the same, except that the extension step at 72°C was 3 min. All PCRs were initiated by a hot start by adding the DNA polymerase at 85°C.
Preparation of Single-Stranded DNA
The duplex or multiplex PCR products were captured on 1 mg of avidin-coated polystyrene particles (Idexx Research Products, Westbrook, ME) in 100 μl of TENT buffer (40 mm Tris-HCl at pH 7.5, 1 mm EDTA, 50 mm NaCl, 0.1% Tween 20) at 22°C for 30 min. The particles were collected by centrifigution for 2 min at 6000g, washed once with 200 μl of TENT buffer, collected again, suspended in 25 μl of TE buffer (10 mm Tris-HCl, 0.1 mm EDTA at pH 8.0), boiled for 2 min, and the supernatant containing the single-stranded DNA (ssDNA) template was collected.
Preparation of RNA
For the minisequencing experiments with RNA as template, 16μl of the duplex or multiplex PCR products was used per transcription reaction. The reactions were carried out at a NTP concentration of 0.5 mm, using 20 units of T7 RNA polymerase (Pharmacia Biotech, Uppsala, Sweden) and 25 units of RNasin enzyme (Promega, Madison, WI) in 25 μl of transcription buffer (20 mm Tris-HCl at pH 8.0, 6 mm MgCl2, 2 mm spermidine, 10 mm NaCl, 5 mm dithiotreitol). For the ASO hybridization experiments, 7.5 μl of the duplex PCR products was used per RNA transcription reaction containing 0.5 mm of ATP, CTP and GTP, 25 μm of UTP, 25 μCi of [32P]UTP (>1000 Ci/mmole, Amersham), 20 units of T7 RNA polymerase, and 20 units of RNasin in 20 μl of transcription buffer. The reactions were allowed to proceed at 37°C for 60 min.
Minisequencing Reactions
For annealing of the ssDNA or RNA to the oligonucleotide arrays, 5.3 μl of the template was added in a final volume of 6.5 μl of 0.2 m NaCl and 0.1% Triton X-100 in TE buffer to each well of the slide, and the reaction was allowed to proceed under a coverslip at 30°C for 15 min. The slides were then rinsed with the annealing buffer and preheated to 60°C. Eight microliters of the minisequencing reaction mixture containing 0.2 pmole (0.3 μCi) of the appropriate [33P]ddNTP (1500 Ci/mmole, AH9530-3, Amersham), 0.2 pmole of each of the corresponding three other unlabeled ddNTPs, 0.6 units of DyNASeq DNA polymerase (Finnzymes) in its buffer (26 mm Tris-HCl at pH 9.5, 6.5 mm MgCl2, 1.8% Triton X-100) preheated to 60°C was added to the wells carrying the annealed templates. The reactions were stopped after 45 sec by washing the slides vigorously in distilled water. The following enzymes were compared in minisequencing reactions with RNA as a template: AMV reverse transcriptase (Promega), RetroTherm RT (Epicentre Technologies, Madison, WI), Tth DNA polymerase (Boehringer Mannheim, Germany). The Tth DNA polymerase has reverse transcriptase activity in the presence of MnCl2, and it was used with the RNA template at the conditions recommended by the manufacturer. With DNA as template, T7 DNA polymerase (Pharmacia Biotech), ThermoSequenase DNA polymerase (Amersham), TaqEXPRESS (GENPAK, Brighton, UK), and DyNASeq DNA polymerase (Finnzymes) were compared. In the reactions, 0.2 pmole of ddNTPs and 0.6 unit of enzyme at the buffer conditions recommended by the supplier were used. The reaction temperature for the AMV reverse transcriptase and T7 DNA polymerase was 50°C, and for the other (thermostable) enzymes it was 60°C.
ASO Hybridization Reactions
The hybridization of 6.5 μl of the labeled ssDNA or RNA templates to the arrays carrying the allele-specific probes was performed in a final volume of 8 μl, in 0.75 m NaCl, 75 mm sodium phosphate (pH 7.5), 1.25 mm EDTA (5× SPE), and 0.1% Triton X-100 at 22°C for 30 min. The slides were washed in 5× SPE at 22°C for 10 min and exposed to an imaging plate and scanned. After scanning, the slides were rewashed in 1× SPE at 22°C for 10 min, followed by exposure and scanning. The ASO slides with RNA templates were washed once more with 0.2× SPE at 22°C for 10 min, exposed, and scanned.
Measurement of the Signals
The minisequencing and ASO slides were exposed to an imaging plate (BAS-MP 2040 S, Fuji, Kanagawa, Japan) for 30 min, and the plate was scanned in a Fuji BAS 1500 Bio-imaging Analyzer. The signals were measured using a Tina 2.09 software package (Raytest, Straubenhardt, Germany) on a microcomputer by outlining each spot (area 2.5 mm2), determining the signal per square millimeter and reducing the average backround signal per square millimeter of the area of the slide without oligonucleotides.
Acknowledgments
We thank Drs Pertti Aula, Kimmo Kontula, Jaakko Leisti, Pirkko Santavuori, and Olli Simell for kindly providing the patient samples and Drs. Hans Söderlund and Lasse Lönnqvist for helpful comments. This work was supported by grants from the Academy of Finland, the Instrumentarium, the Technology Development Center of Finland, the Ulla Hjelt Fond of the Pediatric Research Foundation, and the Rinnekoti Research Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵2 Corresponding author.
-
E-MAIL christine.syvanen{at}ktl.fi; FAX 358-9-4744480.
-
- Received March 3, 1997.
- Accepted April 15, 1997.
- Cold Spring Harbor Laboratory Press








