
Cloning, Characterization, and Copy Number of the Murine Survival Motor Neuron Gene: Homolog of the Spinal Muscular Atrophy-Determining Gene
- Christine J. DiDonato1,
- Xiao-Ning Chen2,
- David Noya2,
- Julie R. Korenberg2,
- Joseph H. Nadeau3, and
- Louise R. Simard1,4
- 1Services de Génétique Médicale et de Neurologie, Centre de Recherche, Hôpital Ste-Justine, Québec, Canada H3T 1C5; 2Medical Genetics, The Cedars-Sinai Medical Center Burns & Allen Research Institute, University of California at Los Angeles School of Medicine, Los Angeles, California 90048; 3Department of Genetics, Case Western Reserve University School of Medicine, Cleveland, Ohio 33106-4955
Abstract
Because of a 500-kb inverted duplication, there are two copies of the survival motor neuron (SMN) gene in humans, cenSMN and telSMN. Both genes produce identical ubiquitously expressed transcripts; however, only mutations in telSMN are responsible for spinal muscular atrophy (SMA), the second most common autosomal recessive childhood disease. We have cloned the murine homolog Smn and mapped the gene to Chromosome 13 within the conserved syntenic region of human chromosome 5q13. We show that the Smn transcript (1.4 kb) is expressed as early as embryonic day 7. In contrast to humans, we found no evidence of alternative splicing. The predicted amino acid sequence between mouse and human SMN is 82% identical, and a putative nuclear localization signal is conserved. FISH data indicate that the duplication of the SMA region observed in humans is not present in the mouse. We also found no evidence of multiple Smn genes using Southern blot hybridization and single-strand conformation analysis. Using these methods, we detected at least four copies of Naipexon 5 clustering distal to Smn. Finally, three biallelic markers were identified within the Smn coding region; two are silent polymorphisms, whereas the third changes a cysteine residue to a tyrosine residue in exon 7. Overall, our results indicate thatSmn is single copy within the mouse genome, which should facilitate gene disruption experiments to create an animal model of SMA.
[The murine Smn cDNA sequence has been submitted to GenBank under accession no. U77714. The Mouse Genome Database accession no. for FISH mapping of Smn andNaip is MGD-INEX-31 and for the genetic mapping of theSmn exon 2b microsatellite marker is MGD-CREX-705.]
Proximal spinal muscular atrophy (SMA) is a common autosomal recessive neuropathy. After cystic fibrosis, SMA is the second most frequent monogenic disease of childhood affecting 1 in 10,000 children (Pearn 1980). The most significant clinical finding is proximal, symmetrical limb and trunk muscle weakness, which results from the loss of α-motor neuron cells in the spinal cord. Because SMA is clinically heterogeneous, the disease has been classified into three groups: type I, type II, and type III, based on age at onset and disease severity (Munsat and Davies 1992). Type I SMA is the most severe form and children generally die before 2 years of age.
The gene responsible for SMA is located within a complex genomic region on chromosome 5q11.2–13.3 (Francis et al. 1993, 1995; Thompson et al. 1993; Brahe et al. 1994; Burghes et al. 1994; DiDonato et al. 1994;McLean et al. 1994; Melki et al. 1994; Theodosiou et al. 1994; Daniels et al. 1995; Lefebvre et al. 1995; Roy et al. 1995a,b; Selig et al. 1995; Wang et al. 1995) that contains a 500-kb inverted duplication (Lefebvre et al. 1995). Within this inverted duplication lies the SMA-determining gene, survival motor neuron (SMN). Consequently, SMN is present in two copies, designated cenSMN or telSMN, and mutations affecting telSMN are responsible for SMA (Lefebvre et al. 1995). Also located within the SMA region is the neuronal apoptosis inhibitory protein (NAIP) gene (Roy et al. 1995b), which by itself is not a SMA-determining gene. Associated with the intact NAIP gene are several NAIP psuedogenes (ϑNAIP) that arose independent of the inverted duplication. Exons 5 and 6 of NAIP are unique to the region and used to distinguish the intact copy of NAIP from ϑNAIP copies. These ϑNAIP genes are clustered and not interdigitated with the SMN copy genes. cenSMN and telSMN are separated by ∼1 Mb of DNA and are virtually identical. They span ∼20 kb and consist of nine exons (1, 2a, 2b–8) (Bürglen et al. 1996). There are four differences within the transcribed sequences of cenSMN and telSMN (Lefebvre et al. 1995; Brahe et al. 1996; Hahnen and Wirth 1996). The first DNA variant is at the third position of codon 280 in exon 7 (TTT = cenSMN; TTC = telSMN) and is a silent polymorphism (Lefebvre et al. 1995); the second variant is a g → a substitution of nucleotide 1155 in the cDNA (exon 8) (Lefebvre et al. 1995); and the third and fourth variants are silent polymorphisms (AgC → AgT, codon 28; CAg → CAA, codon 154) in exons 2a and 3, respectively (Brahe et al. 1996; Hahnen and Wirth 1996).
Several groups showed that the absence of at least telSMN exon 7 occurs in the majority of SMA patients (87%–100%) (Chang et al. 1995;Hahnen et al. 1995; Lefebvre et al. 1995; Rodrigues et al. 1995; van der Steege et al. 1995a; Velasco et al. 1996; DiDonato et al. 1997a;Simard et al. 1997). Additionally, the identification of rare point mutations or small deletions in telSMN provides convincing evidence that SMN is the SMA-determining gene (Bussaglia et al. 1995; Lefebvre et al. 1995; Brahe et al. 1996; Parsons et al. 1996).
The SMN copy genes hybridize to a 1.7-kb transcript that is expressed ubiquitously (Lefebvre et al. 1995; van der Steege et al. 1995b). An additional 4.5-kb transcript cross-hybridizing with the SMN cDNA has been reported (van der Steege et al. 1995b). Analysis of alternatively spliced transcripts predict that different SMN isoforms are produced by cenSMN and telSMN (Gennarelli et al. 1995; Lefebvre et al. 1995). Full-length transcripts and transcripts lacking only exon 7 or exons 5 and 7 are products of the cenSMN gene, whereas telSMN produces predominantly full-length transcripts but also those lacking exon 5 (Gennarelli et al. 1995).
Recently, Liu and Dreyfuss (1996) demonstrated that the 40-kD SMN protein is conserved in vertebrates and interacts with the RGG box region of heterogenous nuclear ribonucleoprotein (hnRNP) U, fibrillarin, itself, and several other novel proteins. Immunolocalization studies in HeLa cells using monoclonal antibodies raised against SMN indicated that SMN is part of a novel nuclear structure termed gems, for Gemini of coiled bodies. These observations raise the possibility that SMN may have a function in RNA metabolism.
To decipher the function of the telSMN protein and its role in SMA etiology, we are using the mouse as a model system. In this paper we describe the first step toward this goal, the cloning and preliminary characterization of Smn, the murine homolog of the SMA-determining gene. We also present the results from a series of experiments showing that Smn is present as a single copy within the mouse genome. This will allow us to characterize theSmn protein more easily and determine its function in normal and disease states. In addition, one can begin to study the differences between cenSMN and telSMN and determine why only mutations in telSMN cause SMA.
RESULTS
Cloning and Sequence Analysis of the Smn cDNA
Smn cDNA clones were isolated from mouse pre-B cell and 7.5-day whole embryo λgt10 libraries. Additional clones were identified in the expressed sequence tag (EST) database. Six different cDNA clones were subjected to further analysis. All were partial or full-length cDNAs that were similar to human SMN (Lefebvre et al. 1995). The longest clone contained a 1250-bp insert comprising the complete coding and 3′-untranslated region (UTR) (Fig.1). Sequence analysis of exon 7 revealed that all clones contained a cytosine at position 825, which is the same nucleotide identified at the corresponding position (nucleotide 840) of the human telSMN copy gene (Lefebvre et al. 1995). The overall nucleotide identity between mouse Smn and human SMN was 83% across the open reading frame. No sequence conservation was observed in exon 8, which contains the 3′ UTR.
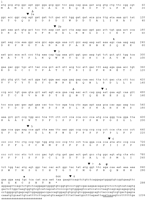
Nucleotide and deduced amino acid sequence of the Smn cDNA. The deduced amino acid sequence beginning with the initiating methionine is given. Numbering of nucleotides and amino acids is shown at the right. (▾) Positions of introns; a potential polyadenylation signal (ATAAA) in the 3′ UTR is underlined; (▵) the position of the DNA variants in exons 2a, 3, and 7 of human SMN. The polymorphic variants identified in SPRET/Ei DNA in exons 2b and 7 are indicated above the Smn sequence. Several other variants were also identified in exon 8 but are not shown because of multiple-base changes. The entire Smn cDNA sequence has been confirmed by comparison with the sequence of genomic DNA from the 129/SvJ strain. The GenBank accession no. is U77714.
The Smn open reading frame encodes a protein of 288 amino acids with a calculated molecular mass of 31.3 kD. Murine Smnis slightly smaller than its human counterpart, as three amino acids in exon 1, two in exon 4, and one in exon 7 are absent (Fig.2A). The predicted mouse protein is 82% identical to human SMN. There are regions that are highly conserved such as the putative 4 amino acid nuclear localization signal (NLS) in exon 2b. Also conserved are the amino acids encoded by exon 2a and the polyproline stretches in exons 4, 5, and 6, whereas other regions have diverged significantly. Figure 2B summarizes the estimated nucleotide and amino acid identities between murine and human SMN for each exon. BLASTp searches of the publicly available databases using full-lengthSmn protein identified its human homolog, aCaenorhabditis elegans protein (P = 3.9 × 10−15), and a hypothetical protein in Schizosaccharomyces pombe(P = 0.011). The 197 amino acid C. elegansprotein (Z81048) is located in chromosome III and has no known function. It shares high identity with regions of the Smnprotein located in exons 2a, 3, and 6. The putative protein was actually identified using GENE FINDER from sequence obtained through the C. elegans genome sequencing project. To date, searches with this protein sequence in the C. elegans EST database have not yielded any positive clones. Additionally, a transgenic strain using the cosmid that contains this putative protein has not yet been produced. Smn also identified a hypothetical 17.4-kD protein in S. pombe (Z54354; P = 0.011). Normally, this would not be considered a very significant result; however, the same regions of Smn exons 2a and 6 were identified. Using the C. elegans protein in a BLASTp search, the 17.4-kD protein was identified (P = 5.9 × 10−5) and the area of similarity between the two proteins corresponding to Smn exons 2a and 6 was extended.

(A) Amino acid comparison of mouse Smn and human SMN. Vertical lines indicate amino acid identity; dots represent gaps that have been introduced to maximize identity. A few possible post-translational modification sites that are conserved between the two species are shown: (Broken underline) Asn glycosylation; (single underline) myristalation; (double underline) cAMP phosphorylation; (▵) PKC phosphorylation; and (*) NLS sequence. (B) Comparison of percent nucleotide and amino acid identity of murine and human SMN.
Characterization of the Smn Transcript
A Northern blot containing poly(A)+ mRNA from fetal and adult mouse tissues was used to determine the size, as well as the temporal and spatial distribution, of Smn transcripts. ASmn cDNA probe that contained exons 2b–8 hybridized to a full-length transcript of ∼1.4 kb in all tissues examined and a 4.4-kb transcript in liver (Fig. 3). Hybridization to fetal tissues indicated that the gene was expressed in the 7-day embryo, the earliest stage analyzed.
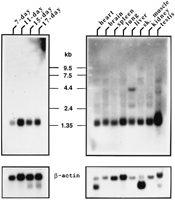
Northern blot hybridization of fetal and adult poly(A)+ mRNA with a partial Smn cDNA. Smn transcripts (∼1.4 kb) are expressed in all tissues and developmental stages analyzed. A 4.4-kb transcript is also present in liver. After hybridization withSmn, Northern blots were stripped and reprobed with β-actin to determine the relative amount of poly(A)+ mRNA in each lane.
To determine whether the full-length transcript is alternatively spliced, Smn cDNA from brain, kidney, liver, and spinal cord was used in RT–PCR (see Methods). In all tissues examined, only full-length Smn PCR products were identified (data not shown) even after transfer and Southern blot hybridization with SmncDNA as probe.
Single-Strand Conformation Analysis
In humans, single-strand conformation analysis (SSCA) of exons 7 and 8 was used to distinguish cenSMN from telSMN (Lefebvre et al. 1995). To identify sequence polymorphisms that might be indicative of gene duplication in the mouse, we performed SSCA for each exon of the Smn gene. Table 1 lists the primer sets, annealing temperatures, magnesium concentrations, and amplicon sizes for each exon. Two different electrophoretic conditions were used to analyze PCR products from 129/SvJ genomic DNA and 129/SvJ genomic subclones (data not shown). The migration pattern of 129/SvJ DNAs for each of the exons analyzed was identical; however, interstrain polymorphisms were observed in exons 2b, 7, and 8 in SPRET/Ei versus 129/SvJ and C57BL/6JEi DNA. Three of these biallelic polymorphisms are within the Smn coding region (exons 2b and 7), and all three strains of mice were homozygous for each SSCA variant (Fig. 1). Figure4 presents the results obtained for exon 2b, which describes two polymorphisms in SPRET/Ei versus 129/SvJ and C57BL/6JEi. The first is a C/T silent polymorphism at nucleotide 231 of the cDNA that was confirmed by direct sequencing as well as sequence from subcloned SPRET/Ei DNA. The second is an (A)19 repeat in SPRET/Ei compared to a tetranucleotide (ATTT)6 repeat in 129/SvJ and C57BL/6JEi. The second polymorphism was used to map the Smn gene.
Novel STSs Used for SSCA and STS Content Mapping

Polymorphism in Smn exon 2b identified by SSCA. An interstrain polymorphism between SPRET/Ei and 129/SvJ or C57BL/6JEi was identified. Sequence analysis identified a tetranucleotide repeat (AAAT)6in 129/SvJ and C57BL/6JEi compared to an adenine repeat (A)19in SPRET/Ei. Additionally, a silent polymorphism in SPRET/Ei DNA was also identified. This is a C/T transversion, and both triplets code for the same amino acid (serine).
Interspecific Backcross Mapping of Smn
The Jackson Laboratory BSS mapping panel [(C57BL/6JEi × SPRET/Ei)F1 × SPRET/Ei)], which has now been typed for >2060 loci (Rowe et al. 1994), was used to determine the chromosomal location of the Smn gene. The (ATTT)6/(A)19 repeat polymorphism in exon 2b was used to genotype 94 backcross DNAs. As shown in Figure 5, the segregation of alleles indicated that Smn is located on Chromosome 13 and maps between D13Bir19 andEfta. Smn cosegregates with 20 other loci (data not shown) on the BSS mapping panel, includingD13Lsd1, which marks the location of the Naip gene (DiDonato et al. 1997b), and D13Mit37. The order of a subset of these loci and their genetic distance in centimorgans is provided in Figure 5A.
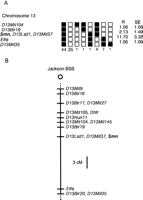
Mapping of Smn to Chromosome 13. (A) Haplotypes from the Jackson Laboratory BSS backcross panel showing part of Chromosome 13 with loci linked to Smn. Loci are listed in order with the most proximal at the top. (▪) The C57BL/6JEi allele; (□) the SPRET/Ei allele. The number of animals with each haplotype is given at the bottom of each column of boxes. The estimated percent recombination (R) between adjacent loci is given at right, with the standard error (SE) of the estimate. Missing typing for markersD13Bir17, D13Mit105, Dhfr,D13Mit145, and D13Bir19 was inferred from the genotypes of the flanking loci where assignment was unambiguous. The Mouse Genome Database (MGD) accession number for genetic mapping ofSmn is MGD-CREX-705. (B) Map showing part of Chromosome 13 from The Jackson BSS backcross panel. The map is depicted with the centromere toward the top. A 3-cM scale bar is shown at right. Loci mapping to the same position are listed in alphabetical order. These data can be obtained from The Jackson Laboratory on World Wide Web sitehttp://www.jax.org/resources/documents/cmdata.
Contiguous Arrays of Bacterial and Yeast Artificial Chromosomes
Genomic clones corresponding to the mouse SMA region were isolated by screening high-density filters of a mouse 129/SvJ BAC library (Genome Systems) with human SMN exons 6–8 and human NAIP exon 5. DNA pools from a 129/SvJ bacterial artificial chromosome (BAC) library (Research Genetics) were screened using a primer set that amplifiedSmn exon 2b. After it was determined that D13Mit146was present in several Smn BAC clones and that it was located proximal of Smn, BAC DNA pools were rescreened via PCR usingD13Mit146 to extend the contig proximally. Overall, these experiments identified six Naip, six Smn, and oneD13Mit146 positive BAC clones. Table 2 shows the sequence-tagged site (STS) content for the minimal number of BACs required to span the region from D13Mit146 toD13Mit37/Naip exon 5. All BACs were also analyzed by restriction digest and Southern blot hybridization using full lengthSmn cDNA and Naip exon 5 as probes. We found no evidence for multiple copies of Smn or gene rearrangements within the clones when compared to genomic DNA (data not shown). However, EcoRI restriction digests of genomic DNA and BAC clones containing Naip exon 5 revealed four distinct copies ofNaip exon 5, that is, 4 unique fragments in genomic DNA. The largest EcoRI fragment (∼10 kb) appeared to be present in two copies. All copies can be accounted for in the various BAC clones (data not shown). Confirmation of these results is provided in the recently published paper by Scharf et al. (1996).
STS Content of BACs and YACs Located BetweenD13Mit36 andD13Mit70
We also identified six yeast artificial chromosomes (YACs) from the MIT database that were positive for markers that cosegregrated withSmn and Naip on the BSS genetic map. This YAC contig, which is ∼1.1 cM in length, contains the region fromD13Mit36 to D13Mit70 and was analyzed for the presence or absence of STSs and simple-sequence length polymorphisms (SSLPs) via PCR (Table 2). The SSLP marker D13Mit146 was positive in YAC clones: 187D4, 380D1, and 334g7 and in BAC clone 321I22 but was negative in all other YACs and BACs tested. This indicated thatD13Mit146 maps distal to D13Mit36 and proximal toSmn and D13Lsd1/Naip. The Smn gene STSs were positive in YACs 187D4, 380D1, and 334g7 and BACs 20g19 and 227n6. The combined BAC and YAC data indicate that if Smn is duplicated, both copy genes would have to map distal to D13Mit146 and proximal of D13Lsd1 (∼180 kb interval). This result is completely inconsistent with its human homolog where cenSMN and telSMN are separated by ∼1 Mb (Lefebvre et al. 1995). Figure 6 presents a physical map of the region surrounding Smn based on STS content.
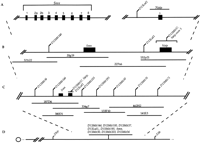
(A) A schematic diagram of the Smn gene and a portion of the Naip gene. The structure of the Smn andNaip genes was established by alignment to the homologous human genes. Note that Naip exon 5 and D13Mit37 are represented multiple times in this area but are shown only once for simplicity. (B) BAC contig of the region containing theSmn and Naip genes. Shown is the minimal number of BAC clones needed to complete a contiguous array spanning the markersD13Mit146–D13Mit37. (C) Contiguous array of YAC clones spanning the region from D13Mit36 to D13Mit70. Shown are only those YACs that were positive with two or more MIT markers mapping to this region. These YACs are negative for all other MIT markers, which suggests that they are not chimeric. The order of the markers from centromere to telomere was established by genetic and physical mapping. (D) Chromosome 13.
Fluorescence in Situ Hybridization Mapping
The 500-kb inverted duplication of the SMA region in humans gave rise to two copies of SMN separated by ∼1 Mb (Lefebvre et al. 1995). We have used fluorescence in situ hybridization (FISH) on mouse metaphase and interphase cells to determine whether this duplication also exists in the mouse and to confirm the localization ofSmn in Chromosome 13. A 100-kb Naip BAC (152P21) that contained one copy of Naip exon 5 and a 180-kb SmnBAC (20g19) were used as probes in FISH. The results indicated that these genes colocalized to mouse Chromosome 13 (Fig.7). From three independent experiments, >30 metaphase and 50 interphase cells were evaluated. In all metaphase cells, signals from Smn and Naip probes were seen clearly on the two chromatids in the region MMU13D1-2.1 in 90% of cells. Dual-color FISH experiments indicated that Smn andNaip signals were close together or overlapping with each other, in random order, in the majority of cells. Only a single copy per chromosome was detected in all of the metaphase and interphase cells analyzed. Thus, the 500-kb inverted duplication observed in humans is not present in mouse. Furthermore, from the interphase study, the Smn and Naip genes are separated by >50 kb.
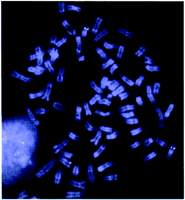
FISH mapping of BAC probes containing the Smn andNaip genes to mouse Chromosome 13. The BAC genetic markerD13Mit300 was used as a hybridization probe to identify mouse Chromosome 13 accurately (D. Noya, X.-N. Chen, B. Birren, K. Devon, J.S. Lee, and J.R. Korenberg, unpubl.). The map assignment was corroborated by chromomycin A3 and distamycin A reverse banding. The differentially labeled Smn (digoxigenin; red signal) andNaip (biotinylated; green signal) probes were cohybridized onto metaphase chromosomes and unstimulated interphase cells prepared from strain 129. The FITC and rhodamine signals were mapped to the region MMU13D1-2.1. The MGD accession number for physical mapping ofSmn and Naip is MGD-INEX-31. It should be noted that multiple copies of a region-specific repeat that contains at leastNaip exon 5 was not resolved by FISH because of the size of the BAC used as probe for the Naip gene.
DISCUSSION
In this paper we present the cloning and characterization of the murine homolog of the human survival motor neuron gene. TheSmn open reading frame (ORF) is 864 bp long and encodes a protein of 288 amino acids with a molecular mass of 31.3 kD. Sequence comparison across the ORF at the nucleotide level indicates that the gene is 83% identical to its human counterpart, but there is no conservation in the 3′UTR (exon 8).
Comparison of the amino acid sequence for murine and human SMN showed 82% identity, suggesting that both proteins have a similar tertiary structure. There is also a 4 amino acid nuclear localization signal (NLS) in exon 2b that is conserved but was not reported in the original cloning of human SMN (Lefebvre et al. 1995). This would explain the staining of gems within the nucleus reported by Liu and coworkers using monoclonal antibodies raised against SMN (Liu and Dreyfuss 1996). Further studies are necessary to determine whether this putative NLS is functional or whether a novel NLS is present within the Smnprotein that directs it to the nucleus. A polymorphism that results in an amino acid substitution (CYS → TYR) at codon 284 in exon 7 of SPRET/Ei mice was identified, which suggests that this cysteine is not crucial to the tertiary and quaternary structure of the protein. Thus, if a missense mutation is identified at this position in humans, one would predict it to be a tolerable substitution, but the phenotypic consequence will most likely depend on the state of the other telSMN gene. Additional comparison of the two predicted proteins indicated that exons 2a and 6 and the polyproline stretches may be functionally significant given their high degree of conservation. Protein alignment also revealed areas of divergence such as exon 4, indicating a lack of selective pressure on this portion of the protein. Finally, a BLASTp search of the publicly available databases using full-lengthSmn protein identified its human homolog, a C. elegans protein of unknown function in chromosome III and a 17.4-kD protein in S. pombe. These proteins, especially the C. elegans protein, share a high degree of identity to portions ofSmn exons 2a, 3, and 6, which indicates an evolutionarily conserved function for these regions. Several point mutations have been reported within telSMN, but none are within these conserved areas (Bussaglia et al. 1995; Lefebvre et al. 1995; Brahe et al. 1996;Parsons et al. 1996). It would be interesting to identify and determine the deleterious effects, if any, that missense mutations might have within these regions of conservation.
Analysis of Smn expression by Northern blot hybridization identified a ubiquitiously expressed 1.4-kb transcript and a 4.4-kb transcript specifically expressed in liver. We show that Smnis expressed prenatally as early as embryonic day 7 in the mouse.Smn differs from the human 1.7-kb transcript (Lefebvre et al. 1995; van der Steege et al. 1995b) by 300 bp, which can be accounted for by the smaller 3′ UTR in the mouse transcript. The 4.4-kb liver-specific transcript could be the same as the 4.5-kb transcript seen by van der Steege et al. (1995b). RT–PCR indicated thatSmn is not alternatively spliced in brain, kidney, liver, or spinal cord. This result contrasts that found in humans, where it has been shown that in muscle and lymphoblasts, cenSMN undergoes alternative splicing of exons 5 and 7 in various combinations (Gennarelli et al. 1995; Lefebvre et al. 1995). Although the major transcript of telSMN is full-length, mRNAs lacking exon 5 are also produced (Gennarelli et al. 1995). The absence of alternative splicing in the mouse suggests that only the full-length Smn product is important for normal α-motor neuron function. If this is true, then only the full-length SMN protein plays a role in SMA etiology, the variant isoforms having no effect. This hypothesis seems plausible given the fact that telSMN, the gene disrupted in all 5q SMA patients, predominantly produces the full-length SMN transcript. However, in humans, the possibility that cenSMN may modify disease severity cannot be ruled out completely, that is, cenSMN copy number and sequence conversion events must be considered (Velasco et al. 1996; Hahnen et al. 1996; van der Steege et al. 1996; DiDonato et al. 1997a). Ultimately, proof for or against this hypothesis will have to await the production of transgenic animals that contain cenSMN or telSMN. One would expect that crossing a telSMN transgenic mouse to a knockoutSmn mouse would rescue the phenotype while crosses with cenSMN transgenics would not, as the major transcript produced by cenSMN is an isoform lacking exon 7.
Four methods were used to analyze Smn copy number within the mouse: linkage analysis, SSCA, FISH, and physical mapping. The exon 2b (A)19/(ATTT)6 polymorphism was used to map theSmn gene by linkage analysis to Chromosome 13. There were no ambiguities in the segregation analysis of backcross DNAs. This is in agreement with Scharf et al. (1996), who used a polymorphism in intron 1 to map Smn on the same panel of backcross DNAs. The results of SSCA indicated that there were no detectable differences between amplified fragments from 129/SvJ genomic DNA and 129/SvJ genomic subclones that contained each of the exons. However, interstrain polymorphisms in exons 2b, 7, and 8 were detected, and in all instances, mice were homozygous for the intragenic interstrain polymorphisms. Furthermore, as there is very little sequence divergence between human cenSMN and telSMN across the coding and noncoding regions (Lefebvre et al. 1995; Bürglen et al. 1996; C. DiDonato and L. Simard, unpubl.) when compared to murine Smn, we postulate that the duplication that gave rise to two SMN genes arose after the divergence of lineages leading to humans and mice. This hypothesis is consistent with our FISH results, where we demonstrated that the large inverted duplication of the human SMA region is not present in mouse. Finally, we determined the physical structure of the Smnregion by creating a YAC and BAC contig that was analyzed by STS content and Southern blot hybridization. Overall, our physical map ofSmn was in agreement with our genetic localization. We found no evidence of gene duplication by Southern blot hybridization. Additionally, STS content mapping was able to resolve several loci that cosegregated genetically and allowed us to determine a relative order of markers from centromere to telomere [cen-D13Mit36–D13Mit146–Smn5′–Smn3′–D13Lsd1–(D13Mit37, Naip exon 5)–D13Mit203–D13Mit195–D13Mit30–D13Mit70–D13Mit71–tel]. Although we detected multiple copies of Naip exon 5 andD13Mit37, we have not ordered them within our contig; therefore, we have placed D13Mit37 and Naip exon 5within parentheses. Proof that multiple copies of Naip exon 5 and D13Mit37 exist is also provided by Scharf et al. (1996), who recently published a physical map of the Lgn1 critical region. It is difficult to compare our physical map with that of Scharf et al. (1996) as we only have two YAC clones in common. However, the maps are consistent with each other in that both position Smnto the same location and the same orientation and that multiple copies of Naip exist. Scharf and coworkers detected differentNaip exon 5 copies by SSCA, whereas we used EcoRI restriction digests and Southern blot hybridization with Naipexon 5 as probe. In both maps, all Naip copies cluster in a region distal to Smn. The clustering of all copies ofNaip is consistent with our FISH results, where a biotinylated (green) signal was confined to a single region on Chromosome 13.
In conclusion, the results reported here integrate the genetic and physical maps of Chromosome MMU13D1-2.1 and confirm that Smnand Naip are part of the large conserved linkage group with human chromosome 5. This report also provides information and resources that will be useful for positionally cloning other genes in this region including Lgn1 (Beckers et al. 1995; Dietrich et al. 1995;Scharf et al. 1996). Most importantly, we have presented our findings on the cloning and characterization of Smn, the murine homolog of the SMA-determining gene. The fact that Smn is single copy will allow us to characterize the Smn protein, decipher its normal function, and produce mice deficient for this gene more easily. Such experiments will provide critical information regarding the role of telSMN in the etiology of proximal SMA and why an almost identical copy gene, cenSMN, cannot compensate completely for the absence of telSMN protein.
METHODS
Isolation of Smn cDNA Clones
Approximately 1 million phage plaques of a λgt10 pre-B cell cDNA library (the kind gift of P. Gros, McGill University, Montréal, Québec, Canada) were plated, transferred to nylon membranes, and hybridized with a 32P-labeled random-primed (Feinberg and Vogelstein 1983) human SMN exon 6–8 (518 bp) probe using standard methods (Sambrook et al. 1989). A λgt10 7.5-day whole embryo cDNA library (the kind gift of B. Hogan, Vanderbilt University Medical Center, Nashville, Tennessee) was screened by PCR usingSmn exon 3 primers according to the method of Israel (1993). IMAGE Consortium (LLNL) cDNA clones 352779 and 419573 (Lennon et al. 1996) were identified by using the human SMN cDNA sequence as a query to search the National Center for Biotechnology Information (NCBI) EST database using the BLASTn program (Altschul et al. 1990). cDNA clones were sequenced on both strands using the double-stranded cycle sequencing system (GIBCO-BRL).
Isolation of Genomic BAC and YAC Clones
Using standard methods, human SMN exon 6–8 and humanNaip exon 5 probes were used to screen high-density 129/SvJ BAC filters (Genome Systems). DNA pools of a 129/SvJ BAC library (Research Genetics) were screened by PCR using primers specific toSmn exon 2b (see Table 1). YAC clones were identified by searching the MIT database for MIT markers spanning the genetic map established by this work. DNAs from BAC and YAC clones were isolated as recommended by Genome Systems.
STS Content Mapping
All STS content mapping was performed by PCR. PCR amplification conditions for MIT markers were according to Dietrich et al. (1992). Novel STSs described in Table 2 were amplified using the conditions described below. YAC and BAC clones were analyzed for the presence or absence of STSs and SSLPs using single-colony picks or DNA obtained from liquid cultures of single colonies.
SSCA
SSCA was performed for each exon of the Smn gene using the primers described in Table 1. PCR was performed in a total volume of 25 μl that contained 50 ng of genomic DNA or 2 ng of subcloned DNA, 20 mm Tris-HCl (pH 8.4), 50 mm KCl, 4 μm each primer, 300 mm dATP, dGTP, and dTTP, 150 mm dCTP, 44 nm [32P]dCTP, and 1 unit of Taq DNA polymerase (BRL). The final MgCl2concentration, annealing temperature, and product size for each exon is described in Table 1. Cycling conditions consisted of an initial 3-min denaturation step at 94°C, followed by 35 cycles of 94°C for 30 sec, annealing for 30 sec, and 72°C for 30 sec in a Perkin Elmer Cetus Thermocycler 1. Resultant PCR products were diluted fivefold with 0.1% SDS and 10 mm EDTA, mixed with an equal volume of loading buffer (95% formamide, 20 mm EDTA, 0.05% bromophenol blue, 0.05% zylene blue), heated to 80°C for 5 min, snap-cooled on ice, and 5 μl was loaded onto a 0.5× MDE (AT Biochem)/0.6× TBE gel. To increase the probability of detecting single-base-pair substitutions, two different electrophoretic conditions were used: room temperature or 4°C at 7 W for 14–17 hr, depending on the size of each exon.
SSCA variants between SPRET/Ei and 129/SvJ, as well as C57BL/6JEi, were identified. Products from 50-μl PCRs were purified using Wizard Magic PCR preps (Promega) and sequenced directly using the double-strand cycle sequencing kit (GIBCO-BRL). These products were also cloned into a plasmid vector using a TA cloning kit (Invitrogen). Four independent clones from each sample were sequenced and the sequences compared to that obtained directly from the PCR product.
Genetic Mapping of Smn
Smn was genetically mapped by PCR analysis of DNA from 94 progeny of the Jackson Laboratory BSS interspecific backcross mapping panel (Rowe et al. 1994). Reaction and amplification conditions for Smn exon 2b primers (Table 1) were identical to those described above. The amplified products were diluted 1:1 with formamide loading buffer, heated at 80°C for 3 min, and 8 μl of each reaction was loaded onto a 6% denaturing polyacrylamide gel and electrophoresed at 2000 V for 3 hr. Gels were dried and exposed to Fuji film at −80°C overnight with two intensifying screens.Smn exon 2b amplified a product of 206 bp, 206 bp, and 199 bp using 129/SvJ, C57BL/6JEi, and SPRET/Ei genomic DNA, respectively.
Northern Blots
Northern blots (Clontech) containing ∼2 μg of poly(A)+mRNA from mouse fetal and adult tissues were hybridized with a radioactive 1126-bp Smn cDNA (exons 2b–8) probe prepared by random priming (Feinberg and Vogelstein 1983). Hybridization was carried out in 7 ml of Rapid Hyb buffer (Clontech) at 65°C for 2 hr according to the manufacturer’s instructions. The filters were washed sequentially at 65°C for 10–20 min in 2× SSC, 0.1% SDS; 1× SSC, 0.1% SDS; 0.5× SSC, 0.1% SDS; 0.2× SSC, 0.1% SDS, and 0.1× SSC, 0.1% SDS, respectively, and monitored with a Geiger counter between buffer changes. The filters were exposed to Fuji film at −80°C with two intensifying screens for varying lengths of time. After exposure, the filters were stripped and rehybridized with β-actin, washed, and exposed for 20 min.
RT–PCR
Tissues from a CD1/Spf female mouse were disrupted by a Polytron homogenizer in Trizol reagent (GIBCO-BRL), and total RNA was isolated according to the manufacturer’s instructions. To analyze theSmn gene for alternatively spliced isoforms, 5 μg of total RNA from liver, kidney, brain, and spinal cord was reverse transcribed using an exon 8 Rev primer (see below) and 200 units of Superscript RT II (GIBCO-BRL) in a total volume of 25 μl according to the manufacturer’s instructions. For each amplification, 3 μl of first-strand cDNA was used. Smn exons 1–4 or 1–7 were amplified using 2 pmoles of forward and reverse primers in a total volume of 25 μl and 1 unit of Taq polymerase (GIBCO-BRL). Amplification conditions were as follows: an initial 3-min denaturation step at 94°C, followed by 35 cycles of 94°C for 30 sec; 56°C for 30 sec; and 72°C for 40 sec in a Perkin Elmer Cetus Thermocycler 1. The resulting PCR products were electrophoresed in a 2% agarose/1× TBE gel. Smn exon 1–7 RT–PCR products were transferred to a Hybond N+ nylon membrane (Amersham), hybridized with a labeled Smn exon 1–2b probe and washed at high stringency in 0.1× SSC, 0.1% SDS, at 65°C for 45 min. The blot was exposed to Kodak X-OMAT film overnight at −80°C with two intensifying screens. Only the 860-bp PCR product corresponding to full-length Smn was identified. PCR primers (5′ → 3′) are as follows: exon 8 (reverse), gCACATTTgTgCTCAgTCACg; exon 1 (forward), ATggCgATGggCAgTggC; exon 4 (forward), AATgAAAgTCAAgTTTCCACA; exon 7 (reverse), gTATgTgAgCACTTTCCTTC.
FISH Mapping of Smn and Naip BAC Clones
DNAs from mouse BACs containing either the Smn (20g19) or Naip (152P21) genes were labeled with digoxigenin-11–dUTP and biotin-14–dATP, respectively, using a nick translation system (GIBCO-BRL). FISH analysis was performed essentially as described byKorenberg and Chen (1996). Briefly, in 10 μl of the hybridization mixture (50% formamide, 10% dextran sulfate; and 2× SSC), 100 ng each of biotinylated and digoxigenin-labeled DNA probes were mixed with 3 μg of human Cot 1 DNA, 4 μg of mouse Cot 1 DNA (GIBCO-BRL), and 3 μg of sonicated salmon sperm DNA to suppress cross-hybridization with repetitive sequences. The DNA probes were applied to denatured mouse chromosome slides prepared from female mouse spleen cells (strain 129) using a modification of the method described by Boyle et al. (1990) and Zhu et al. (1995). An independent control experiment was done that involved hybridization of the probes to mouse lymphocytes, where the majority of cells were assumed to be in G1 as the cultures were not treated with concanavalin A. Three washes were performed in buffer containing 2× SSC and 50% formamide for 5 min at 40°C, followed by three additional washes in 1× SSC for 5 min at 45°C. Hybridization signals were detected with avidin-conjugated fluoroscein isothiocyanate (Vector Labs) and sheep anti-digoxigenin–rhodamine. Amplification of FITC signals was achieved by using biotinylated–anti-avidin. Chromomycin A3 and distamycin A were used to counterstain individual chromosomes. The images were captured and stored using the Photometrics Cooled-CCD camera (CH250) and BDS image analysis software (ONCOR Imaging, Inc.).
Acknowledgments
Special thanks go to Ken Morgan for helpful discussions and critical reading of the manuscript. We also thank Mary Barter and Lucy Rowe at The Jackson Laboratory for analysis of the interspecific backcross data. This work was supported by grants from the Medical Research Council (MRC) of Canada to L.R.S., The Department of Energy (DE-FG03-92ER-61402), and the Molecular Genetic Basis of Williams Syndrome (National Institutes of Health Grant 1-P01-HD-33113-01A) to J.R.K. L.R.S. is a Fonds de la Recherche en Santé du Québec Scholar, and J.R.K. holds the Geri and Richard Brawerman Chair in Molecular Genetics.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL simardlo{at}ere.umontreal.ca; FAX (514) 345-4766.
-
- Received January 23, 1997.
- Accepted February 19, 1997.
- Cold Spring Harbor Laboratory Press








