
Genomic Organization of the Human PEX Gene Mutated in X-Linked Dominant Hypophosphatemic Rickets
- Fiona Francis1,8,
- Tim M. Strom2,
- Steffen Hennig1,
- Annett Böddrich1,
- Bettina Lorenz2,
- Oliver Brandau2,
- Klaus L. Mohnike3,
- Michele Cagnoli3,
- Christina Steffens1,
- Sven Klages1,
- Katja Borzym1,
- Thomas Pohl4,
- Claudine Oudet5,
- Michael J. Econs6,
- Peter S.N. Rowe7,
- Richard Reinhardt1,
- Thomas Meitinger2, and
- Hans Lehrach1
- 1Max-Planck Institut für Molekulare Genetik, Berlin 14195, Germany; 2Abteilung Medizinische Genetik, Kinderpoliklinik der Ludwig-Maximilians-Universität, München 80336, Germany; 3Zentrum für Kinderheilkunde, Otto-von-Guericke Universität, Magdeburg 39112, Germany; 4Gesellschaft für Analyse-Technik und Consulting mbH (GATC), Konstanz D-78467, Germany; 5Institut de Génétique et de Biologie Moleculaire et Cellulaire (IGBMC), 67404 Illkirch, France; 6Department of Medicine, Duke University Medical Center, Durham, North Carolina 27710; 7University College London, Department of Medicine, Middlesex Hospital, London W1N 8AA, UK
Abstract
X-linked dominant hypophosphatemic rickets (HYP) is the most common form of hereditary rickets. Recently we have cloned thePEX gene and shown it to be mutated and deleted in HYP individuals. We have now completely sequenced a 243-kb genomic region containing PEX and have identified all intron–exon boundary sequences. We show that PEX, homologous to members of a neutral endopeptidase family, has an exon organization that is very similar to neprilysin. We have performed an extensive mutation analysis examining all 22 PEX coding exons in 29 familial and 14 sporadic cases of hypophosphatemia. Sequence changes include missense, frameshift, nonsense, and splice site mutations and intragenic deletions. A mutation was found in 25 (86%) of the 29 familial cases and 8 (57%) of the 14 sporadic cases. Our data provide the first evidence that most of the familial and also a large number of the sporadic cases of hypophosphatemia are caused by loss-of-function mutations in PEX.
[The sequence data described in this paper have been submitted to GenBank under accession nos.Y08111–Y08132 and Y10196.]
X-linked dominant hypophosphatemic rickets [HYP; MIM 307800 (Mendelian inheritance inman number); McKusick 1994] has an incidence of 1 in 20,000 individuals and is the most common form of hypophosphatemia. The main physiological traits of the disease are a leak of phosphate from the kidney causing low phosphate levels in the blood and defective bone mineralization. Patients exhibit rickets and osteomalacia, lower extremity deformities, short stature, bone pain, dental abnormalities, and abnormal vitamin D metabolism. Several other less common disorders of inherited renal phosphate wasting also exist, including an autosomal dominant form (ADHR; McKusick 1994, MIM 193100; Econs and McEnery 1997) and hereditary hypophosphatemic rickets with hypercalciuria (HHRH;McKusick 1994, MIM 241530), which shows a complex inheritance pattern (Tieder et al. 1987). An additional tumor-induced form of hypophosphatemia exists, oncogenic hypophosphatemic osteomalacia, in which removal of the tumor leads to a return in normal phosphate levels (Fukomoto et al. 1979; Lobaugh et al. 1984; Weidner et al. 1985). These additional forms of the disease suggest that phosphate homeostasis is a complex process involving multiple gene products.
Recently we cloned a candidate gene, PEX, for the X-linked dominant form of hypophosphatemic rickets, localized to the human Xp22 region (HYP Consortium 1995). PEX has homologies to a family of zinc metalloproteases that includes neprilysin (NEP;D’Adamio et al. 1989), the Kell antigen (KELL; Lee et al. 1991) and endothelin-converting enzymes 1 and 2 (ECE-1 andECE-2; Schmidt et al. 1994; Xu et al. 1994; Emoto and Yanagisawa 1995). NEP is known to inactivate a wide variety of peptide hormones, whereas ECE-1 and ECE-2 process inactive big endothelin 1 to its active form. A substrate forPEX has not yet been identified, and hence the role of this endopeptidase in the pathophysiology of HYP is not clear.
Two mouse mutations, Hyp (Eicher et al. 1976) and Gy(Lyon et al. 1986), map to the syntenic region of the mouse X chromosome, and affected mice exhibit hypophosphatemic rickets.Gy mice have some additional disease features to Hypmice that include circling behavior, inner ear abnormalities, sterility, and reduced viability. Therefore it has been suggested that two different, closely situated mouse genes are involved in hypophosphatemia. Recently, Du et al. (1996) reported the isolation and characterization of the mouse Pex gene, and demonstrated its expression in osteoblasts by Northern blot analysis. We have detected deletions in this gene in Hyp and Gy mice (Strom et al. 1997), although the phenotypic differences observed in Gymice remain unexplained.
The analysis of the genomic region containing the PEX gene is essential for the further understanding of the pathophysiology of hypophosphatemia in both humans and Gy and Hyp mice. Initially we reported only a partial cDNA sequence of the humanPEX gene (GenBank accession no. U60475; HYP Consortium 1995); in this study we have characterized the complete PEX gene structure. We have found that PEX, in common with neprilysin, is composed of multiple small exons spread over a large genomic region. We have identified 22 exons spanning 220 kb of genomic sequence, and 17 out of the 22 exons are <130 bp in size. We have performed an extensive mutation analysis in individuals with familial or sporadic cases of hypophosphatemia. Our data provide evidence that most of the familial and also a large number of the sporadic cases are caused by loss-of-function mutations in PEX.
RESULTS
Primary Structure of the PEX Gene Locus
We have completely sequenced 243 kb of genomic DNA (EMBL accession no. Y10196; Fig. 1) spanning the region encompassed by the markers DXS8254 on the telomeric side (nucleotide positions 8260–8442 bp) and DXS1683 on the centromeric side (nucleotide positions 228147–228300 bp). Twenty-two PEX coding exons are present within this region, and in total the gene covers ∼220 kb. A detailed analysis of this sequence has been performed by use of a variety of gene prediction programs and by screening the nonredundant and dbEST databases (Fig. 1). The splice acceptor and splice donor sequences in the flanking introns of PEX were found to conform to the GT–AG rule (Table 1). Nineteen of the 22PEX exons were predicted by FEXH (Solovyev et al. 1994), 18 by GRAIL, (Uberbacher and Mural 1991), and 7 by GeneID (Guigo et al. 1992). Fourteen of the 22 PEX exons (exons 1–9, 11–14, and 18) were consistently recognized by both GRAIL and FEXH. Notably, GeneID predicted only a cluster of exons in the middle of the gene (exons 6–11 and 13). FEXH recognized 35 additional (non-PEX) exons on the same strand, and GRAIL predicted an additional 27, although only 4 exons were predicted in common by both programs. GeneID predicted less exons in general across the whole region, only 13 additional exons were predicted on the forward strand. Of these, four were also predicted by FEXH, and one was predicted by both additional programs. On the reverse strand, GRAIL predicted 35 exons, and FEXH predicted 33. Seven of these were predicted by both programs. GeneID was not used to predict exons on the reverse strand.
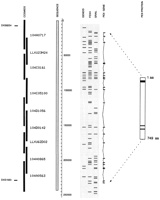
Genomic sequence data encompassing the PEX gene region in Xp22.1. This data has been entered into an ACeDB database, to assimilate better the results from various analysis programs. The scale bar indicates the length of the sequence in base pairs. In the shaded box to the left of the scale bar is the contiguous genomic sequence (one contig of ∼243 kb). Cosmid clones are black bars atleft. In total, the sequence was derived from nine overlapping cosmids. The predicted protein structure of PEX is represented schematically at right. Within this, the uppermost black box represents a transmembrane domain, and the two thinner black boxes represent conserved zinc-binding motifs (HEXXH and ENXADXGG), containing the three zinc-binding ligands (H, H, and E). ThePEX gene consists of 22 exons spanning 220 kb. The genomic organization of PEX is indicated to the left of thePEX protein. The results for the forward strand from three different gene prediction programs are depicted in the shaded box to the right of the scale bar. Comparing the results of these three programs with the known exons from the PEX gene, it can be seen that Grail predicts 18 of the 22 PEX exons (and 27 additional exons), whereas FEXH predicts 19 of the 22 PEXexons (and 35 additional exons). Of the additional exons predicted (non-PEX), only four are predicted in common between the two programs. This genomic sequence contains the markers DXS8254 (at position 8.2 kb) and DXS1683 (at position 228 kb). Nucleotides 1–28835 were found to overlap with a sequence already submitted to GenBank (accession no. U73024).
PEX Gene Structure Information
Examination of the genomic sequence upstream of the first exon of our cDNA sequence published previously, identified seven additional nucleotides likely to be the 5′ coding end of PEX(ATGGAAG). These nucleotides are identical between mouse and human as shown by a comparison to mouse 5′ rapid amplification of cDNA ends (RACE) product sequences (Strom et al. 1997). This comparison also showed high conservation (82% identity over 530 nucleotides) in the region upstream of the ATGGAAG sequence. This region, when translated, contains a number of stop codons and is presumably the 5′ untranslated region (UTR). Additional evidence for this was revealed by database searches that identified a highly significant match to an expressed sequence tag (EST) containing the 5′ end of a rat incisor cDNA clone (GenBank accession no. R47026). This cDNA clone is the rat homolog of PEX and contains ∼470 nucleotides upstream of the putative start methionine, and the complete PEX coding sequence followed by ∼2.5 kb of 3′ UTR (data not shown). Analysis of the sequence surrounding the first in-frame ATG in man, mouse, and rat suggests that PEX lacks a Kozak consensus sequence, which might be an indication that it is highly regulated at the post-transcriptional level (Kozak 1991). Rare cutting enzyme sites are also absent upstream of this exon, although GRAIL detects a GC content of 54%. Interestingly, the GRAIL program predicts only a single promoter region in the first 80 kb of genomic sequence, and this is localized 748 bp before the beginning of PEX exon 2. However, the PEX/Pex 5′ RACE and cDNA results are not consistent with the use of this promoter.
At the 3′ coding end of the PEX gene, an additional 327 nucleotides have been identified extending the human sequence published previously and containing exons 19–22. Exon 19 contains a sequence motif (ENXADXGG; see Fig. 1) that is known to be highly conserved in the neutral endopeptidase family, which includes PEX. This motif contains a glutamic acid residue that is thought to be the third zinc-binding ligand (Le Moual et al. 1991) and an aspartic acid residue that has been shown in NEP to be essential for catalysis (Le Moual et al 1994). A stop codon is present in exon 22, following amino acid residue 749, and this region is highly conserved in the neutral endopeptidase family. The 3′ UTR extends for a further 233 nucleotides before reaching a putative polyadenylation signal (AAUAAA). Recently, Du et al. (1996) have reported a transcript size of 6.6 kb for the mouse Pex gene, which includes a long 3′ UTR, although Northern blot results for the human gene have not yet been reported. Examination of the downstream human genomic sequence detects six further AATAAA or ATTAAA sequences over the following 4.2 kb: These are 389, 1141, 1937, 2497, 3337, and 3388 nucleotides downstream from the stop codon. In each of the seven cases the approximate size of the seven possible transcripts would be 2.9, 3.1, 3.9, 4.9, 5.2, 6.1, and 6.3 kb, respectively. The polymorphic DXS1683 microsatellite marker (Econs et al. 1994) is contained in this region beginning at a position 1256 nucleotides downstream from the stop codon.
PEX and NEP (D’Adamio et al. 1989) are fairly similar in their exon organization (Fig. 2). Each gene is composed of 22 small coding exons; in each case the first coding exon is thought to contain the short amino terminus of the protein followed by a predicted transmembrane domain. PEX exon 5 is larger than the others (227 bp) and the equivalent residues of NEP are contained in two smaller exons (96 and 119 bp) separated by a small intron (108 bp). Similarly, NEP exon 19, which contains the HEXXH zinc-binding motif characteristic of this family of endopeptidases, is 120 bp in size, whereas the equivalent residues in PEX are encompassed in two exons of 55 bp and 68 bp in size. Apart from these two regions of difference in coding exon organization, the remaining PEXexons follow very closely the NEP coding exons; various conserved features such as cysteine residues and amino acids thought to be involved in substrate binding are positioned in similar regions of equivalent exons. Particularly conserved between these genes are the patterns of codon breakage at the splice junctions (Fig.2).

Similarity of PEX and NEP genomic structures. ThePEX and NEP coding exons are represented as boxes, and the sizes of the exons are written below each. The numbering of the exons is 1–22 for PEX representing all known exons, and 3–24 for NEP, which has two 5′ exons (not shown) containing solely untranslated sequences. The first exon in each case begins with a start methionine (ATG), and the last exon ends with a stop codon (*). Seven of the equivalent exons are identical in size between the genes (PEX exons 3, 10, 11, 14, 15, 19, and 21 are the same as the equivalent exons in the NEP genes 5, 13, 14, 17, 18, 21, and 23). Above exon boxes are represented various conserved features: (TM) transmembrane domain; (C) cysteine residue; (Z) pentapeptide zinc-binding motif (HEXXH); (E) second zinc-binding motif (ENXADXGG). The three exons following the transmembrane domain exon contain five cysteine residues similarly spaced between PEXand NEP (amino acid residues 54, 59, 77, 85, and 142 inPEX, and 57, 62, 80, 88, and 143 in NEP). One cysteine residue is present in the middle of the gene at position 406 in PEX, and equivalent position 410 in NEP. Similarly, toward the end of the gene cysteine residues are present at 617, 693, 733, and 746 positions in PEX and equivalent positions of 620, 694, 734, and 746 in NEP. A fingerprint for each gene has been generated by classification of the intron interruption of the reading frame at the end of each exon (Sharp 1981): introns lying in between codons (phase 0), introns interrupting a codon between the first and second base (phase 1), and introns interrupting a codon between the second and third base (phase 2). The fingerprints forPEX and NEP are almost identical.
The PEX gene has large introns (Table 1), and contained in several of these (principally introns 3, 12, and 15) are clusters of predicted exons that have been recognized by more than one gene prediction program. This may suggest that there are additional genes in the region. These exons have not, however, detected any significant matches in dbEST and nonredundant database screens.
Additional Features of the Sequence
At the distal end of the sequenced region, our sequence overlaps ∼29 kb with that produced by another group (GenBank accession no.U73024). A detailed comparison of the overlapping sequences shows that they are very similar. It was possible to detect 62 regions of nucleotide difference (single base pair changes or insertions or deletions of one or more nucleotide residue), although because the respective cosmids are derived from different genomic sources, the majority of the nucleotide differences are likely to be polymorphisms. Thirty-four of these differences appear in Alu/Line repeat elements or in regions of low complexity. Two of these nucleotide differences were consistent with our data, in a region of overlap between a cosmid derived from the Lawrence Livermore library (LLXU24M23) and a cosmid derived from the Imperial Cancer Research Fund (ICRF) library (ICRFc104A0717).
Other polymorphisms detected include a trinucleotide repeat (TAA) that was detected at nucleotide position 35790. Thirteen copies of this repeat were present in cosmid ICRFc104A0717, and 14 copies in cosmid LLXU24M23. Dinucleotide repeats (GT) were evident in the sequence at positions 92021, 152026, and 161611, although it is not known whether these are polymorphic. A 25-nucleotide tandem repeat was detected in the overlap region of cosmids ICRFc104H0658 and ICRFc104A0635 (nucleotide positions 203970–203995). Sequenced shotgun clones from both cosmids were found to have one to four copies of this repeat that has the sequence CATACAGCGCTGTATGATTTATTAT.
Mutations
The entire PEX coding sequence was analyzed for mutations in 29 familial and 14 sporadic cases of hypophosphatemia. These individuals were European, originating mainly from Germany (see Methods). Intron–exon boundary sequences for the 22 PEX exons were used to design primers for single-strand conformation polymorphism (SSCP) analysis (Table 2).
Primers for Mutation Analysis
PCR analysis revealed seven deletions in male patients comprising one or more exons. Specifically, it was not possible to amplify exons 4, 5, 6, 9, 10 and 11, 12, 17–22, and 19–22 from the individuals 3941, 4205, 7754, 5761, 4098, 4004, and 7756, respectively (see Table3). The majority of these deletions were confirmed by detection of absent or changed restriction fragments on Southern blots (individuals 3941, 4205, 7754, 5761, and 4098). In individuals 3941 and 4205, deletions were found to be ∼17 kb and 6 kb, respectively, as we have reported previously (HYP Consortium 1995). A mechanism that might cause gene deletions involves the presence of direct repeats at deletion breakpoints, possibly leading to illegitimate recombination. In some cases Alu repeats have been implicated in this process (Lehrman et al. 1985; Luzi et al. 1995), in other cases short repeat sequences (Canning and Dryja 1989; Kornreich et al. 1990). Knowledge of the complete genomic sequence and repeat distribution of thePEX gene will facilitate further precise mapping of patient deletions, in order to investigate the mechanisms involved.
PEX Mutations in HYP Families
SSCP screening identified 26 mutations in male and female individuals: 16 of these were frameshift or stop mutations, 8 were missense mutations, and the remaining 2 were splice site mutations (Table 3). Mutations generating premature stop codons were found in exons 1 (R20X), 8 (R291X), 12 (W456X), 20 (W660X), and 22 (R747X). Interestingly, this latter stop mutation (shown in Fig.3) is only two amino acid residues away from the end of the coding sequence, and this residue has been shown in NEPto be involved in substrate interaction (Beaumont et al. 1991). Frameshift mutations occurred in exons 6 (682delTC; previously reported in HYP Consortium 1995), 10 (1134delT), 14 (1559delG and 1571insC), 18 (1783insTGAT and 1831delTT), 20 (1991insTGAC), and 21 (2093delC). The insertions in exons 18 and 20 are duplication events of TGAT and TGAC nucleotides, respectively.
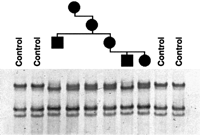
Segregation of the stop mutation R747X in family 316. Amplification products of exon 22 were analyzed by SSCP. Individuals in the pedigree from left to right are 4266 (male), 4264 (female), 4265 (female), 4063 (female), 4064 (male), and 4065 (female).
Four different types of missense mutation were observed: Cys to Arg in exon 3 (C85R), Pro to Leu in exon 15 (P534L), Gly to Arg in exon 17 (G579R), and Arg to Pro in exon 19 (R651P). Mutations were found scattered throughout the entire gene as depicted schematically in Figure 4, and the majority were unique, with the exceptions of R20X, P534L, G579R, and R747X, which were found in two, two, four (shown in Fig. 5), and three unrelated cases, respectively. Population-based studies (120 alleles tested) were performed in the cases of the missense (P534L, G579R, and R651P) and splice-site (splice donor site exon 16) mutations to test the possibility that the changes seen were polymorphic. In each case no further individuals with base changes were observed, suggesting these are true mutations. For further verification segregation of the mutation with the disease was tested in some of the families by either SSCP or restriction site change (Fig. 3; Table 3). No polymorphisms were detected in the coding sequence of PEX, although a polymorphic CA repeat (DXS1683) was present in what is potentially the 3′ UTR. In addition, the following intronic polymorphisms were identified: A C/T polymorphism was detected 10 nucleotides upstream of the beginning of PEX exon 18; 37 alleles were found to have a C and 33 a T. Similarly, 23 nucleotides before the start of exon 10, one allele out of 70 tested (in individual 4108), was found to have a T instead of a C in this position. These polymorphisms were detected by SSCP and sequencing.
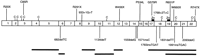
Mutations in the PEX gene. PEX exons are represented as boxes numbered 1–22, and the different mutations detected are drawn schematically above and below the gene. Stop mutations, missense mutations, and splice site mutations are represented above the gene, and frameshift mutations and deletions (thick black lines) below. Conserved cysteine residues (C) and zinc-binding motifs (Zn) are also represented.
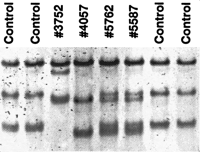
SSCP analysis of exon 17 after amplification of genomic DNA with intronic primers. The aberrant bands of patient 3752 (male) were found to have a G to C transition at nucleotide position 1735; patients 4057 (male), 5762(female), and 5587 (female) were found to have a G to A transition at nucleotide position 1735 giving rise to a missense mutation (G579R). The SSCP gels were run with 10% glycerine (see Methods).
The SSCP detection rate in the male sporadic and familial cases was 80%. Mutations or deletions have not yet been identified in 2 of 19 males with positive family history (individuals 3628 and 4295) and 1 out of 3 sporadic male cases (individual 3789). Cosegregation of the disease and PEX in these familial cases has not yet been tested. In addition, the 5′ and 3′ UTRs have not yet been examined. In females we were not able to detect a mutation in 2 out of 10 familial cases (individuals 3980 and 7784) and 5 out of 11 sporadic cases (2040, 3616, 4223, 7755, and 8343). The overall detection rate in females was 67%. This lower detection rate in females is likely to be attributed to difficulties in detecting large deletions by PCR, because the nondeleted allele will always produce an amplification product. Quantitative Southern blot analysis in these individuals may identify deletions undetected previously and is being investigated.
DISCUSSION
The PEX gene is of medical interest because it is mutated in HYP individuals and deleted in Hyp and Gy mice (Strom et al. 1997). The role that PEX plays in phosphate homeostasis and bone mineralization is not clear. Toward the further characterization of this gene, we have examined its genomic structure and performed an extensive mutation analysis in individuals with X-linked and sporadic hypophosphatemia.
Three different exon recognition programs were used to scan the genomic sequence, and all of the PEX exons were predicted by at least one program, including predictions of parts of the first and last exons. In this analysis, GeneID was least able to recognize exons in the human genomic sequence, as has been described by others (Solovyev et al. 1994). The GRAIL and Genefinder programs were fairly consistent in their ability to detect the PEX exons, and notably the exons that were missed by either program were small in size.PEX has large introns (on average ∼10 kb), and hence it might be expected that other genes lie within these. There were, however, no significant dbEST or other database matches corresponding to predicted exons in these regions, and hence further experiments are required to test their validity.
The genomic region spanned by PEX is substantially larger than that described for NEP, which was found to cover ∼80 kb (D’Adamio et al. 1989). Nevertheless, NEP has a very similar exon organization, including similarity in exon size and highly conserved patterns of codon breakage at intron–exon boundary positions. This is not unusual for members of multigene families because missplicing events and reading frameshifts are expected to be selected against (Brown et al. 1995). In two cases in the coding portions of PEX and NEP, the exon organization differs: PEX exon 5 is relatively large (227 bp) and the equivalent region in NEP is encompassed by two small exons with an intervening intron; conversely exons 16 and 17 of PEXare encompassed by one larger exon in NEP. Intron insertions within exons (or deletions between exons) of gene family members have been reported previously (Rogers 1985, 1990), although the mechanism and reason for this is unclear (Doolittle and Stoltzfus 1993; Stoltzfus et al. 1994).
Knowledge of the gene structure of PEX will facilitate dissection of the hereditary forms of hypophosphatemia. Thirty-three mutations and deletions of PEX have been detected in HYP individuals. This accounts for 25 (86%) of the 29 familial cases and 8 (57%) of the 14 sporadic cases. The SSCP detection rate in male individuals was 80%, which is in good accordance with other studies (Grompe 1993; Ravnik-Glavac et al. 1994). To date, only a small number of families with an autosomal dominant mode of inheritance (ADHR) have also been described. Some features of ADHR include delayed onset of penetrance, and some affected individuals have been shown to regain their ability to reabsorb phosphate (Econs and McEnery 1997). These penetrance issues add to the difficulty in establishing a diagnosis. In light of this difficulty it is likely that ADHR is more common than previously thought and mutations in the gene responsible may explain some apparently sporadic cases that do not have PEX mutations.
Four different types of missense mutation were observed in the present work: C85R, P543L, G579R, and R651P. The cysteine residue at position 85 in PEX is highly conserved within all members of the neutral endopeptidase family (Xu et al. 1994; HYP Consortium 1995). A missense mutation involving such a residue could disrupt correct protein folding by loss of an intramolecular disulphide bond. The proline to leucine exchange (P534L) is likely to affect the local hydrophobicity of the PEX gene product, because leucine is an aliphatic residue. This substituted proline is adjacent to a conserved asparagine residue that in NEP has been shown by site-directed mutagenesis to be involved in substrate binding (Asn542;Dion et al. 1995). The arginine to proline exchange (R651P) is likely to affect the charge of PEX, because arginine is positively charged and proline is not. This latter mutation is situated in a highly conserved region two residues beyond the ENXADXGG motif, which has been shown in NEP to be involved in zinc binding and catalysis (Le Moual et al. 1991). Similarly, the glycine to arginine change (G579R) is situated one residue before the HEXXH motif. The deletions identified in the mouse models for HYP are also likely to have a dramatic effect on the function of the Pex gene (Strom et al. 1997). Gy mice have been shown to have a deletion at the 5′ end encompassing the first three exons, and Hypmice have a deletion at the 3′ end encompassing the last seven exons. In summary, a wide spectrum of different mutations distributed throughout the gene and likely to cause loss of function or disruption of the levels of PEX or Pex have been identified.
PEX seems to be of low abundance in most tissues as shown by the difficulties in obtaining Northern blot results (HYP Consortium 1995), in identifying cDNA clones (Strom et al. 1997), and by the lack of PEX-related ESTs in the databases. Northern blot analysis, however, has shown higher levels of mRNA expression in primary bone cultures derived from mouse calvaria (Du et al. 1996). In addition, one rat EST of PEX was identified, derived from an incisor cDNA library. Therefore it seems that PEX/Pex may be more highly expressed in bone and teeth. Previous experiments performed byEcarot-Charrier et al. (1988) showed an intrinsic defect inHyp osteoblasts by transplantation of these mutant cells into normal mice, and independent studies performed by Shields et al. (1990)suggested a specific defect in the development of secondary dentin in odontoblasts of HYP patients. These experiments suggest thatPEX plays an important role in these tissues. Because the substrate of PEX is not yet known, it is not possible to say how loss of function of PEX causes these bone and teeth defects and a renal phosphate leak. It has been shown previously that decreased phosphate reabsoption in the kidneys of Hyp mice is attributable to decreased levels of the renal type II sodium phosphate cotransporter (Npt2; Tenenhouse et al. 1994). The genomic structure of human NPT2 has been described recently (Hartmann et al. 1996), which will aid studies aimed at questioning the effect ofPEX and its substrate in the regulation of transcription ofNPT2.
The 5′ UTR of PEX is unusual because it does not contain a translation start site conforming to the Kozak consensus sequence (Kozak 1987). Particularly at the −3 position, a purine residue is found in 97% of vertebrate mRNAs, whereas in PEX a C nucleotide has been identified. In addition, the sequence upstream from an ATG start methionine is usually devoid of T residues, whereas inPEX these occur at positions −2, −4, and −5. It has been proposed that genes with unfavorable initiation sites encode potent regulatory proteins, which are therefore highly regulated at the translation level (Kozak 1991). Alternatively in some genes two different start methionines can be used to generate different protein products from the same gene (Kastner et al. 1990). This mechanism is thought to provide additional flexibility in the control of gene expression and may happen in a tissue-specific manner (Kozak et al. 1991). It will be necessary to examine the PEX gene product in different tissues by use of specific antibodies to find out whether this is the case. The GRAIL program predicts a promoter region upstream of PEX exon 2, which is interesting because, if used, this would presumably produce a secreted protein without the amino terminus and potential transmembrane domain. Nevertheless, this seems unlikely because 5′ RACE experiments with primers in this region have produced sequences that include PEX exon 1 from both lymphoblast and bone RNAs (HYP Consortium 1995; and Du et al. 1996).
Examination of the mutations found in unrelated families shows that in the majority of cases hypophosphatemia can be correlated with a mutation in PEX. These mutations are likely to cause a loss of function, but HYP is inherited in a dominant fashion, and it is believed there are no differences in severity of the disease between males and females (Whyte et al. 1996). To explain this phenomenon we speculated previously that PEX is subject to random X inactivation (HYP Consortium 1995). In this case normal males and females would be expected to have equal levels of the gene product, affected males none, and affected females a reduced amount that is below a threshhold required for normal activity. Such a sensitivity to amounts of PEX could be fitting for a gene that may be highly regulated at the translation level. Obviously further experiments are required to assess the levels of PEX and its substrate in target tissues and to fully elucidate the role of PEX in phosphate homeostasis and the pathophysiology of HYP.
METHODS
Cosmid Contig
A cosmid contig was completed in the PEX gene region (Fig. 1) by screening two different X chromosome-specific cosmid libraries. Cosmids 104A0717, 104C0161, 104C05100, 104D1056, 104D0142, 104H0865, and 104A0563 were obtained from the ICRF cosmid library (Nizetic et al. 1991), and cosmids LLXU24M23 and LLXU62D02 were obtained from the Lawrence Livermore (LLOXNCC01) library (Fig. 1). An additional five cosmids distal to cosmid 104A0717 extend this contig (data not shown), although these were not sequenced in this project.
Sequencing Strategy
The nine overlapping cosmids were sequenced using the shotgun sequencing approach: Libraries were constructed by ligation of sonicated or sheared cosmid DNA to blunt-ended linearized plasmid DNA (pGATC or pUC18). Sequencing templates were prepared by PCR amplification of random shotgun clone inserts, and were sequenced from one side only using ABI dye primer chemistry. Approximately 600–800 reads were processed and assembled, and gap closure strategies were then performed, including sequencing from the reverse side of a shotgun clone, direct primer walking on cosmid DNA, and sequencing of PCR products generated across the gaps. Overlapping regions of the cosmids were sequenced only once; however, the sequence was finished to a high degree of accuracy according to international standards [Human Genome Organization (HUGO), unpubl.]. Specifically, all regions were double-stranded or sequenced with an alternative chemistry; an attempt was made to resolve all sequence-related problems, such as compressions and repeats; and all regions were covered by sequence from more than one subclone. The contiguous sequence was verified by comparison to restriction maps of the individual cosmids or comparison to restriction fragment digest patterns.
The 243 kb of genomic sequence is derived from the following cosmids: 1–41742 (ICRFc104A0717), 41743–48745 (LLXU24M23), 48746–86185 (ICRFc104C0161), 86186–109335 (ICRFc104C05100), 109336–149529 (ICRFc104D1056), 149530–170606 (ICRFc104D0142), 170607–178656 (LLXU62D02), 178657–218362 (ICRFc104H0865), and 218363–242825 (ICRFc104A0635). This sequencing project led to the detection of four nucleotide errors that were present in our original published sequence of the human PEX cDNA (GenBank accession no. U60475), leading to three amino acid differences: codon 363 was GAC (Asp) and should be GCC (Ala); codon 403 was AGG (Arg) and should be TGG (Trp); and codon 641 (at the end of the previous sequence) was GCG (Ala) and should be GGA (Gly).
Assembly and Analysis of Sequence
Sequencing reads were processed and assembled using a variety of programs in the Staden package (Dear and Staden 1991; Bonfield et al. 1995). The processing included masking of sequencing vector and cosmid vector sequences, assessment of quality, clipping of unreliable sequence, and a prescreen for repeat elements. Sequences were assembled using xgap, and edited using xgap and gap4 editors. Escherichia coli contaminating sequences were identified using FASTA and BLAST programs from the GCG package to screen the prokaryotic database of EMBL.
The exon prediction programs used were GRAIL v. 1.3c (Uberbacher and Mural 1991); the Genefinder programs (Solovyev et al. 1994) FEXH, HEXON, and FGENEH; and GENEID (Guigo et al. 1992). Repeat elements were detected and masked prior to database searches by use of REPEAT MASKER (A. Schmidt, unpubl.) and a primate-specific-repeat family database (Repbase; Jurka 1994). Low-complexity regions were masked by screening a database of simple repeats (Simple.db) by use of BLASTN and parsing the output through XBLAST (Claverie and States 1993). Database searches were performed with BLASTN and BLASTX programs (Altschul et al. 1990) to screen the nonredundant nucleotide dbEST and protein databases, respectively. To perform an exhaustive search, the sequence was divided into smaller pieces using Seqsplit and the search results recombined using Blastunsplit (Sonnhammer and Durbin 1994). The output of the BLAST programs was filtered using MSPcrunch (Sonnhammer and Durbin 1994). The sequence data and results of the analysis programs were imported into ACeDB v4.1 [Richard Durbin, Medical Research Council (MRC), Cambridge, UK; Jean Thierry-Mieg, Centre National de la Recherche Scientifique (CNRS), Montpellier, France].
Mutation Screening
DNAs of 29 familial and 14 sporadic cases with hypophosphatemia were collected from different pediatric endocrine and nephrologic units. Most patients are of German origin with the following exceptions: individuals 8817, 3941, and 3628 are Belgian, Balkan, and Italian, respectively. Individuals 7754, 7755, and 7756 are Swiss in origin.
The 22 exons of PEX were amplified with intronic primers (Table 2) and screened by SSCP. PCR was performed with 100 ng of genomic DNA, 0.6 μm of primers, 160 μmdNTP, 0.1 unit of Taq polymerase, 50 mm KCl, 10 mm Tris-HCl (pH 8.3), 1.5 mm MgCl2, and 0.01% (wt/vol) gelatine in a total volume of 50 μl. Cycling profiles included an initial denaturation step (94°C for 5 min) followed by 30 cycles with denaturation for 30 sec at 94°C, annealing at the exon specific temperature for 30 sec, extension at 72°C for 40 sec, and a final extension step of 72°C for 5 min. PCR product length varied from 190 to 370 bp. Amplified fragments from all exons were analyzed by SSCP using Hydrolink (AT Biochem) gels at 20°C with and without glycerol. Staining was performed with Cyber Green and detection performed with a FluorImager (Molecular Dynamics).
Variant bands were reamplified and used for direct sequencing with both the sense and antisense primer using cycle sequencing on Applied Biosystems 377 PRISM automated sequencers.
Acknowledgments
We are indebted to the clinicians and patients who were involved in this study. We thank Michaela Seeger, Kerstin Schmidt, and Roman Pawlik for sequencing and oligonucleotide synthesis, and Yoshihiko Yamada for the rat cDNA clone. This work was supported by grants from the Commission of the European Communities, the Deutsches Forschungsgemeinschaft, and the Friedrich-Baur-Stiftung. F.F. was supported by the Peter und Traudl Engelhorn Stiftung.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵8 Corresponding author.
-
E-MAIL francis{at}mpimg-berlin-dahlem.mpg.de; FAX +49 30 8413 1380.
-
- Received January 14, 1997.
- Accepted April 1, 1997.
- Cold Spring Harbor Laboratory Press








