
An STS-Based Radiation Hybrid Map of the Human Genome
- Elizabeth A. Stewart,
- Kathleen B. McKusick,
- Amita Aggarwal,
- Ewa Bajorek,
- Shannon Brady,
- Angela Chu,
- Nicole Fang,
- David Hadley,
- Mark Harris,
- Sami Hussain1,
- Richard Lee,
- Annu Maratukulam,
- Kyle O’Connor2,
- Shanti Perkins,
- Mark Piercy,
- Fawn Qin,
- Tim Reif,
- Carla Sanders,
- Xiaohong She,
- Wei-Lin Sun,
- Poroshat Tabar,
- Susan Voyticky,
- Sid Cowles3,
- Jian-Bing Fan1,
- Chris Mader,
- John Quackenbush,
- Richard M. Myers, and
- David R. Cox4
Abstract
We have constructed a physical map of the human genome by using a panel of 83 whole genome radiation hybrids (the Stanford G3 panel) in conjunction with 10,478 sequence-tagged sites (STSs) derived from random genomic DNA sequences, previously mapped genetic markers, and expressed sequences. Of these STSs, 5049 are framework markers that fall into 1766 high-confidence bins. An additional 945 STSs are indistinguishable in their map location from one or more of the framework markers. These 5994 mapped STSs have an average spacing of 500 kb. An additional 4484 STSs are positioned with respect to the framework markers. Comparison of the orders of markers on this map with orders derived from independent meiotic and YAC STS-content maps indicates that the error rate in defining high-confidence bins is <5%. Analysis of 322 random cDNAs indicates that the map covers the vast majority of the human genome. This STS-based radiation hybrid map of the human genome brings us one step closer to the goal of a physical map containing 30,000 unique ordered landmarks with an average marker spacing of 100 kb.
A physical map of the human genome consisting of 30,000 unique, ordered landmarks at an average spacing of 100 kb is an important goal of the U.S. Human Genome Project. The construction of such a map has been greatly facilitated in recent years by the development of sequence-tagged sites (STSs) as genomic landmarks (Olson et al. 1989). Each STS is defined by a short segment of human DNA sequence and is assayed by means of PCR (Green and Olson 1990; Goold et al. 1993). Common sets of such sequence-based markers can be readily screened against any DNA source and therefore can be used to integrate maps constructed by different mapping methods.
A variety of mapping reagents has been used to determine the order of STSs in the human genome and to estimate the distance between these landmarks. Yeast artificial chromosome (YAC) libraries containing large human DNA inserts have been used to construct STS-based maps of individual human chromosomes as well as a map of the entire human genome (Chumakov et al. 1992, 1995; Doggett et al. 1995; Gemmill et al. 1995; Hudson et al. 1995). However, certain GC-rich regions of the human genome appear to be poorly represented in YAC libraries (Chumakov et al. 1992). In addition, no widely accepted procedures exist to assess the relative confidence in alternative marker orders for such YAC STS-content maps. Finally, high rates of chimerism and rearrangement in YAC clones make estimates of distance between the STSs and determination of order in such maps imprecise.
Radiation hybrid (RH) mapping is a somatic cell genetic approach that is well-suited for the construction of high-resolution, long-range contiguous maps of the human genome. In an early version of this approach, a Chinese hamster somatic cell hybrid containing hamster chromosomes and a single human chromosome of interest is lethally irradiated, breaking each chromosome into fragments whose size depends on the dose of irradiation. After irradiation, the human and hamster chromosomal fragments rapidly rejoin with one another, resulting in complex chromosomal rearrangements. Such an irradiated cell is rescued by fusion with a nonirradiated hamster cell under conditions where only somatic cell hybrids between the irradiated and nonirradiated cells can form viable colonies. In such “radiation hybrids”, human chromosomal fragments are often inserted into the middle of hamster chromosomes, resulting in stable mitotic segregation for most human fragments in the viable colonies. The observation that random fragments of human chromosomes can be retained in RHs in the absence of selection for human chromosomal material provides the basis for RH mapping (Benham et al. 1989; Cox et al. 1990). DNA isolated from a panel of 80–100 such independent RHs provides a mapping reagent for ordering STSs and for determining the distances between them in the human genome. In this approach, the frequency of irradiation-induced breakage between two markers is used as a measure of distance, and marker order is determined in a manner analogous to meiotic linkage mapping (Cox et al. 1990). As in the case of meiotic linkage mapping, the relative confidence in alternative marker orders in RH maps can be assessed by using standard maximum likelihood statistical methods (Boehnke et al. 1991; Lunetta and Boehnke 1994). However, unlike meiotic linkage mapping, which requires polymorphic markers for map construction, RH mapping can integrate polymorphic and nonpolymorphic markers, such as STSs derived from expressed sequences (ESTs; Adams et al. 1991). An additional advantage of RH mapping is that panels for constructing maps at very different levels of resolution can be generated by experimentally manipulating the dose of irradiation. A number of STS-based RH maps of individual human chromosomes have been constructed (Cox et al. 1990; James et al. 1994; Raeymaekers et al. 1995; Shaw et al. 1995).
Despite the utility of chromosome-specific RH panels, the construction of a high-resolution map of the human genome would be greatly facilitated by the availability of a “whole-genome” panel of RHs, formed by fusing a diploid irradiated human cell with a hamster cell, as originally envisioned by Goss and Harris (1975). We and others have demonstrated that unselected human chromosomal fragments representative of the genome are stably retained in diploid whole-genome RHs and that such hybrids can be used to construct maps (Walter et al. 1994; D.R. Cox, K. O’Connor, S. Hebert, M. Harris, R. Lee, B. Stewart, G. DiSibio, M. Boehnke, K. Lange, R. Goold, and R.M. Myers, unpubl.). In addition, we have analyzed issues of experimental design and error detection related to the use of whole-genome RHs and have developed computational methods for the analysis of data derived from such diploid hybrids (Lange et al. 1995; Lunetta et al. 1995, 1996). At present, three different hamster–human whole genome RH panels have been constructed and are available to the scientific community (Research Genetics, Huntsville, AL). All three panels were formed by fusing irradiated human diploid male cells with Chinese hamster cells. However, the dose of irradiation used to construct each panel was very different, resulting in complementary reagents that can be used to construct maps over a wide range of resolution. The Genebridge 4 panel, developed in the laboratory of Dr. Peter Goodfellow, was generated with 3000 rads of X rays and consists of 93 hybrids (Gyapay et al. 1996). This panel has been used by several groups to construct STS-based RH maps of the human genome (Hudson et al. 1995; Gyapay et al. 1996). The relatively low dose of irradiation used to generate the Genebridge 4 panel allows the construction of continuous maps when these hybrids are scored with as few as 400 STSs spanning the genome. However, this immediate advantage limits the ultimate resolution of the maps that can be generated with these reagents. The G3 radiation hybrid panel, generated at the Stanford Human Genome Center with 10,000 rads of irradiation, allows for the construction of radiation hybrid maps of a higher level of resolution and is described in detail in this report. Recently, both the Genebridge 4 and the G3 panels have been used to integrate human genes with markers on the human meiotic map (Schuler et al. 1996). The third panel, the TNG RH panel, was generated at the Stanford Human Genome Center with 50,000 rads of irradiation and can be used to order markers at 50-kb resolution (Lunetta et al. 1996).
Here, we report the use of the G3 RH panel to construct an STS-based RH map of the human genome containing more than 10,000 loci covering the majority of the human genome.
RESULTS
STS Development
Our strategy for developing STSs involved the analysis of DNA sequence to eliminate vector sequences, repetitive DNA sequences, and duplicate sequences that occurred previously in the data set. Oligonucleotides for PCR amplification were chosen with the PRIMER program (M.J. Daly, S. Lincoln, and E.S. Lander, pers. comm.), under conditions that allow a single set of PCR conditions to be used for the majority of the STSs and that generate PCR products 100–300 bp in length. These products were assayed by ethidium bromide staining following agarose gel electrophoresis. An STS was judged successful when the primers produced a distinct PCR product of the expected size from total human DNA and failed to produce a product of this size from either hamster or mouse genomic DNA. Each STS that met these criteria was then screened against National Institute of General Medical Sciences (NIGMS) polychromosomal hybrid panel 1 and/or NIGMS hybrid mapping panel 2 (Drwinga et al. 1993; DuBois and Naylor 1993). Approximately 80% of STSs could be assigned to a single human chromosome. Such STSs, considered “unique markers,” were used for subsequent RH mapping experiments. Approximately 50% of all sequences for which primers were designed resulted in a unique STS.
The 10,478 STSs that met these stringent criteria and were placed on the RH map fell into three different categories (Table1):
Sources of STSs for the RH Maps
1. Genomic DNA sequences. We generated 2550 STSs from human genomic DNA clones sequenced at the Stanford Human Genome Center. An additional 513 STSs derived from genomic DNA sequence were developed in other laboratories, including 259 STSs from the Whitehead–MIT Genome Center and 254 STSs from a variety of sources.
2. Genetic markers. A total of 3541 polymorphic markers were used. Of these, 2702 were developed at Genethon (Dib et al. 1996), 713 were developed at the Cooperative Human Linkage Center (CHLC; Murray et al. 1994), and 126 were developed at the University of Utah (Litt et al. 1995).
3. Expressed sequences. A total of 3874 STSs from expressed sequences were used. Of these, 2859 were developed at the Stanford Human Genome Center from ESTs in dbEST (2415 STSs), from the Unigene collection in GenBank (212 STSs), and from a variety of other sources (232 STSs). An additional 1015 STSs derived from expressed sequences were developed in other laboratories, including 592 STSs from the Whitehead Institute for Biomedical Research/MIT Center for Genome Research, 159 STSs from The Institute for Genomic Research, 137 STSs from Genethon, and 127 STSs from Dr. K. Matsubara (Osaka University, Japan).
Data Generation and Characterization of the G3 RH Panel
A diploid male human lymphoblastoid cell line was irradiated with 10,000 rads and fused with thymidine kinase-deficient A3 hamster cells. Eighty-three independent RH clones were isolated following selection in HAT medium. Each RH cell line was expanded by growth in roller bottles, and a single large preparation of genomic DNA was isolated from each hybrid. The 83 RH DNA preparations, along with genomic DNA from the donor human lymphoblastoid cells and the recipient A3 hamster cells, constitute the Stanford G3 Radiation Hybrid Panel. This panel of DNAs is commercially available from Research Genetics (Huntsville, AL). All of the mapping described in the paper was performed with these DNA preparations.
The presence or absence of a specific STS in each radiation hybrid was determined by performing PCR amplification in a 96-well format, followed by agarose gel electrophoresis and ethidium staining. Each 96-well microtiter plate contained two positive control wells of human genomic DNA, two negative control wells of hamster DNA, one well for each of the 83 RH DNA samples, and nine wells that served as water controls. The PCR products from each microtiter plate were analyzed on a single four-tiered agarose gel. Because all reactions were carried out in duplicate, the assay of a single STS required 192 PCR assays and the electrophoresis of two agarose gels. Gel images produced by UV transillumination were captured with a CCD camera. Gel images were scored for the presence of PCR products of the expected size in a semiautomated fashion on a UNIX workstation by using a modified version of DNA/GUI (Drury et al. 1992). A hybrid was scored for an STS if the duplicate wells both gave positive or negative results. However, if the duplicate wells were discordant, the data point was not used. The mean discordancy rate for each STS was slightly less than 1%; STSs with a discordancy rate greater than 8% were discarded. A group of five individuals at the Stanford Human Genome Center typically assayed between 150 and 200 STSs in duplicate per week.
The average retention frequency of STSs mapped by using the G3 panel was 16%, with 87% of the STSs demonstrating a retention frequency between 10% and 30%. As expected, the haploid X chromosome had a retention frequency approximately half that of the autosomes. However, an unexpected significantly increased retention was observed for markers on the haploid Y chromosome and remains unexplained. With the exception of the sex chromosomes, the average marker retention frequency was quite similar from chromosome to chromosome, with no significant differences noted (Table 2). Variation in marker retention frequency along the length of each chromosome was minimal except in the immediate vicinity of the centromere, where the retention frequency increased dramatically on many chromosomes, as noted previously (James et al. 1994; Gyapay et al. 1996) and illustrated in Figure 1. In addition, the retention frequency in the immediate vicinity of the thymidine kinase gene on chromosome 17 approached 100%, which was expected because this human enzyme marker was used to select for the hybrid cell lines.
Summary of RH Map Data

Retention frequencies of markers in RHs. Depicted is the distribution of the retention frequencies of the STSs on the comprehensive RH map for human chromosome 11. Each STS is represented as a bar of constant, arbitrary length. The frequencies range from 0.06 to 0.42. Average retention frequencies for all of the chromosomes are included in Table2.
Map Construction
We wrote a computer program (SAMAPPER) that allows for the semiautomated construction of RH maps containing hundreds of markers by using data derived from diploid whole-genome RHs (K. McKusick and D.R. Cox, unpubl.). This program uses standard maximum likelihood methods (Boehnke et al. 1991; Lange et al. 1995), estimating the overall likelihood of a particular marker order by summing the pairwise likelihood of adjacent markers. Unlike other mapping strategies that define a set of well-ordered framework markers and place all other markers relative to these framework loci, our mapping approach initially determines the most likely order of all markers on a chromosome and then considers groups of 20–35 linked markers simultaneously to determine the most likely marker order within each linkage group. Although we did not use a fixed set of framework markers to build the map, an important aspect of our mapping strategy was the use of 1381 STSs, previously ordered on the Genethon meiotic linkage map, to help establish long-range continuity and to determine the relative orders of linkage groups. We used simulated annealing to search extensively the likelihood space and identify the order with the highest likelihood among all those examined. We defined this as the “comprehensive order” or high confidence order. We then considered the set of all orders examined with likelihoods not less than one-thousandth that of the comprehensive order and used this set of orders to identify groups of markers that do not overlap in the entire set of orders. We defined such nonoverlapping marker groups as 1000:1 bins, or high-confidence bins.
At the time of map construction, we had scored 8196 STSs on the G3 RH panel. Of these, 1181 had a scoring pattern on the RHs identical to one or more STSs and were defined as duplicates. Only the set of 7015 STSs with unique RH scoring patterns was used in map construction. Maps were constructed one chromosome at a time. Initially, linkage groups were formed such that all markers in the group had a lod score of 6 or greater with at least one other marker in the linkage group. All such lod 6 linkage groups for a particular chromosome that contained at least one of the Genethon STSs described above were used for map construction. This represented 5545 of the 7015 STSs. The linkage groups for each chromosome were then combined into a single group per chromosome, and all of the markers for a single chromosome were ordered by using a simulated annealing algorithm. In general, this procedure regenerated the original linkage groups and, at the same time, established long-range continuity of the map. Occasionally, prior knowledge of the meiotic position of Genethon markers on the linkage map was used to identify and correct “false joins” between linkage groups. Upon establishing the long-range order of all markers on a chromosome, the comprehensive order of a set of 30 markers in the middle of the chromosome was determined as described above, and 1000:1 bins for that set of markers were defined. This procedure was repeated for adjacent overlapping sets of 20–35 markers until a comprehensive order and 1000:1 bin lines had been defined for the entire chromosome. Finally, the overall length of a chromosomal map was calculated following removal of each marker from the map one at a time. In those cases where removal of a marker reduced the map length by >20 cR10,000 [(cR) centiRay], the marker was defined as an expander and was permanently removed from the map. Following removal of an expander, the comprehensive order of markers and the 1000:1 bin lines for the chromosome were determined anew, and the map was again tested for expanders. No more than four such cycles per chromosome were required to remove all expanders. Of the 5545 markers used to construct the maps, 496 STSs (∼9%) were removed as expanders, with 392 of these 496 expander STSs removed in the first cycle. This process resulted in a comprehensive RH map of the human genome consisting of 5049 ordered STSs, defining 1766 1000:1 bins (Fig. 2; Table2). In addition, 945 of the duplicate STSs, defined above, had an RH scoring pattern indistinguishable from one of the 5049 STSs on the comprehensive map, giving a total of 5994 mapped STSs.

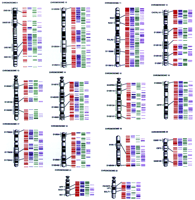
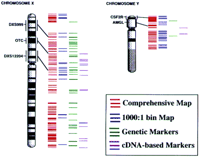
RH maps of the human genome. The four sets of colored lines to the right of the ideogram, from left to right, correspond to the markers on the comprehensive RH map (red lines), the high-confidence bin lines (blue lines), the genetic markers within the comprehensive map (green lines), and the expressed sequences within the comprehensive map (purple lines) for each chromosome. The lines are drawn according to their relative positions on the maps. A small fraction of the markers had been mapped previously by cytogenetic techniques. Some of these markers are indicated by their names and by a black bar that links the RH comprehensive map with the chromosome ideogram. Although the RH maps are depicted as extending to the telomeres, no specific telomeric markers were included in this study.
Those STSs that were not able to be incorporated into the map by using our algorithm, including markers that were not part of a lod 6 linkage group and expanders, were placed in the interval on the map giving the highest likelihood. Markers placed on the map in this fashion are said to be positioned rather than mapped. Since constructing the 5994 marker map, we have scored >2000 additional STSs on the G3 RH panel. Each of these markers, the majority of which were derived from cDNAs, was also positioned on the map. In total, our RH map of the human genome consists of 10,478 STSs, 5994 that have been mapped and 4484 that are positioned with respect to these mapped markers (Table 2).
Description of the Map
The RH map has a total length of 103,303 cR10,000, with an average spacing between adjacent markers of 17 cR10,000, corresponding to a physical distance of 500 kb (Table 2). Because 945 of the 5994 mapped STSs are assigned to positions identical to one or more STSs and do not contribute to map resolution, the average spacing between adjacent markers is slightly less than the effective resolution of the map. Of the 5970 intervals defined by adjacent markers on the map, 112 have an interval size of 90 cR10,000 or greater. In an effort to determine the expected distribution of distances between adjacent markers on the map, assuming a random distribution of both markers and RH breakpoints, we used a method described previously (Cox et al. 1994b). In Figure3, this expected distribution of interval distances is compared with the observed distribution of distances between adjacent markers on the map. Whereas the observed number of very small intervals is decreased and the observed number of very large intervals is increased over expectation, in general, the observed distribution of interval sizes is similar to the expectation for randomly spaced markers and RH breakpoints.
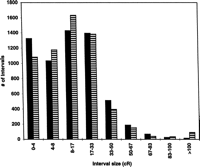
Assessment of the randomness of marker distribution and irradiation breakage. The observed distribution of the distances between neighboring markers on the RH map is shown as striped bars. This is compared with the expected distribution of interval sizes, as shown in black bars, calculated assuming a random distribution of both markers and RH breakpoints as described previously (Cox et al. 1994).
The accuracy of the map was assessed by comparing the order of 578 Genethon markers on both the radiation map and the Genethon meiotic linkage map. It is important to note that no prior mapping information regarding this subset of Genethon markers was used to assist in the establishment of marker order during the process of RH map construction. Of the 578 markers, 46 (8%) violated a 1000:1 bin line on the RH map and a 1000:1 order on the meiotic linkage map, indicating a 4% error rate with respect to the definition of 1000:1 bins. Comparison of the comprehensive order of the 578 loci on both the RH and the linkage maps showed that 185 of 578 (32%) loci are discrepant. Most discrepancies were quite minor, representing inversions of adjacent loci. Comparison of the order of 140 additional STSs scored on both the RH map and a YAC STS-content map of the short arm of human chromosome 4 resulted in similar findings (data not shown). Further studies are required to determine which of the observed discrepancies in marker order represent errors in the RH map, as opposed to errors in the genetic and/or YAC maps.
In an effort to assess the fraction of the human genome represented by the 5994 marker RH map, we scored 322 randomly selected STSs derived from expressed sequences on the G3 hybrid panel and determined their linkage with respect to the 5994 markers. Of the 322 STSs, 239 (74%) were linked with one or more of the mapped markers with a two-point lod score of ⩾6.0, 49 (15%) were linked with one or more markers with a two-point lod score between 5.99 and 3.0, and 34 markers (11%) were linked with a two-point lod score of <3.0. All loci with lod scores of 6.0 or greater were assigned to the “correct” chromosome, as determined by using the NIGMS hybrid mapping panels. Loci with the highest two-point lod score between 5.99 and 3.0 were not always assigned to a chromosome consistent with the NIGMS hybrid panel results. However, when the correct chromosomal assignment of such loci was assumed, each such STS could be localized to a single consistent region of the genome over 95% of the time even when the maximal two-point lod score with a mapped marker was as low as 3.0 (data not shown). Assuming that the 322 STSs represent a random sample of the human genome, these results suggest that the RH map covers the vast majority of the human genome and that with prior knowledge of chromosomal assignment, at least 89% of random STSs can be positioned with respect to the 5994 mapped markers on the RH map.
Complete RH map data, including STS sequences, chromosomal assignments, RH scoring patterns, marker retention frequencies, two-point lod scores, and distances for all 10,478 mapped and positioned STSs can be accessed via the Stanford Human Genome Center World Wide Web server (http://www-shgc.stanford.edu). If an STS of interest is not among the presently mapped or positioned STSs, it is a straightforward matter for an investigator to determine the G3 RH scoring pattern of that STS, given the public availability of the G3 panel of RH DNAs. Such a scoring pattern can then be used to position the STS on the G3 RH map in the vast majority of cases, as described above. We have developed an “RHServer” that facilitates this process for the scientific community, reporting the most likely position of an STS given its scoring pattern on the G3 panel. The RHServer can be accessed via the Stanford Human Genome Center World Wide Web site.
DISCUSSION
The STS-based RH map of the human genome reported here contributes to the goal of a physical map containing 30,000 unique ordered landmarks with an average marker spacing of 100-kb. Direct comparison of our map with other whole-genome maps, including the recently published transcript map of the human genome, is difficult given the limited number of common STSs shared between these maps and the fact that a variety of mapping reagents have been used to construct the various maps. With the exception of ∼3500 markers ordered previously on the human meiotic linkage map that can be used to integrate our map with the RH–YAC map generated by the Whitehead–MIT Genome Center, these two maps of the human genome have <1000 STSs in common (Hudson et al. 1995). Similarly, of the 16,000 STSs that comprise the transcript map of the human genome (Schuler et al. 1996), only 2875 STSs are shared in common with the RH map reported here. In addition, because the transcript map was constructed by integrating RH data from a number of different laboratories, placing markers into intervals defined by ∼1000 markers ordered on the human gentic linkage map, the 3-Mb resolution of the transcipt map does not reflect the resolving power of either the G3 panel or the Genebridge 4 panel of RHs.
Combining the map reported here with the Whitehead RH–YAC map and the transcript map results in ∼31,000 distinct STSs. Although this number of STSs is sufficient to define a physical map of the human genome at 100 kb average spacing, the challenge remains to order definitively these existing STSs at this level of resolution.
The Stanford G3 RH mapping panel described here consists of 83 hybrids with an average retention frequency of 16% and with a relationship between physical distance and irradiation-induced breakage of 29 kb per cR10,000. Thus, the Stanford G3 panel is analogous to a library of human chromosomal fragments of 3-Mb average size constituting a 12.5-fold redundant representation of the human genome. Viewed in this way, it is clear that the theoretical limit of mapping resolution possible with this panel is 3 Mb/12.5, or 240-kb resolution. Maps constructed using the G3 hybrids under conditions where the number of radiation-induced breaks is rate limiting confirm these theoretical estimates of maximal map resolution (Matise et al. 1996). A random distribution of 12,500 STSs across the human genome would result in an average spacing of 240 kb between adjacent markers. Thus, by continuing to score STSs on the G3 panel followed by reconstruction of the map as described in this paper, we can expect to improve the resolution of the present G3 RH map.
In comparison to the Stanford G3 panel, the Genebridge 4 panel consists of 93 hybrids with an average retention frequency of ∼30% and with a relationship between physical distance and irradiation-induced breakage of ∼300 kb per cR3000 (Gyapay et al. 1996). Thus, the theoretical limit of mapping resolution with this panel is 30 Mb/27.9, or slightly greater than 1-Mb resolution, representing 3000 STSs distributed randomly across the genome. Maps constructed using the Genebridge 4 panel under conditions where the number of radiation-induced breaks is rate limiting confirm these theoretical estimates of maximal map resolution (Matise et al. 1996). Typing >6000–9000 markers on the Genbridge 4 panel will not improve the resolution of the map significantly. At present, >13,000 unique expressed sequences have been scored on the Genebridge 4 panel (Schuler et al. 1996).
Given the above considerations, combined with the fact that the present G3 RH map contains sufficient markers to span the vast majority of the genome, the G3 panel, rather than the Genebridge 4 panel, appears to be the mapping reagent of choice for future radiation mapping studies involving expressed sequences and/or genomic DNA sequences.
Despite the fact that the G3 panel has the potential to provide maps of higher resolution than the Genebridge 4 panel, it is clear that even the G3 mapping panel does not have sufficient power to order markers spaced at 100 kb with high confidence. Other mapping reagents are needed to reach this mapping goal. One such reagent is the TNG panel of whole-genome RHs generated at the Stanford Human Genome Center. This reagent contains sufficient radiation-induced chromosomal breaks to generate high-confidence 100-kb maps (Lunetta et al. 1996). Further studies with these TNG hybrids, along with the development of statistical methods to integrate RH data sets generated with different mapping panels, should result in the high-resolution, high-confidence maps required to complete the sequence of the human genome in a timely and cost-effective manner.
METHODS
STS Development
STSs were developed from a variety of sources as described in Results. A standard set of PCR conditions was used for both the initial test on human and hamster DNA and the chromosomal assignment test on the NIGMS Somatic Cell Hybrid Mapping panels for almost all of the STSs. Reactions were in 10 μl and contained 25 ng of genomic DNA, 2.33 μl water, 1.0 μl 10× buffer (200 mm Tris-HCl at pH 8.3, 500 mm KCl, 25 mm MgCl2), 0.2 mm of each dNTP (Pharmacia), 1.0 μl of a solution containing 10 μl of each oligonucleotide primer, and 0.35 unit ofTaq DNA polymerase (Perkin Elmer Roche). PCR amplification was performed in either 9600 Perkin Elmer Thermocyclers or MJ Research DNA Engine Tetrad cyclers and used an initial denaturing step at 94°C for 90 sec, followed by 30 cycles at 94°C for 15 sec, 62°C for 23 sec, and 72°C for 30 sec. A final extension step of 72°C for 210 sec was performed and samples were maintained at 4°C until agarose gel electrophoresis.
Automated sample pipetting was used for most of the STS development and RH mapping assays performed in this study. Genomic DNA samples were aliquoted into PCR plates by using a Hydra 96 Microdispenser (Robbins Scientific Corp.). All other components of the PCR assays were dispensed by using an eight-channel MicroLab2200 (Hamilton Co.).
After PCR amplification, 5 μl of loading dye (10% Ficoll-400, 0.1m EDTA at pH 8.0, 0.025% bromophenol blue) was added to each sample, and 12 μl of the mixture was electrophoresed on 3% SeaPlaque agarose gels in 1× TBE and examined by ethidium bromide staining.MspI-digested pBR322 was used as a size standard. Bands were visualized by UV transillumination, and images were captured with a 640 × 480-pixel CCD camera (Alpha Innotech, Corp.). Captured images were transferred to a UNIX workstation, and thermal paper hard copies were stored in notebooks.
Further details concerning assay conditions for the STSs developed for this study can be obtained from the Stanford Human Genome Center (http://www-shgc.stanford.edu) or from dbSTS at GenBank (http://www.ncbi.nlm.nih.gov).
Generation of the G3 Panel of RHs
The G3 panel of RHs was constructed from a diploid male human lymphoblastoid cell line, RM, and a thymidine kinase-deficient Chinese hamster cell line, A3 (a daughter cell line of A23; Westerveld et al. 1971). Briefly, the RM lymphoblastoid cell line was irradiated with 10,000 rads of X rays and fused to the A3 cell line in the presence of 50% (wt/vol) polyethelene glycol. The fused cells were grown under HAT selection for 10–14 days. Two or three well-separated hybrids were picked from each plate and cultured for DNA isolation.
RH Assays
RH assays were performed for each STS as follows. Reactions were carried out in duplicate in two 96-well PCR plates with genomic DNA from two positive control samples (RM), two negative control samples (A3), and 83 RHs (Stanford G3 panel). The remaining nine wells in each plate contained water instead of genomic DNA. Reaction conditions and gel electrophoresis for the majority of the STSs were as described above for STS development. Each set of 96 samples was electrophoresed on a single 10 × 15-cm agarose gel containing four tiers of 26 wells each.
After capturing gel images as described above, data were analyzed on UNIX workstations running a modified version of DNA/GUI (developed by K. Clark and H. Donis-Keller, Washington University School of Medicine, St. Louis, MO). Each gel from the duplicate pair was scored independently in a semi-automated manner for the presence or absence of PCR products of the expected size. A hybrid was scored as positive for an STS only if a band of the expected size was present on each of the duplicate gels. Discrepancies between the two gels for an STS were designated as “not scored” in subsequent analysis. Data from duplicate gel pairs with more than seven discrepancies were discarded.
Acknowledgments
We thank Drs. Michael Boehnke, Kenneth Lange, and Kathryn Lunetta for valuable discussions during the course of this work, Drs. Mark Boguski and Greg Schuler for providing Unigene DNA sequences, Drs. Kenichi Matsubara, Craig Venter, Mihael Polymeropolous, James Sikela, Tom Hudson, Eric Lander, Ray White, and Charles Auffray for providing a portion of the STSs that were used in this study, and Dr. Kevin Clark and Dr. Helen Donis-Keller for generously providing DNA/GUI. We acknowledge the support of Sandoz Pharmaceuticals for providing a portion of the oligonucleotides used in this study. This work was supported by a center grant from the National Center for Human Genome Research (GESTEC 1P50 HG 00206 to R.M.M. and D.R.C.) and SERCA award National Institutes of Health grant HG00007 (to J.Q.)
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
Present address: 1Affymetrix, Inc., Santa Clara, California 95051; 2Millenium Pharmaceuticals, Inc., Cambridge, Massachusetts 02139-4815; 3Incyte Pharmaceuticals, Inc., Palo Alto, California 94394.
-
↵4 Corresponding author.
-
E-MAIL cox{at}shgc.stanford.edu; FAX (415) 725-8058.
-
- Received November 13, 1996.
- Accepted March 7, 1997.
- Cold Spring Harbor Laboratory Press








