
Zooming in on the Human–Mouse Comparative Map: Genome Conservation Re-examined on a High-Resolution Scale
- Ethan A. Carver1,2,3 and
- Lisa Stubbs1,3,4
Abstract
Over the past decade, conservation of genetic linkage groups has been shown in mammals and used to great advantage, fueling significant exchanges of gene mapping and functional information especially between the genomes of humans and mice. As human physical maps increase in resolution from chromosome bands to nucleotide sequence, comparative alignments of mouse and human regions have revealed striking similarities and surprising differences between the genomes of these two best-mapped mammalian species. Whereas, at present, very few mouse and human regions have been compared on the physical level, existing studies provide intriguing insights to genome evolution, including the observation of recent duplications and deletions of genes that may play significant roles in defining some of the biological differences between the two species. Although high-resolution conserved marker-based maps are currently available only for human and mouse, a variety of new methods and resources are speeding the development of comparative maps of additional organisms. These advances mark the first step toward establishment of the human genome as a reference map for vertebrate species, providing evolutionary and functional annotation to human sequence and vast new resources for genetic analysis of a variety of commercially, medically, and ecologically important animal models.
Conservation of genomic organization in different mammals has been postulated since the early 1900s (Haldane 1927), and since that time conserved genetic linkage groups have been documented in a variety of vertebrate species (for summary, see Andersson et al. 1997; Jones et al. 1997). Although significant conservation of synteny (a frequently misused word that simply describes chromosomal linkage) is observed throughout the mammalian orders, no two genomes have been compared and contrasted as extensively as those of human and the laboratory mouse. Currently, ∼200 linkage groups are known to be conserved between the two species (DeBry and Seldin 1996; Eppig and Nadeau 1996), and although the pace of discovery has slowed considerably in recent years, new conserved linkage groups are still being reported (Dinulos et al. 1996; Stubbs et al. 1996; Watkins-Chow et al. 1996).
Because mouse chromosomes represent the most thoroughly studied of all nonhuman vertebrate genomes, mouse–human conserved synteny relationships have become the gold standard of mammalian comparative maps. It is now evident that mouse chromosomes do not provide the simplest models for human comparison: Rodent chromosomes have undergone an unusually high number of genomic rearrangements per unit of evolutionary time (Graves 1996). Nevertheless, the degree to which gene content and order has been conserved is considerable, and the availability of detailed comparative maps has sparked a remarkable interchange of genetic, genomic, and functional information between the mouse and human genomes over the past several years. Most notably, an increasing number of human disease genes are discovered through association with mouse mutations producing similar phenotypic effects (for review, see Bedell et al. 1997; Montague et al. 1997).
Certain human disease genes, and, for example, quantitative trait loci, might indeed be discovered most efficiently by use of information derived from mapped mouse mutations. The real advantages of comparative genomics, however, are provided to animal researchers who gain access to superior cDNA, genomic clone, DNA sequence, and other resources through surrogate use of the human map. In the best cases, the distinct sets of advantages provided by each species are combined to serve a common purpose. The power of human–mouse comparative mapping data is tapped most efficiently through consolidation of mouse genetics and human genome resources to create a single, unified system that aids gene discovery and functional analysis in both species. As genetic and genomic tools become more sophisticated, an increasing number of gene-discovery strategies based on the merger of human, mouse, and other mammalian maps are being reported (e.g., Grobet et al. 1997;Kambadur et al. 1997; Lembertas et al. 1997).
The successful use of such map-merging strategies requires that relationships between conserved genomic regions have been well-defined. Despite the wealth of mapping information available for both species, however, syntenic relationships remain poorly defined or even uncharted within a surprising number of mouse and human regions (Figs.1 and2). Alignment of human and mouse maps is complicated by several factors, including practical differences in the details of human and mouse map construction. For example, a surprisingly small number of orthologous gene pairs have been accurately mapped in both species (Eppig and Nadeau 1995). Whereas most human genes have been assigned through cytogenetic or other physical methods, the majority of mouse genes have been mapped by use of genetic techniques [Copeland et al. 1993; Mouse Genome Database (MGD) 1997, as reflected in the different formats of Figs. 1 and 2]. The fact that recombination breakpoints are not evenly distributed in most regions complicates alignment of these two types of data, especially cross-species. Furthermore, mapping data available for both species are often subject to large margins of error—cytogenetic assignments often span several chromosome bands, for example, whereas most existing mouse maps combine data derived from many sources in a useful, but highly error-prone best fit consensus.

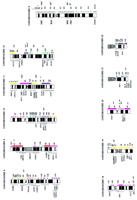
Human–mouse comparative map. A comparative map of the human genome and the homologous murine regions. An ideogram of the human karyotype is shown, with murine homology regions indicated to the right of each map. Solid homology regions, defined by numerous conserved markers that have been mapped with good accuracy in both species, are indicated by solid bars. Hatched bars denote regions to which fewer markers have been mapped and for which human and/or mouse map assignments have been established with less accuracy. Asterisks (*) indicate the approximate positions of single markers that have been mapped to both species—several asterisks observed adjacent to a human band indicate that a small number of markers have been mapped to that region, but those markers are known to map close together over a limited interval and/or are mapped at low levels of accuracy. Each murine homology region is color-coded to correspond to specific mouse chromosomes (see also Fig. 2). The cytogenetic positions determined for several human diseases, represented by locus symbols, are indicated to theleft of each human map. A selection of locus symbols corresponding to mouse mutations that are known to map within a particular homology region are included at far right of the map in italics. An asterisk denotes the human gene or mouse mutant has been cloned; underlining indicates that human diseases and murine model, aligned in parallel positions on the map, are known to be associated with orthologous genes. For reference information, see Fig.2.
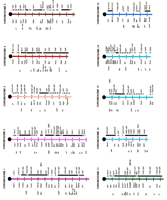
Mouse–human comparative map. A map of the mouse genome and corresponding human homology regions. A map of the mouse genome is illustrated, with human homology regions indicated to theright of each map. The length of the chromosomes are delineated by crosshatch bars, which are spaced at 10-cM intervals along the mouse map. Human homology segments are labeled with the cytogenetic bands to which they map, as shown in Fig. 1. Conserved markers are placed to the left of the mouse map to provide anchor points for each homologous segment. Symbols and markings are identical to those described in the legend of Fig. 1. The positions of cloned mouse mutations, at left, and human diseases, atright of each map, are indicated by underlining and/or asterisks, are shown both here and in Fig. 1 for completeness and to aid in cross-referencing the two maps. Positions of additional mouse mutations, many of which are not cloned or solidly anchored to the human map, are also listed. On-line resources that aided the construction of Fig. 1 and this figure included MGD: The Mouse Genome Database (http://www.informatics.jax.org/mgd.html), Davis Human/Mouse Homology Map (http://www.ncbi.nlm.nih.gov/Homology/), and OMIM—Online Mendelian Inheritance in Man (http://www.ncbi.nlm.nih.gov/Omim/). Information regarding phenotypes, cloned genes, and other mutations that are not represented here can be obtained directly from these sources.
Nevertheless, when human and mouse maps are compared on the whole genome scale, a number of strong and solid regions of conserved synteny are clearly observed (Figs. 1 and 2). As recent data have shown, continuous and simply aligned homologous segments are exceptions rather than the rule: Many or most syntenically homologous regions are interrupted by insertions, transpositions, deletions, inversions, and other types of rearrangements. Several large syntenically homologous mouse and human regions have now been compared in detail, and together these studies are beginning to provide an intriguing new snapshot of genome evolution developed on a high-resolution scale.
Specific Homology Groups: Some Detailed Examples
Aligning High-Resolution Mouse Genetic and Human Physical Maps
The existing human–mouse comparative maps are derived largely from alignment of mouse interspecific backcross (IB) maps with human genes localized by fluorescent in situ hybridization (FISH). Whereas high resolution IB maps permit gene order and spacing to be established with considerable accuracy, FISH mapping generally permits positions of related human genes to be defined only on the level of chromosome bands or, at best, subbands (e.g., Koizumi et al. 1995; Pilz et al. 1995; MGD 1997; the bulk of the data summarized in Figs. 1 and 2 are of this type). At this level of resolution, the most dramatic evolutionary rearrangements can be identified, but subtle internal rearrangments remain uncertain or undetected.
As an increasing number of comparative alignments are anchored to high resolution human physical maps, the fact that significant rearrangements have occurred within many or most syntenically homologous regions is increasingly apparent. Alignment of a genetic map of MmuX with the physical map of a 20-Mb segment of human Xp21-Xpcen (Willard et al. 1994), for example, revealed that a 4-Mb segment had been transposed to a position located 40 cM distal of other Xp21-related loci in mouse. Further analysis showed that sequences within a 9-Mb region, containing conserved genes Pfc andXk, are inverted in order in MmuX relative to human Xp (Figs.1 and 2; Blair et al. 1994, 1995). Another detailed study of this type has concerned the organization of a large interval of Mmu11 and related regions of H5q23–q35. Contrary to early expectations, the human 5q/Mmu11 homology region is not contiguous; two segments containing human homologs of Mmu11 genes are separated in H5q, interrupted by an interval of homology to Mmu13. The Mmu13-related region is also not contiguous, but is divided into two separate segments interrupted by insertion sequences related to Mmu18. A minimum of four microrearrangments would be required to account for the relative organizations of the ∼10-cM region of Mmu11 and related genes in H5q (Watkins-Chow et al. 1997).
Alignment of a cosmid-based physical map of H19q (Ashworth et al. 1995) with genetic maps of related mouse regions has also been reported recently (Stubbs et al. 1996). IB analysis of 42 conserved human 19q-linked genes defined the locations of three transposed chromosomal segments and one inversion in mouse relative to H19q. Two of the transposed segments have been moved to different positions within the same conserved linkage group in Mmu7, whereas one segment comprises an isolated region of syntenic homology between H19q and Mmu17 (Figs. 1and 2). More recent studies have shown an additional nested inversion within the larger inverted region (Kim et al. 1997). Interestingly, the ends of evolutionary rearrangements involve adjacent genomic segments that are clustered at just two sites. Aside from these five internal rearrangements, gene content, order, and spacing are extremely well conserved throughout the length of the H19q/Mmu7 homology region (Stubbs et al. 1996).
Aligning Parallel Sets of Mouse and Human Physical Maps
To date, physical maps have been generated within only a few homologous regions of human and mouse. Despite the small number of existing examples, evidence of several different types of evolutionary histories have been documented in comparative physical mapping studies completed to date. These different types of relationships are illustrated by examples summarized below.
LINKAGE GROUPS THAT ARE HIGHLY CONSERVED ON THE PHYSICAL LEVEL
The construction of long-range restriction maps spanning 13 and 11 Mb, respectively, of homologous regions of mouse and human chromosomes 1 (Oakey et al. 1992) has provided an unusually detailed look at gene organization within a large and stable region of conserved synteny. Gene content, order, and spacing were found to be nearly identical in human and mouse throughout the length of this region, and significant rearrangements were not detected. This extraordinary level of similarity, documented on the level of restriction fragment lengths, remains the best example of gene and linkage conservation documented to date. Physical maps of several shorter parallel mouse and human regions have also been reported, including 2-Mb regions of H1p/Mmu3 (Kingsmore et al. 1989), 2-Mb regions around the mouse and human XISTX-inactivation center regions (Cooper et al. 1993; Lafreniere et al. 1993), and a 500-kb region of H11p15.4/Mmu7 (Sellar et al. 1994; Stubbs et al. 1994). Physical maps of human chromosome Xq28 and a related 3-Mb segment of MmuX have also been compared (de Gouyon et al. 1996). In each of these reported cases, mouse and human regions were observed to be virtually identical in content, order, and physical spacing of homologous genes.
A CASE OF RAPID EVOLUTION: MOUSE AND HUMAN PSEUDOAUTOSOMAL REGIONS
Because most X-linked genes are subject to X inactivation, it has long been presumed that rearrangements that moved genes from the X chromosome to an autosome would be highly deleterious (Ohno 1967). Genes located in the most terminal portion of human Xp and the distal tip of MmuX, which correspond to the pseudoautosomal regions (PAR) of both species, are not inactivated. Because of the role the PAR plays in X–Y pairing, fertility, and other essential functions, however, syntenic conservation within this region logically would be predicted. Surprisingly, mouse and human PAR represent the most highly diverged homology segments examined to date (Reid et al. 1995; Kipling et al. 1996; for review, see Blaschke and Rappold, this issue). In contrast to other X-linked loci, mouse counterparts of human PAR genes are not all located on MmuX. Four human PAR genes, including CSF2RA, IL3RA, CLCN4, and SHOX (also called PHOG) have been mapped to autosomes, including Mmu19, Mmu14, Mmu7, and Mmu3, respectively (Fig. 1; Disteche et al. 1992; Palmer et al. 1995; Ellison et al. 1996; Rao et al. 1997; Blaschke and Rappold, this issue). The wide dispersal of these genes in mouse seems even more extraordinary given their close physical linkage in the human PAR: For example, humanCSF2RA and IL3RA are located within 100 Kb of each other (Reid et al. 1995). The Clcn4 gene is PAR-linked inMus spretus, a close cousin of the laboratory mouse, indicating that transposition of this gene to Mmu7 has occurred very recently (Palmer et al. 1995). The fact that murine orthologs ofSHOX, a homeobox gene associated with short stature in human XO (Turner’s syndrome) females (Rao et al. 1997), and other PAR genes are not X-linked explain at least some of the striking phenotypic differences observed in Turner’s syndrome patients compared with XO mice (Ashworth et al. 1991; Blaschke and Rappold, this issue).
THE MAJOR HISTOCOMPATIBILITY COMPLEX: THREE GROUPS OF TIGHTLY LINKED, RELATED GENES BEAR THE IMPRINT OF DIFFERENT EVOLUTIONARY PRESSURES
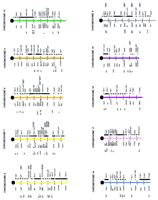
One of the earliest studied (Weiss et al. 1984; Hardy et al. 1986; Steinmetz et al. 1986), best mapped, and most gene-rich regions of mammalian genomes, the major histocompatibility complex (MHC) provides an excellent illustration of different types of syntenic relationships that are encountered when human and mouse regions are examined in detail. Physical maps aligning the proximally positioned mouse and human class II gene regions were first to be reported (Hansen and Trowsdale 1990). The 900-kb human class II region is approximately three times the length of the related murine interval, attributable to selective duplication of certain DP, DQ, and DR gene family members (Fig. 3A). An additional difference involves the insertion of a 60-kb interval containing mouse class I genes with no direct human counterparts, H2k and K2, in the proximal region of the MHC in mouse. These H2k and K2are thought to have arisen by duplication of more distally locatedQa genes and moved to the class II region via intrachromosomal recombination events (Weiss et al. 1984).

Comparative map of human and mouse MHC regions. Comparative alignment of the class II (A), class III (B), and class I (C) gene regions of the MHC regions of human chromosome 6 and mouse chromosome 17. Data are taken from published studies, as described in the text (Hansen and Trowsdale 1990; Amadou et al. 1995;Peelman et al. 1996). Conserved sequences are indicated by a solid box, whereas human genes that do not detect true orthologs in mouse, or vice versa—e.g., representing recent gene duplications or highly diverged but functionally homologous sequences—are indicated by open boxes. Orthologous gene pairs are connected by broken lines.
In contrast, the centrally located class III regions of humans and mice are very similar in structure (Fig. 3B). Physical maps of the porcine class III region have also been constructed and aligned with maps of human and mouse. Variable expansions/duplications of a region containing cytochrome P450 genes of the CYP21C group and duplications of a small number of class III genes were the only notable differences between the three species (Peelman et al. 1996). Maps of the most distally located MHC region, containing class I genes HLA-A, HLA-B, HLA-C, HLA-D, HLA-E, and HLA-F in human, and genes in mouse that are functionally related but cannot be recognized as orthologs on the basis of sequence or structure, has been presented recently (Amadou et al. 1995). Interestingly, the class I loci and neighboring genes of other types, including OTF3, TCTEX-5, MOG, and HSRI, which do recognize clear murine orthologs, occupy similar physical positions in the mouse (Fig. 3C).
T-CELL RECEPTOR GENE FAMILIES
Extensive physical maps have also been constructed within the T-cell receptor gene family clusters, TCRA/D, TCRB, and TCRG (for summary, see Hood et al. 1993). As observed for the MHC class II regions, the primary differences observed between mouse and human T-cell receptor gene clusters are gene duplications that have occurred relatively recently in evolutionary time. Whereas mouse and human α/δ complexes are very similar in structure and gene alignment, the human TCRB region is considerably larger than the related murine region (800 vs. 500 kb), as a result of repeated duplications of a few, specific Vβ segments in primates. In contrast, the mouseTcrg region is twice the length of the related human interval (400 vs. 200 kb in humans) due to gene duplications specifically seen in the rodent, but not primate lineage. Mouse and human TCRA/D clusters are very similar in structure; conservation on the level of DNA sequence has also been documented for portions of this region (Koop et al. 1992; see below).
HUMAN AND MOUSE REGIONS COMPARED AT THE LEVEL OF DNA SEQUENCE
The most detailed look at human–mouse genomic conservation is provided by the limited amount of comparative genomic sequencing documented to date. Sequence data obtained from a 94.6-kb region of the mouse T-cell receptor α/δ gene cluster region (Koop et al. 1992), aligned with that generated from the parallel human TCRA/TCRD region, remains the largest segment of human–mouse comparative DNA sequence published to the present time. Unlike other human and mouse regions that have been sequenced in parallel (Lamerdin et al. 1995, 1996; Oeltjen et al. 1997), noncoding portions of the T-cell receptor gene regions also displayed an overall high level of sequence conservation (66% average, vs. 66%–79% similarity for exons in this region). Nevertheless, a relatively high level of conservation of certain intronic and intergenic regions permitted the identification of conserved regulatory elements in the TCRA/TCRD region (Koop et al. 1994).
Another large region that has been sequenced in both species is a 93/89-kb interval surrounding the Bruton’s tyrosine kinase (BTK) gene in human and mouse, respectively (Oeltjen et al. 1997). These studies showed colinearity not only of five genes, but also conservation of the approximate sizes and overall organization of exons, introns, and intergenic regions in human and mouse. Highly conserved noncoding sequences were also observed near and between exons of most BTK region genes. Two conserved sequence elements located within the BTK transcription unit were demonstrated to function as tissue-specific silencers of BTK in T cells; sequence conservation has also been used by other authors to identify elements serving regulatory functions that are conserved in human and mouse (Januzzi et al. 1992; Renucci et al. 1992). In fact, because regulatory sequences are so often conserved in both position and sequence in human and mouse, comparative sequence alignments currently represent the most efficient means of identifying promoters, enhancers, silencers, and other regulatory elements.
Homology Region Borders
Have syntenic rearrangements occurred entirely at random, or are certain regions especially prone to chromosome translocations, inversions, and transpositions that have distinguished the genomes of humans and mice? A correlation between sequence content and genetic instability has been noted at several common sites of DNA rearrangement in the human population: A significant fraction lie within regions occupied by locally reiterated sequences (e.g., Stoppa-Lyonnet et al. 1990; Saleh et al. 1992; Rabbitts 1994; Dutly and Schnitzel 1996; van Deutekom et al. 1996). Exactly how repeated sequences might serve to facilitate rearrangement is uncertain, but strong evidence of illegitimate inter- and intrachromosomal recombination events has been obtained in several cases (e.g., Kulozik et al. 1992; Dutly and Schnitzel 1996; van Deutekom et al. 1996; Kehrer-Sawatski et al. 1997).
That repeated sequences might also be associated with evolutionary rearrangement events has been suggested by several studies. For example, the breakpoint of t (X;1) (p11; q21) translocations associated with papillary renal cell carcinoma (RCC) were mapped to a small region of Hq121 between SPTA1 and a clustered gene family, includingCD1C, CD1B, CD1A, CD1D, CACY, and at least four other members (Weterman et al. 1996). Interestingly, the boundary between two segments of H1q21 that are related to Mmu1 and Mmu3, respectively, is located between SPTA1 and CD1C, a region of <200-kb (Oakey et al. 1992). A syntenic breakpoint has also been mapped in the MHC class I gene region, within a tandemly organized family of genes including B30.2, MOG, BT, and RFP(Amadou et al. 1996). B30.2 and Mog remain linked to class I genes in mouse, but Btn and Rfp have been separated from family members to form a separate region of H6p homology on Mmu13 (Fig. 3C). The Mmu17 border region also contains the breakpoint of one of the four inversions that distinguish mouset-haplotypes (Hamvas et al. 1997). Neither the inversion breakpoint, nor the syntenic border, has been mapped with enough precision to claim correspondence, but the coincident location of these sites is provocative nonetheless.
In another study, comparisons between related regions of Mmu7 and H19q permitted the borders of several evolutionary rearrangements to be localized. One rearrangement has involved the transposition of clustered pregnancy specific glycoprotein (PSG) gene family members to a more proximal position in Mmu7, whereas related carcinoembryonic antigen (CEA) family genes (located adjacent to PSG genes in H19q), have remained in the same position in human and mouse (Fig.4). The border of two additional transposed segments, located adjacent to each other in H19q13.3–13.4, are solidly occupied by tandemly clustered zinc finger-containing (ZNF) genes (Ashworth et al. 1995; Stubbs et al. 1996, J. Kim, M. Shannon, L. Ashworth, and L. Stubbs, unpubl.). At least two other breakpoints of H19q13.4/Mmu7 conserved synteny are located in regions packed with ZNF gene families, which are present in particular abundance on H19 (Fig.4; Ashworth et al. 1995; Stubbs et al. 1996; Kim et al. 1997; Shannon et al. 1997). Of interest in this regard are observations that ZNF gene clusters are also frequently involved in translocations associated with human cancer (Rabbitts 1994 and references therein). The significance and generality of these early observations cannot be addressed directly at present, but should be clarified in the near future as additional syntenic breakpoints are mapped and sequence alignments of large mouse and human genomic regions begin to emerge.
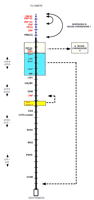
Positions and borders of the major evolutionary rearrangements between human 19q and mouse chromosome 7. Comparative map of human chromosome 19q and related regions in the mouse. The physical map of human 19q is represented, showing the positions of segments that are rearranged in order in mouse. Only a few anchor markers are presented for simplicity. Each crosshatch bar in the map represents a distance of 1 Mb in the human physical map. Colored boxes surround regions that are transposed in order in mouse relative to human; broken arrows indicate the positions to which they have been moved in the mouse genomes. Blue hatching on the backbone of the map indicates the extent of a large inversion in Mmu7 relative to H19q13.4; a smaller inversion, involving genes related to ZNF132, ZNF134, and related clustered zinc finger gene family members, comprise a second, nested inversion within this larger region. Genes located at the borders of each rearranged segment are indicated in red. A number of zinc fingers containing genes have been identified on chromosome 19q and in related regions of Mmu7; some have been characterized and are indicated by specific locus names (e.g., ZNF134); clusters of other genes that have not been completely sequenced or assigned locus names are indicated simply as ZNF on this map. Data are compiled from published studies (Ashworth et al. 1995;Stubbs et al. 1996; Kim et al. 1997; Shannon et al. 1997); additional information can be obtained from the original references.
Toward a Reference Map for Vertebrate Species
Comparative mapping data have provided direct links between genetic maps of human and mouse, providing a surrogate genetic resource to speed the identification of human disease genes and to pinpoint the positions of health-related loci that are difficult to trace in the human population (for review, see Bedell et al. 1996). Our current understanding of the similarities and differences between the two genomes also permits researchers to tap directly into human gene mapping and DNA sequence data for analysis of mouse mutations, quantitative variances, and inherited susceptibilities long before similar resources can be recreated for a second mammalian species. Comparative maps of the genomes of other animals—especially commercially valuable livestock species and long-standing medical models such as the rat—have recently begun to emerge, and despite their relatively low level of resolution and completion, are serving valuable roles in the genetic analysis of these animals (for summary, see Andersson et al. 1997; Grobet et al. 1997; Kambadur et al. 1997). Because the genetics of most of these species is poorly characterized relative to that of human or mouse, establishment of direct links to gene mapping, sequence, and functional information via the comparative map would be especially valuable.
Fortunately, several new technologies are now available to speed the first stages of comparative map development for additional vertebrate genomes. For example, human chromosome paints have been used successfully to detect homologous segments in metaphase chromosome spreads of other species (Scherthan et al. 1994; Weinberg and Stanyon 1995). Zoo FISH methods are relatively insensitive—detecting only the relatively large and contiguous homology segments—and are incapable of distinguishing rearrangements within homology regions. Cross-species FISH experiments, however, have the advantage of being rapid, simple, and applicable on a genome-wide scale. Zoo FISH also provides one of the only methods by which basic syntenic homology data can be obtained without the development of specific conserved markers or specialized mapping systems for each new species. Another promising method that permits mapping data to be extrapolated cross-species uses universal primer pairs for PCR amplification of conserved anchor loci (Comparative anchor tagged sequences, or CATS; Lyons et al. 1997). CATS primers are designed to maximize chances that products will extend across intron–exon boundaries and are, therefore, often polymorphic in length. This property makes CATS markers useful for a variety of purposes, including interspecific crosses, somatic cell hybrid mapping, clone selection, and physical mapping experiments. Simple sequence repeat markers developed for use in one genome can also often be used to generate polymorphic markers for genetic and physical mapping of related species (Moore et al. 1991; Hino et al. 1993). EST sequencing projects also serve to provide new conserved markers that can be used for both hybridization- and PCR-based mapping schemes to anchor the maps of human and mouse genomes of other species.
Although genetic mapping systems, like the interspecific crosses that have revolutionized mouse genetics, will not be created easily enough or exploited rapidly enough to yield benefits for research with other vertebrate species, radiation hybrid technology may provide strategies for gene mapping in at least some uncharted genomes (Lunetta et al. 1995, 1996; Gyapay et al. 1996; Schmitt et al. 1996). Together, these new technologies hold great promise in eventually linking human genetics and DNA sequence to functional information derived from experimental species ranging from chicken to rat, and in permitting the upcoming wave of human sequence and mapping data to mark the birth of a unified reference map for vertebrate genomes.
Acknowledgments
We thank Mark Shannon and Linda Ashworth for critical comments on the manuscript. This work was supported by a grant from the U.S. Department of Energy (under contract DE-AC05-96OR22464 with Lockheed-Martin Energy Research, Inc. and contract W-7405-ENG-48 with Lawrence Livermore National Laboratory).








