
Steady-State Transcript Levels of the Porphobilinogen Deaminase Gene in Patients with Acute Intermittent Porphyria
Abstract
PCR-based solid-phase minisequencing method was used to analyze the steady-state mRNA levels of the porphobilinogen deaminase gene in eight patients with acute intermittent porphyria. The patients had the earlier characterized mutations 517C → T (R173W), 518G → A (R173Q), 673C → G (R225G), 673C → T (R225X), 713T → G (L278P), and 1073delA (frame shift). All mutations, except the missense mutation 517C → T in exon 10, affected the steady-state transcript levels of the mutant allele. The mutant mRNA levels in lymphocytes varied from 5% to 95% of the corresponding wild-type allele levels. In contrast to the CRIM-negative mutation 517C → T, the CRIM-positive mutation in the same codon 518G → A resulted in reduction of the steady-state transcript level of the mutant allele to 65% of that of the normal allele. The two mutations, 673C → G or T, affecting the same nucleotide in exon 12 also differed considerably in their effect on mRNA levels: The transcript level of the allele with a missense mutation was decreased to 80% of that of the normal allele, whereas a nonsense mutation at the same position resulted in a dramatic decrease (fivefold) in the levels of the mutant transcript. Our data showed large variations between the levels of mutant transcript in AIP patients and these variations did not correlate either to CRIM class, to the location of the disease causing mutation in the PBGD gene, or to the clinical phenotype of AIP.
[The sequence data described in this paper have been submitted to EMBL under accession nos. AJ002126–AJ002137.]
Acute intermittent porphyria (AIP) is a metabolic disease with an autosomal dominant pattern of inheritance (Kappas et al. 1995). The disease is caused by a partial deficiency of the third enzyme, porphobilinogen deaminase (PBGD; also known as hydroxymethylbilane synthase; EC4.3.1.8) in the heme biosynthetic pathway. Ten to twenty percent of individuals with the enzyme defect experience occasional acute attacks, which consist of abdominal pain and various neuropsychiatric symptoms, but up to 50% of AIP patients may have milder symptoms (Kauppinen and Mustajoki 1992; Kappas et al. 1995).
The PBGD gene has been sequenced and thoroughly characterized (Raich et al. 1986; Grandchamp et al. 1987; Chretien et al. 1988; Lee 1991; Namba et al. 1991); it is assigned to chromosome 11q24 and contains 15 exons. The size of the gene is 10 kb, of which 1.3 kb represents coding sequence. Two tissue-specific isoforms have been characterized. The two transcripts arise from two separate promoters via alternative splicing of exons 1 and 2. The mRNA of the housekeeping (nonerythropoietic) isoform contains exons 1 and 3–15 encoding an enzyme of 361 amino acids, whereas the erythroid isoform is encoded by exons 2–15, thus lacking the first 17 amino acids of the amino terminus.
Previously, AIP has been divided into two main subtypes according to the ratio of the PBGD polypeptide concentration and enzyme activity in erythrocytes (cross-reacting immunologic material; CRIM-negative and -positive subtypes) (Anderson et al. 1981). Thus, in CRIM-negative patients the amount of immunoreactive protein has corresponded to the enzyme-activity and in CRIM-positive patients an inactive protein is detected. Recent studies on the PBGD gene have revealed >100 mutations in AIP patients (Deybach and Puy 1995; Kauppinen et al. 1995) showing the heterogeneity of AIP at the molecular level despite relatively uniform clinical manifestations.
To clarify the effects of different mutations on the transcription and initial molecular events in the pathogenesis of AIP, we have monitored the steady-state mRNA levels of the mutant and wild-type alleles of PBGD in patients’ lymphoblast cell cultures. In this communication we report the effect of six disease-associated mutations on the steady-state transcript level of the PBGD gene. The allele-specific transcript levels were determined by a quantitative PCR-based solid-phase minisequencing method (SPMS) (Syvänen et al. 1990,Karttunen et al. 1996).
RESULTS
Six different AIP mutations of previously described Finnish patients were chosen for this study (Table 1) (Kauppinen et al. 1995). Two point mutations, 517C → T (R173W) and 518G → A (R173Q), affect the same codon in exon 10. In addition, two point mutations hit the same nucleotide in exon 12: a 673C → G (R225G) transversion and a 673C → T (R225X) transition creating a premature termination codon. The missense mutation 713T → G results in a substitution of one amino acid (L278P). The sixth mutation analyzed is a deletion in exon 15, 1073delA, resulting in a frame shift, which removes the termination codon without creating a new one before the polyadenylation signal. The mutation 518G → A is found among CRIM-positive patients, whereas patients carrying the other mutations are CRIM negative (Mustajoki and Desnick 1985).
Characteristics of AIP Mutations in the PBGD Gene
To monitor the steady-state transcript level of wild-type and mutant alleles, we have used the specific detection of the nucleotide present at the mutation site in the patients’ DNA and the corresponding cDNA by PCR amplification and SPMS analysis (Fig. 1) (Karttunen et al. 1996). Table 2 shows the calculated ratios between the two sequences present and the relative proportion of mutant mRNA compared with the wild type. As an example, the raw data of one of the mutations analyzed (673C → T) is shown in Table3.
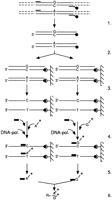
Schematic presentation of the steps in the SPMS method (modified fromSyvänen et al. 1993), mutation 673C → T as an example. (1) PCR with one biotinylated (•) and one unbiotinylated primer. (2) Affinity capture of the biotinylated PCR product on streptavidin-coated microtiter wells. (3) Washing and denaturation. (4) Primer extension reaction. All PCR reactions were aliquoted in eight separate wells. The tritium-labeled nucleotide representing the normal allele (G*) was added in four wells, and four wells contained the tritium-labeled nucleotide representing the mutant allele (A*). Depending on the sequences present, the labeled nucleotide will hybridize next to the minisequencing primer. The nucleotides not incorporated will be washed out. (▪) Detection primer; (A*/G*) [3H]dATP/[3H]dGTP; (DNA-pol.) DNA polymerase. (5) Measurement of the incorporated label. (6) Calculation of the result.
The Results of SPMS Analysis of Steady-State PBGD mRNA Levels in AIP Patients
The Solid-Phase Minisequencing Analysis of Steady-State mRNA Levels in Three Subjects with the Mutation 673C → T in the PBGD Gene
All mutations, except the missense mutation 517C → T in exon 10, affect the steady-state transcript levels of the mutant allele. The mutant mRNA levels in lymphocytes varied from 5% to 95% of the corresponding wild-type allelic mRNAs (Table 2). In contrast to the CRIM-negative mutation 517C → T, the CRIM-positive mutation in the same codon (518G → A) results in reduction of the steady-state transcript level of the mutant allele to 65% of that of the normal allele.
The two mutations affecting the same nucleotide in exon 12 differ considerably in their effect on mRNA levels: The missense mutation at nucleotide 673C does not dramatically affect the level of the mutant transcript (mean 80% of that of normal), whereas in the case of the nonsense mutation, the level of the mutant transcript is reduced fivefold compared with that of the normal allele. The total RNA samples of three different members of the latter family, who carry the same nonsense mutation, have the same amount of mutant transcript (Table 3). In this family, the clinical picture varies among the affected individuals: The mother has been asymptomatic throughout her life, one of the daughters has had mild symptoms but no attacks, whereas the other daughter has experienced acute attacks. Thus, the phenotype does not correlate with the amount of mutant transcript in this family.
The missense mutation (713T → G) in exon 12 results in a dramatic reduction in mutant mRNA and almost a total loss of the steady-state level of the mutant allele was detected (Table 2). In contrast, the loss of the termination codon in exon 15 caused by a deletion (1073delA) reduces the mutant allele transcript level to 70% of that of the normal allele (Table 2).
In the case of mutations 517C → T and 1073delA, the mutant transcript, the levels of which were originally relatively high, was degraded during storage more rapidly than the normal allele (Table 2). The levels of normal allele were not altered when stored in 70% ethanol at −80°C. In comparison, in the case of the mutation 673C → G, the relative amount of the mutant transcript was not affected when stored under similar conditions.
DISCUSSION
In this report we have studied the effect of six different AIP point mutations on the steady-state transcript levels of the PBGD gene. In the majority of cases, the mutations resulted in a significant decrease in the amount of mutant mRNA. Of special interest were the mRNA levels of alleles carrying either a nonsense and a missense mutation at the same codon. In the case of the missense mutation 673C → G, the transcript level of the mutant allele was not reduced considerably, but the nonsense mutation 673C → T in the same nucleotide resulted in a dramatic reduction in the amount of mutant transcript. This suggests that a premature termination codon is the causative agent for the reduction in the mutant transcript level.
Previously, the presence of a premature termination codon has been shown to reduce the steady-state mRNA levels in the case of several unrelated genes, but this is not the rule (for review, see Cooper 1993). Mechanisms by which nonsense mutations reduce the amount of mutant transcript have been clarified recently (Carter et al. 1996). In this study, several nonsense mutations in different positions of the T-cell receptor-β gene were analyzed, and the authors concluded that at least one intron downstream of the nonsense codon was required to trigger down-regulation of the premature termination codon-containing allele. Moreover, this phenomenon occurs in the nucleus post-transcriptionally. The mechanism by which the premature termination codon is scanned in the nucleus is still an enigma, because, at present, the cytoplasmic ribosome is the only known entity that can scan codons.
The mutant transcript levels of the three clinically different patients with the same nonsense mutation at nucleotide 673C were similar to each other, and thus, there was no correlation between the phenotype and the mutant mRNA levels in lymphocytes. The individual variation in excretion of porphyrin precursors and clinical manifestations may result from differences in tissue-specific expression or the interference of other factors, such as environment and other genes. Genes encoding metabolic enzymes of the liver may be important in the pathogenesis of AIP, because several factors such as drugs, alcohol, and hormonal changes are known to induce symptoms (Kappas et al. 1995).
The effect of missense mutations on transcript levels has characteristically been reported to be nonsignificant, whereas nonsense mutations often cause a dramatic reduction in the transcript levels of the same gene (Mustafa et al. 1995; Karttunen et al. 1996; Ploos van Amstel et al. 1996). Our results show, however, that missense mutations can also reduce the abundance of mutant mRNA.
Several mechanisms for the variation in mRNA stability in vivo have been proposed (for review, see Ross 1995, 1996). Stability of mutant mRNA can be related to the altered binding site of a protein that stabilizes mRNA. Furthermore, some endoribonucleases recognize specific sequences in mRNAs and degrade them, and the mutation may create a novel cleavage site. The secondary structure of RNA can also be modified by a mutation and thus, it becomes more unstable.
Direct sequence analyses of the PCR amplified cDNA samples, which were reverse transcribed from the total RNA extracted from the patients’ lymphoblast cell lines, actually provide rough estimates of the transcript levels of the mutant alleles (Kauppinen et al. 1995). The results are in good agreement with results obtained by SPMS analysis: Those mutations that remained undetectable after direct sequencing of the cDNA had also low steady-state transcript levels of the mutant allele in SPMS analysis.
The CRIM activity was determined previously by rocket-immunoelectrophoresis in which the polyclonal rabbit anti-human-PBGD antibody was used against PBGD protein obtained from lysed erythrocytes (Anderson et al. 1981, Mustajoki and Desnick 1985). On the basis of these results, in the samples of the CRIM-positive patients with the mutation 518G → A, the PBGD enzyme concentration was 1.6–1.8 times higher than expected from the PBGD activity measurements (CRIM ratio). This suggested that the amount of inactive enzyme was 60%–80% that of the active enzyme. The amount of mutant mRNA of this patient was 65% that of the normal allele, which agrees well with the CRIM ratio. This also suggests that the mutant mRNA is efficiently translated to an enzymatically inactive protein and, furthermore, the stability of the mutant polypeptide is comparable with that of the normal enzyme. In the case of CRIM-negative mutations, however, the transcript levels did not correlate with the amount of PBGD protein. The transcript levels of the mutant 517C → T, 673C → G, and 1073delA alleles were relatively high compared with that of the normal allele. In contrast to this, no inactive protein was detected, and the patients were classified as CRIM negative. This has been proposed to be caused by the instability of mutant mRNA, insufficient translation, or rapid intracellular decay of the mutant polypeptide (Mustajoki and Desnick 1985).
METHODS
AIP patients
The patient material comprised of eight AIP patients representing both CRIM-negative and -positive subtypes of AIP (Mustajoki and Desnick 1985; Kauppinen et al. 1992). All of them have been characterized previously by a PBGD gene mutation (Kauppinen et al. 1995). Of the patients, four have had acute attacks (518G → A, 673C → T, 713T → G, and 1073delA), three have been symptom-free (517C → T, 673C → G, 673C → T), and one patient has had mild symptoms (673C → T).
DNA and RNA extraction, and cDNA synthesis
Leukocyte DNA was released from the venous blood samples with EDTA as the anticoagulant as described by Higuchi (1989). Total RNA was extracted from patients’ lymphoblastoid cell lines by use of the guanidium isothiocyanate method (Chirgwin et al. 1979). Three parallel extractions of total RNA were performed from two different cell lines, except in the case of mutation 713T → G, in which only two independently isolated RNA samples were available. All RNA samples were stored in 70% ethanol at −80°C. Complementary DNA was synthesized from 2–5 μg of total RNA with 37.5 units of Moloney murine leukemia virus (M-MuLV) Reverse Transcriptase (New England Biolabs, USA) with 0.5 units of RNase inhibitor (RNAsin, Promega, Wisconsin, USA), and dNTPs at 0.2 mm concentrations in a total volume of 15 μl of the enzyme buffer. Random hexanucleotide mix (Hexanucleotide Mixture, Boehringer Mannheim, Germany) was used when synthesizing the cDNA of patients having mutations 517C → T, 518G → A, 713T → G, and 1073delA, whereas 20 pmoles of specific primer 5 (Table 4) was used in the samples with the 673C mutations. The reaction mixture was incubated for 10 min at room temperature and for 60 min at 42°C. Five independent reverse transcription (RT) reactions were performed from each RNA sample.
Oligonucleotides Used in the RT, PCR, and SPMS Reactions
PCR amplification
Fifteen μl of RT product or 10 μl of lysed leukocyte sample was used as the template for PCR (Mullis and Faloona 1987). Five parallel PCR reactions were performed from each RNA or DNA sample. The PCR reaction mixture contained 100 pmoles of the unbiotinylated primer and 20 pmoles of the biotinylated primer (see Table 3), dNTPs at 0.2 mm concentrations, and 3 units of DNA polymerase (Dynazyme, Finnzymes, Finland) in 100 μl of the enzyme buffer. The temperature profile for the PCR reactions was 2 min at 94°C for the first denaturation step, followed by 30 sec at 94°C, 30 sec at 58°C, and 30 sec at 72°C for 30 cycles.
SPMS analysis and calculation of allele ratios
Figure 1 shows a schematic of the steps of SPMS, which were performed as described previously (Karttunen et al. 1996). For each minisequencing reaction, 10 μl of the PCR product and 40 μl of 20 mm sodium phosphate buffer, at pH 7.5, including 0.1% Tween 20 were added to streptavidin-coated microtiter wells (Combiplate 8, Labsystems, Finland) and incubated at 37°C for 90 min. The wells were washed three times with 200 μl of 40 mm Tris-HCl (pH 8.8), 1 mm EDTA, 50 mm NaCl, and 0.1% Tween 20 and denatured twice with 100 μl of 50 mm NaOH for 5 min at room temperature. Fifty microliters of minisequencing solution [10 pmoles primer, 0.6 units of DNA polymerase (Dynazyme, Finnzymes, Finland) in its buffer and 0.2 μl of [3H]dNTP (specific nucleotides and their activities given in Table 4)] was added to each well. All PCR reactions were aliquoted in eight separate wells. The tritium-labeled nucleotide representing the normal allele was added in four wells, and four wells contained the tritium-labeled nucleotide representing the mutant allele. The plates were incubated at 50°C for 10 min. Depending on the sequences present, the number of the labeled nucleotide hybridized next to the minisequencing primer varies. The nucleotides not incorporated were washed out as described above. The primer hybridized with labeled nucleotide was denatured with 60 μl of 50 mm NaOH for 5 min at room temperature. The radioactivity eluted with NaOH was counted in a liquid scintillation counter (Microbeta 1450, Pharmacia, Sweden).
For each PCR product, four parallel reactions were performed to analyze both the wild-type and the mutant nucleotide, respectively. After the SPMS reactions, the mean activity for both alleles was calculated. The activity ratios reflecting the amounts of the mutant and normal sequences present in the original RNA were correlated to the ratio obtained from the identical analysis of the heterozygous genomic DNA sample that served as a 1 : 1 standard.
Acknowledgments
Dr. Ann-Christine Syvänen is acknowledged for the review of the this manuscript. This study was supported by grants from The Aarne Koskelo Foundation, The Research Funds, and The Clinical Research Institute of the Helsinki University Central Hospital, University of Helsinki, and the Academy of Finland.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL sami.mustajoki{at}helsinki.fi; FAX 358 9 471 4012.
-
- Received August 27, 1997.
- Accepted October 6, 1997.
- Cold Spring Harbor Laboratory Press








