
Abstract
Evolutionary rate covariation (ERC) is an established comparative genomics method that identifies sets of genes sharing patterns of sequence evolution, which suggests shared function. Whereas many functional predictions of ERC have been empirically validated, its predictive power has hitherto been limited by its inability to tackle the large numbers of species in contemporary comparative genomics data sets. This study introduces ERC2.0, an enhanced methodology for studying ERC across phylogenies with hundreds of species and tens of thousands of genes. ERC2.0 improves upon previous iterations of ERC in algorithm speed, normalizing for heteroskedasticity, and normalizing correlations via Fisher transformations. These improvements have resulted in greater statistical power to predict biological function. In exemplar yeast and mammalian data sets, we demonstrate that the predictive power of ERC2.0 is improved relative to the previous method, ERC1.0, and that further improvements are obtained by using larger yeast and mammalian phylogenies. We attribute the improvements to both the larger data sets and improved rate normalization. We demonstrate that ERC2.0 has high predictive accuracy for known annotations and can predict the functions of genes in nonmodel systems. Our findings underscore the potential for ERC2.0 to be used as a single-pass computational tool in candidate gene screening and functional predictions.
The omics era has greatly increased the number of sequenced genomes (Hotaling et al. 2021). With this influx of information, network and system biologists have developed approaches to study gene and protein interactions from a broader perspective and at a much larger scale. Networks of such interactions can be reconstructed based on gene coexpression, semantic similarity, and other experimental methods. Such networks have become commonly used to select disease gene candidates (Chen et al. 2009; Sun et al. 2010; Paredes-Sánchez et al. 2015) and to predict protein function (Sharan et al. 2007; Zhu et al. 2010; Xiong et al. 2014; Saha et al. 2019). However, there are still limitations. These methods require many contextual details to be understood, such as the environment from which the organism/cells were collected, the phenotypes being expressed, and the developmental stage from which the information is captured. The requirement for this a priori knowledge limits many studies to studying only model systems. By studying protein interactions through a phylogenetic lens, we can remove the need for these details and incorporate nonmodel species into the analyses. Understanding that gene expression, physical interactions, and epistatic interactions all potentially respond to the selection pressures on a gene, we can study patterns of evolution across species to find genes and proteins that have shared functions and processes. In this study, we propose an improved evolutionary method to build systems biology–level networks that allow for analysis of both model and nonmodel species.
Evolutionary selective pressures act on every species across different developmental stages, environments, and phenotypes. Thus, we can treat each species as its own experiment conducted across millions of years. Adding more observations in a traditional bench experiment increases the power to detect patterns. Similarly, adding more species to a phylogeny increases the statistical power to detect evolutionary patterns. These patterns can be used to identify genes that have been subjected to shared selective pressures. Because genes that participate in a function together (i.e., cofunctional genes) experience many of the same selective pressures (Goh et al. 2000; Pazos and Valencia 2001; Lovell and Robertson 2010; Clark et al. 2012), changes in those pressures are expected to change their relative evolutionary rates (RERs) in parallel. Thus, by identifying genes with correlated changes in branch-specific RERs, functional relationships between genes can be inferred.
Evolutionary rate covariation (ERC) is a measure that quantifies the correlation of the RERs of a pair of genes across all branches in a phylogeny, both internal and external. Thus, implementations of ERC require that gene trees have congruent species topologies (branching patterns). ERC is not to be confused with simple correlations between the average rates of genes; rather, ERC examines how rates change between species and whether those changes correlate with other genes.
Calculating the ERC for each pair of orthologous genes across a phylogeny allows for the generation of an evolutionarily derived cofunctional map of the cell, which in turn can be used to predict functional relationships and interactions (Goh et al. 2000; Pazos and Valencia 2001; Ramani and Marcotte 2003; Yosef et al. 2009; Clark et al. 2012). ERC has been calculated on a wide variety of taxonomic groups, including mammals (Priedigkeit et al. 2015; Kowalczyk et al. 2021), fungi (Clark et al. 2012, 2013; Steenwyk et al. 2022; Little et al. 2024), Drosophila (Findlay et al. 2014; Raza et al. 2019), and plants (Forsythe et al. 2021).
In principle, high ERC values allow the detection of any type of functional relationships, whether underpinned by physical interactions, such as protein complexes (Little et al. 2024), or nonphysical interactions, such as enzymatic pathways (Findlay et al. 2014; Brunette et al. 2019; Raza et al. 2019). ERC has been used to successfully identify novel interactors in protein pathways that were then experimentally validated (Findlay et al. 2014; Brunette et al. 2019; Raza et al. 2019). It has also been used to screen for candidate genes in disease networks (Priedigkeit et al. 2015), identify relaxation of constraint in meiotic proteins in clonally reproducing yeast (Clark et al. 2013), and determine the intracellular localization of poorly characterized zinc transporters (Kowalczyk et al. 2021). The success of ERC as a screening method demonstrates the efficacy and usefulness of the method across phyla and biological scopes. However, the ERC methodology used in the studies above was only performed on a maximum of 63 species.
Here, we introduce a new open source software package, ERC2.0. This software makes it possible, for the first time, to calculate ERC across hundreds of species and tens of thousands of genes. Whereas previous methods used Pearson's correlations and branch lengths not corrected for heteroskedasticity, ERC2.0 makes improvements to employ Fisher transformation and a new branch length normalization step, each of which we demonstrate to improve statistical power. Our case studies on data sets of yeast and mammal species (Supplemental Files 1, 2) show the improved predictive power of ERC2.0 relative to our previous implementation, ERC1.0 (Clark et al. 2012).
Results
The SMC5/6 complex demonstrates the correlation between high ERC values and functional relatedness
ERC allows us to compare the RERs of each branch, internal and external, of a phylogeny and to identify genes with shared patterns across a set of species (Fig. 1A). A high ERC correlation value is an evolutionary signature of cofunction, as is demonstrated by the RERs of SMC5 and SMC6, two genes whose protein products are known to form a complex (r = 0.78) (Fig. 1B). Although SMC5 and SMC6 show high ERC, the expectation is that most gene pairs are not cofunctional and so would not be acted upon by shared selective pressures. To illustrate, proteins that are not functionally related tend to have correlation values closer to zero, as seen when SMC5 (r = 0.15) or SMC6 (r = 0.06) is compared to a nonrelated gene PEX6 (Fig. 1A,B).
SMC5/6 complex shows significantly elevated ERC in both yeast and mammal phylogenies. (A) Gene trees showing the branch lengths for SMC6, SMC5, and PEX6, respectively, across 18 yeast species. Representative species, S. cerevisiae and K. polysporus, are highlighted in purple and red, respectively, in all three trees. (B) Scatter plots comparing the relative evolutionary rates for SMC6 × SMC5, SMC5 × PEX6, and SMC6 × PEX6 using the same 18 yeast species as shown in A with the representative species, S. cerevisiae and K. polysporus, shown in a purple and red dot, respectively. A linear regression line with 95% confidence interval is shown in black and gray, respectively. The Pearson's correlation for each comparison is shown in the top left corner of the plot. (C) Cartoon schematic of the SMC5/6 complex. An asterisk indicates a yeast-specific complex member. (D) Fisher-transformed ERC (FtERC) values for all pairs within the SMC5/6 complex for a yeast data set of 343 species (left; gray) and mammal data set of 120 species (right; gray) with size-matched random gene pair samples for each data set (white). The data point with the highest FtERC in both data sets is between SMC5 and SMC6 (blue dot). Both data sets had significantly higher ERC—(**) P < 0.001, (****) P < 0.00001—than the sample of randomly selected gene pairs from the entire genome. Panels C and D created in BioRender (Little et al. 2024; https://www.biorender.com).
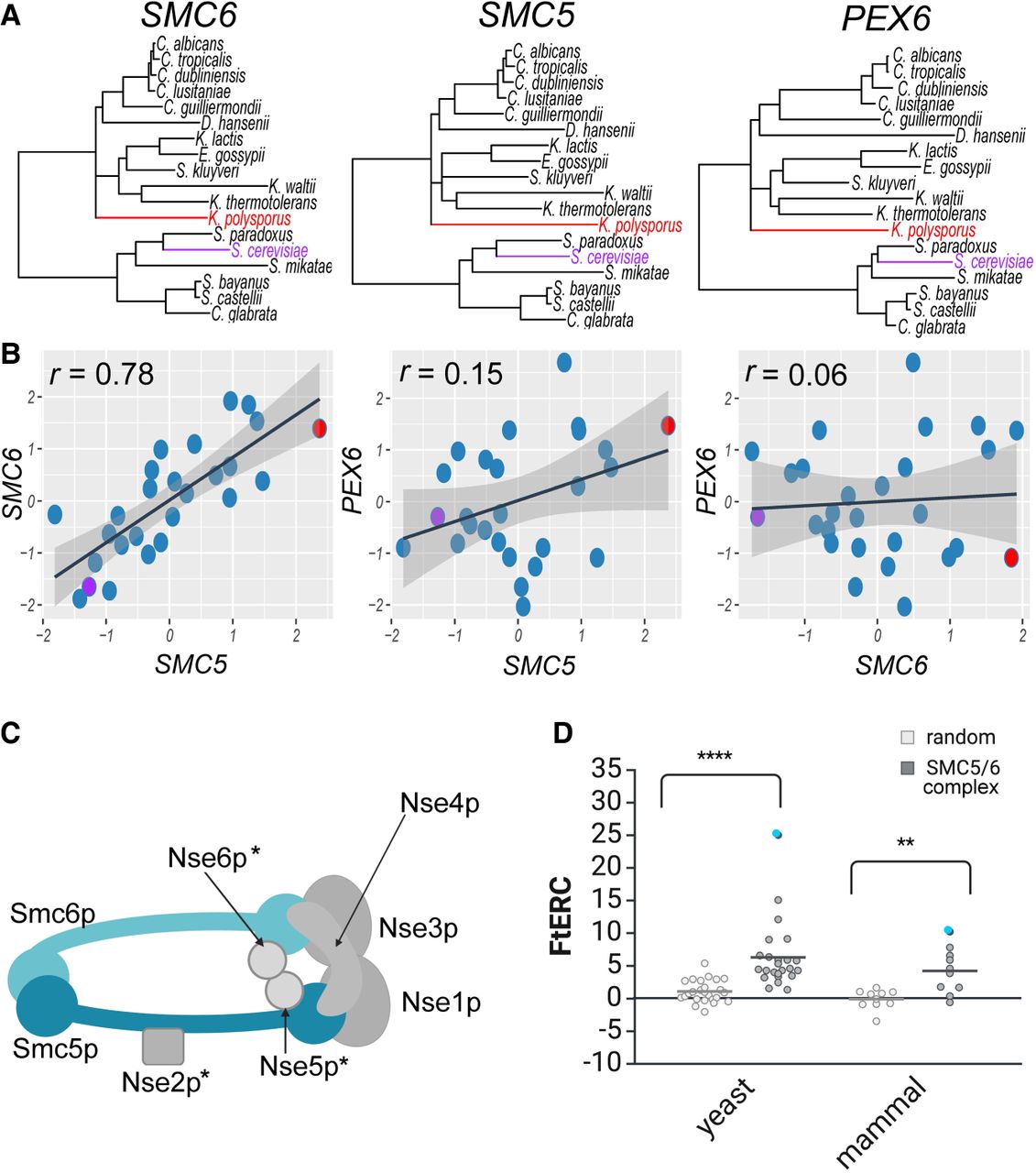
In addition to the high ERC between SMC5 and SMC6, ERC values between all pairs of proteins in the SMC5/6 complex are positive and elevated in a data set using 343 yeast species (Shen et al. 2018) and for almost all pairs in a data set using 120 mammal species (Fig. 1C,D; Supplemental Data [see Data access]; Hecker and Hiller 2020). These observations reflect the cofunctional relationship of these proteins, as they have experienced the same changes in selective pressures over evolutionary time. In general, the ERC scores are higher for the yeast data set than for the mammals, likely owing to the greater amount of evolutionary divergence among the yeast species. These results demonstrate how ERC matrices are useful as a mineable resource for evolutionary screening for new functional relationships in various taxa.
Fisher transformation normalizes correlations and reduces variance across diverse branch counts
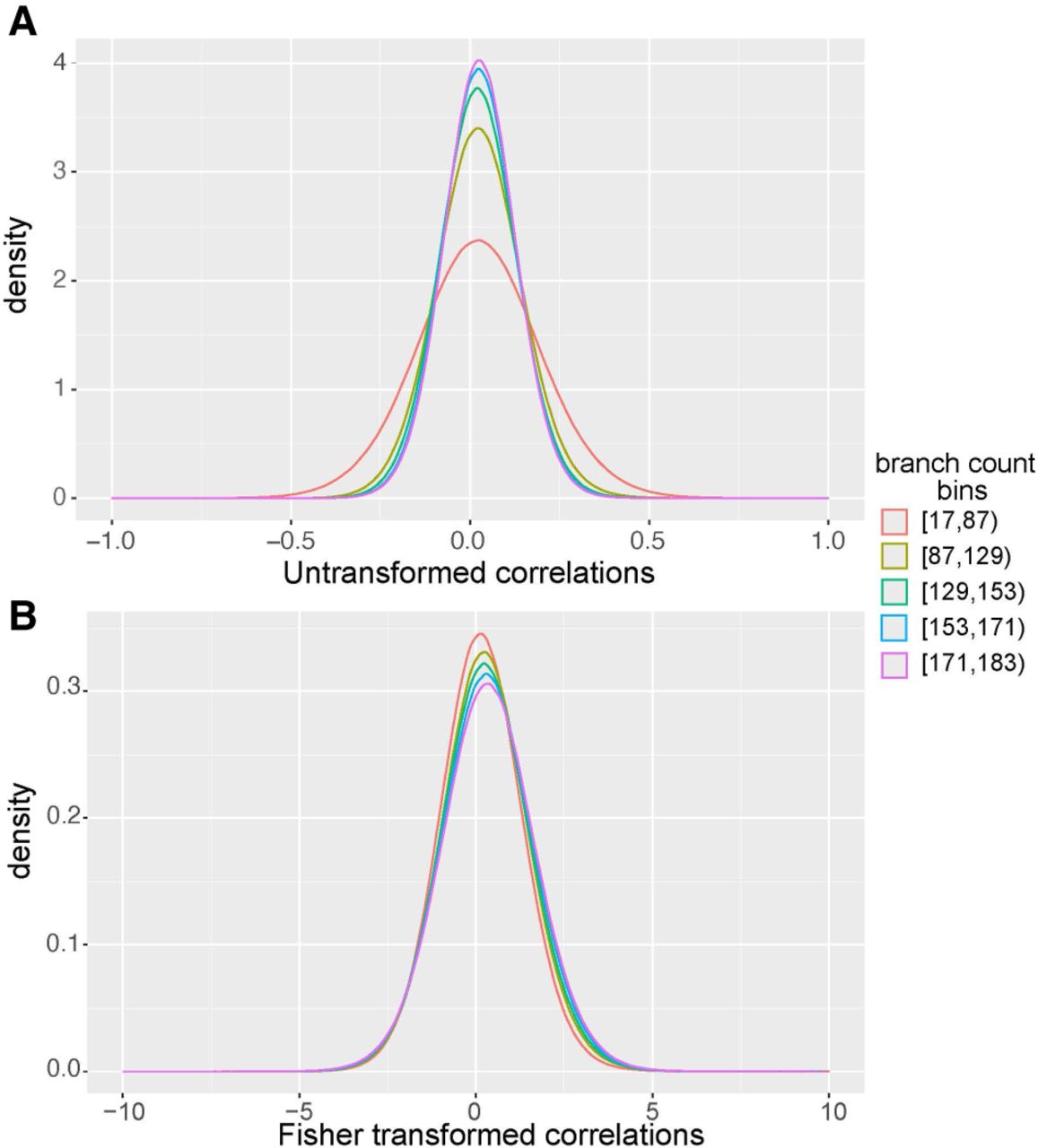
Previous iterations of ERC have been used to validate protein interactions (Clark et al. 2012), identify candidate genes within pathways (Findlay et al. 2014), and build gene-based disease networks (Priedigkeit et al. 2015) using the Pearson correlation coefficient. However, in larger data sets, such as a data set of 120 mammals (Hecker and Hiller 2020), the number of branches contributing to the correlation can vary when a gene is not present in every species. This creates two issues. First, the correlations cannot be compared across data sets because of the discrepancy in the number of data points contributing to each score. Second, with fewer branches, the distribution of correlation coefficients exhibits a greater variance (Fig. 2A).
Fisher transformation normalizes variance across 183 million gene pairs with different branch counts. (A) Untransformed Pearson's correlations for gene pairs calculated on 120 mammals. Gene pairs were sorted into equally sized bins based on the number of branches that contributed to the ERC score. (B) Fisher-transformed correlations for gene pairs calculated on 120 mammals. Gene pairs were sorted into equally sized bins based on the number of branches that contributed to the ERC score. This transformation allows scores to be meaningfully compared across taxa. Using Fisher-transformed values results in a more consistent variance and higher predictive power than untransformed correlation coefficients (Supplemental Fig. S2).
To normalize the correlation coefficients for the number of branches that went into the calculation, we introduce a Fisher transformation (Fisher 1915) to ERC values using the following equation:
Faster: ERC2.0 allows for computationally tractable calculations of hundreds of species
Increasing the numbers of species and genes permits investigating novel interactions but requires a tractable compute time. ERC2.0 uses a new data structure to speed the computation of all correlations between all genes. A major challenge in computing all pairwise correlations is that it requires pruning each gene's tree so that their species set matches; most genes are missing several species owing to evolutionary or technical reasons. For this reason, ERC2.0 stores all trees in a format that includes all possible subtrees. Together with a rapid indexing system found in the getClusterList function and efficient C-based tree operations in the TreeTools package (Smith 2019), the retrieval time for matching trees for each gene pair was greatly decreased, which makes data sets of much larger size accessible (Supplemental Table S1). Although ERC1.0 takes >2 h to calculate ERCs between 500 genes for 343 species on 10 CPUs (Supplemental Table S1), ERC2.0 with this new methodology completes the same task in 4 min.
Removal of heteroskedasticity in branch rates improves power
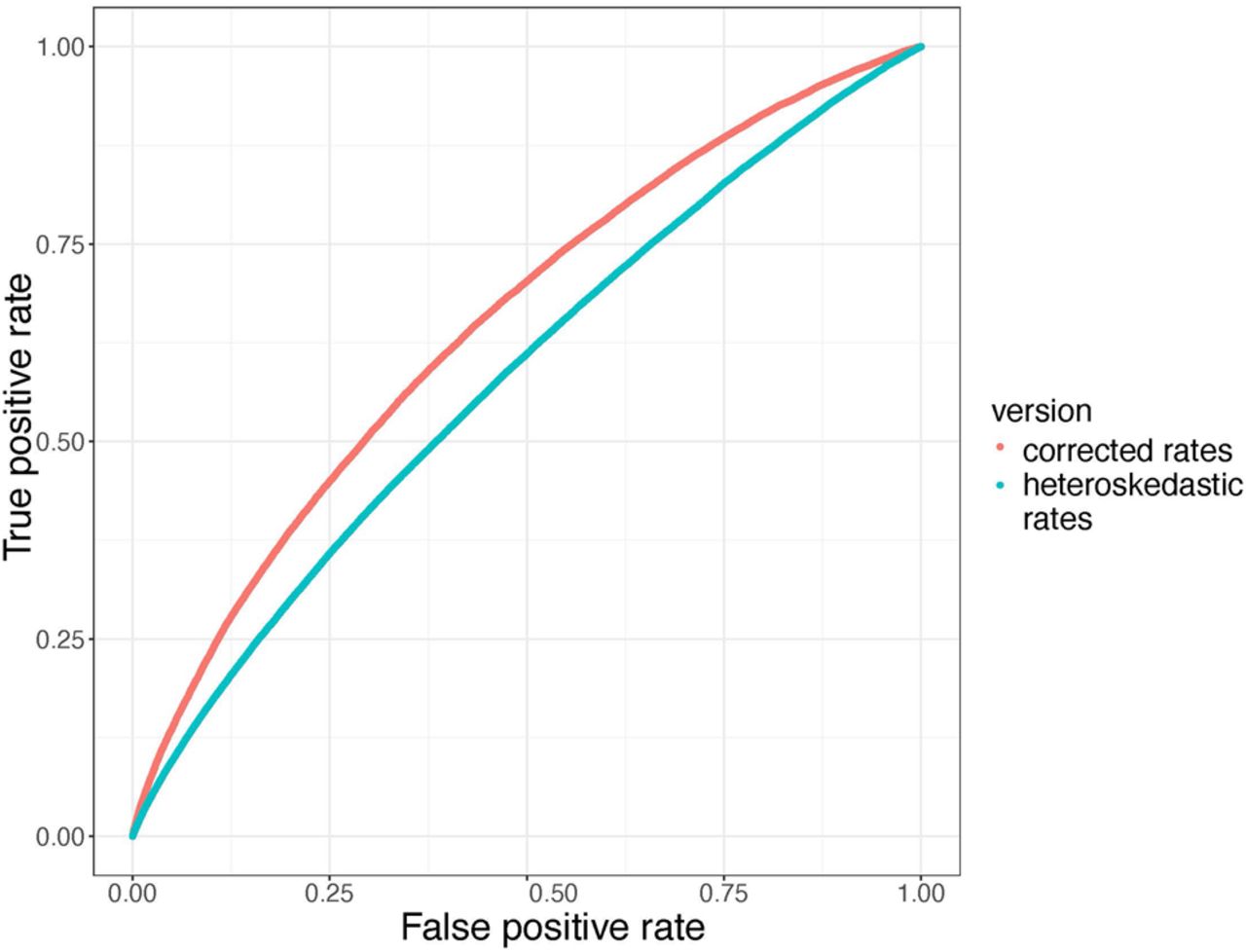
Heteroskedasticity is a statistical phenomenon in which there is an unequal variance in the residuals across a range of dependent variables, in this case average branch length. Heteroskedasticity violates assumptions made on linear regressions and reduces confidence in our inference of rate shifts. An additional improvement is that RERs on each gene tree branch are now calculated using a method that removes heteroskedasticity from the resulting rates, as was developed for the RERconverge package (Partha et al. 2019). Because these rates no longer have a variance that scales with their magnitudes, they are better suited in theory for application in statistical tests, such as linear correlations (ERCs). In practice, we measured a clear increase in power when branch RERs were corrected for heteroskedasticity (ERC2.0) compared with uncorrected (ERC1.0), as seen by the improvement in area under the ROC curve (ROC-AUC; 0.652 to 0.581, respectively) (Fig. 3). To make these comparisons, we used the STRING database as a truth set, where true positives are STRING pairs with combined scores greater than 700, considered to be a high confidence threshold by STRING.
Removal of heteroskedasticity improves ERC predictive power. Receiver operator characteristic (ROC) curves contrasting true-positive and false-positive rates demonstrate higher power in ERC2.0 values calculated using corrected rates (red curve; AUC = 0.652) compared with ERC1.0 values using uncorrected, heteroskedastic rates (blue curve; AUC = 0.581). The true-positive set are interactions from STRING with combined scores that are more than 700. ERC scores were calculated on a phylogeny of 18 yeast species.
Better: ERC2.0 outperforms previous ERC methodology and improves with larger data sets
The faster methodology can calculate ERC scores for tens of thousands of genes across hundreds of species (Supplemental Fig. S1). To evaluate the increase in power when more species are added, we expanded the analysis of a previous ERC study that used only 18 species of yeast (Clark et al. 2013). We hypothesized that increased species counts would improve the biological relevance of ERC scores and, therefore, predictive power.
To test this, we used logistic regression modeling to test how well ERC can be used to predict which genes belong to a given functional annotation (Methods). Logistic regression modeling allows us to test the practicality of using ERC scores to predict gene function for binary outputs in which a gene either belongs to an annotation or does not. The logistic regression model then uses the ERC values between all genes to select which genes or “features” are most important for accurately predicting the membership in a particular functional annotation. We used 10-fold cross-validation with an elastic net penalty and the ROC-AUC as the evaluation measurement to optimize the model. This resulted in a set of ROC curves across different regularization parameters that allows us to choose the model with the best fit for an annotation term.
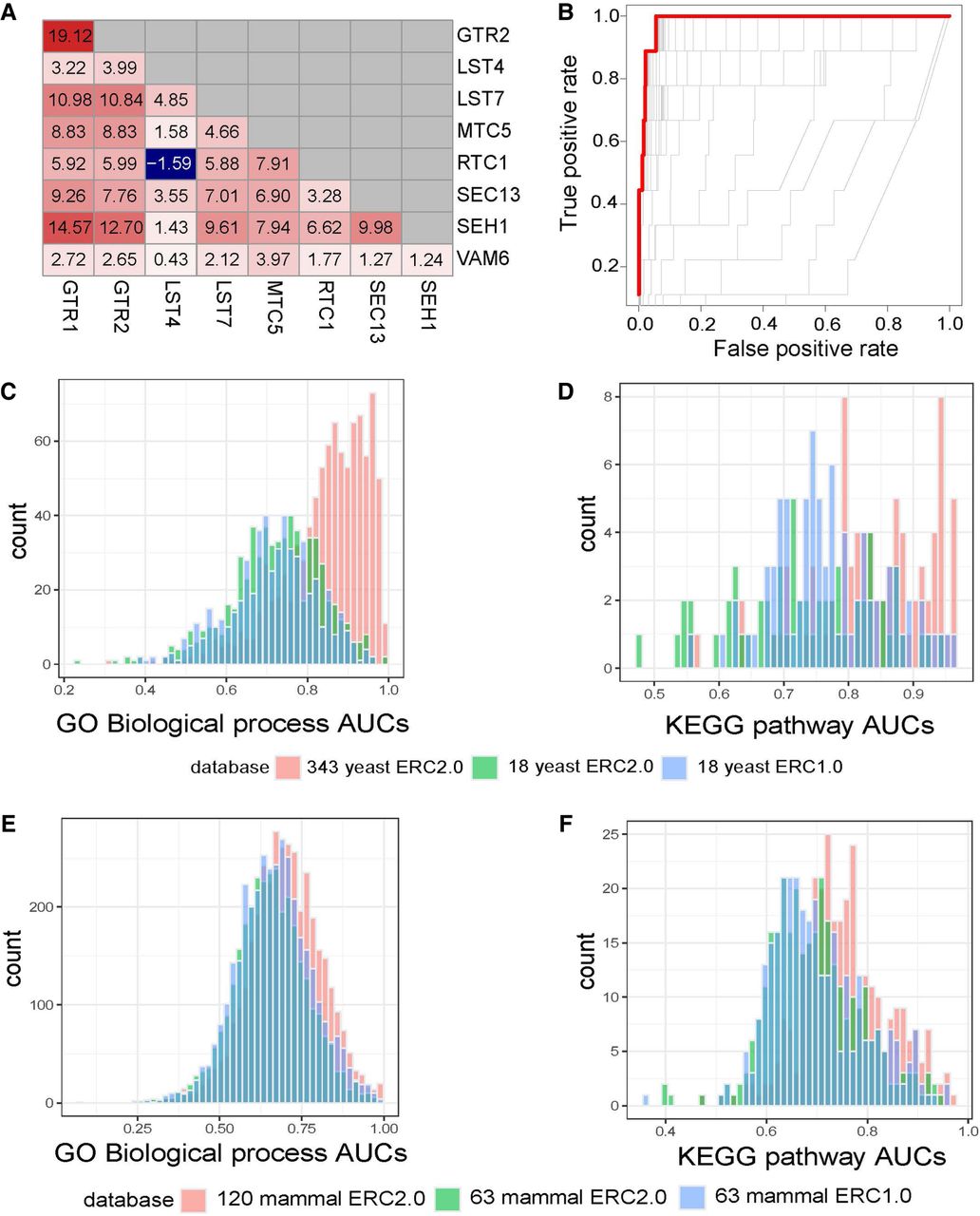
We began by querying the broad TORC1 signaling pathway, which has elevated ERC (Fig. 4A). For this pathway, ERC2.0 calculated on 343 yeast species produced a ROC-AUC of 0.988, meaning it had very little error in predicting members of that pathway (Fig. 4B). We then looked at more annotations to test how well ERC can predict pathways and processes globally. We used two data sets for yeast and mammals found in yeastEnrichr (Kuleshov et al. 2016) or Enrichr (Kuleshov et al. 2016), respectively. We again used ROC-AUC to quantify the performance of each ERC data set to predict two different sets of annotations (Fig. 4C–F). In yeast, we used KEGG pathways (Kanehisa 2019) and GO biological processes (GO-BP) (The Gene Ontology Consortium 2000). For both sets, ERC2.0 run on 343 yeast species had a significantly higher average ROC-AUC than both the ERC2.0 run on 18 species and the ERC1.0 on 18 species. Although the average ROC-AUC for the ERC2.0 run on 18 species was not significantly higher than for the ERC1.0 run on 18 species, it was still higher for GO-BP (0.72 vs. 0.71, respectively) and KEGG (0.77 vs. 0.73). Even more promising, the average ROC-AUC for ERC2.0 run on 343 yeast species is greater than 0.8 for both GO-BP and KEGG, indicating that the ERC2.0 data set has a high potential for predicting new genes within the annotated functional terms.
ERC2.0 improves predictive potential for GO biological processes (GO-BP) and KEGG pathways. (A) Pairwise yeast ERC scores for members of the biological process pathway, “positive regulation of TORC1.” Red colors indicate a higher FtERC; blue indicates a lower FtERC. (B) ROC curves for each model generated with cv.glmnet for “positive regulation of TORC1.” The red line indicates the model with the highest ROC-AUC of 0.988. (C,D) cv.glmnet validation of GO biological processes (C; n = 545), and KEGG pathways (D; n = 49) using ERC2.0 on 343 yeast species (red), ERC2.0 on 18 yeast (green), and ERC1.0 on 18 yeast (blue). There is a significant difference in means between 343 yeast ERC2.0 and both 18 yeast ERC2.0 and ERC1.0 for both GO-BP (P < 2.2 × 10−16, P < 2.2 × 10−16, respectively) and KEGG pathways (P < 2.2 × 10−16, P = 1.089 × 10−8, respectively). (E,F) cv.glmnet validation of GO biological processes (E; n = 5807) and KEGG pathways (F; n = 320) using ERC2.0 on 120 mammal species (red), ERC2.0 on 63 mammal species (green), and ERC1.0 on 63 mammal species (blue). There is a significant difference in means between 120 mammal ERC2.0 and both 63 mammal ERC2.0 and ERC1.0 for both GO-BP (P < 2.2 × 10−16, P = 1.147 × 10−5, respectively) and KEGG pathways (P = 1.259 × 10−15, P = 1.486 × 10−12). Each count is the highest AUC (s = lambda.min) predicted by glmnet.cv.
Because the difference in ROC-AUC between the original 18 yeast data set and the current 343 yeast data set is large, we also performed ROC analysis in intermediate data sets by subsetting the 343 species data set in increments of 49 species (Supplemental Fig. S3). We found a plateau as the number of species reached about 100. We hypothesize that the initial increase is because of clade-specific interactions as species are added and that the plateau is because of the saturation of annotated genes in the data set. This same pattern is not observed in the mammal data set; however, we suspect this is because of the lower number of species in the mammal phylogeny.
Both GO-BP and KEGG were also tested on the three iterations of mammal ERC. Similar to the yeast data sets, the larger 120 mammal data set run with ERC2.0 had a significantly higher mean AUC than either of the 63 mammal data sets run with ERC2.0 or ERC1.0. Although the 120 mammal ERC2.0 data set showed a lower average ROC-AUC for both data sets than yeast with the average AUCs at 0.70 and 0.77 for GO-BP and KEGG, respectively, there are still terms that have a ROC-AUC above 0.9. This shows that there are annotation terms that ERC2.0 is especially primed to predict in mammals.
We also compared the predictive power of ERC to the current literature base as captured in STRING (Szklarczyk et al. 2019). We used lambda.1se AUCs for the same two yeast annotation data sets as mentioned above to compare models trained using ERC values to models trained using STRING confidence scores (Supplemental Fig. S4). We limited the ERC analysis to only genes that are also present in STRING. The AUCs for the STRING-trained models were greater than the ERC AUCs for 1147/1166 of the annotations, with a mean AUC of 0.95.
We next asked whether the combination of ERC and STRING scores would improve the STRING model performance (Supplemental Fig. S4). Combining them improved the AUC of 1363 annotation terms compared with STRING alone, indicating net improvement upon adding ERC. The greatest improvement was for the GO-BP term “peptide transport (GO:0015833),” which improved from a STRING AUC of 0.742 and ERC AUC of 0.723 to a combined AUC of 0.914. Although ERC is unable to outperform the entirety of the STRING data set, the improvement of some annotation terms, as well as the relatively high average AUCs for ERC alone, show that there is novel information being captured by this tool and that there may be some benefit to adding ERC scores as a piece of evidence in the STRING combined scores. Furthermore, we envision that ERC2.0 will provide rapid and highly accurate functional prediction when applied to nonmodel organisms lacking the extensive experimental data sets found in yeasts and mammals, and it can be done using sequence information alone for a relatively minuscule cost.
ERC2.0 networks best predict organism-level cluster interactions in mammals compared to cellular processes in yeast
Given the comparatively low ROC-AUCs for the mammalian data sets, we investigated the overall structure of the mammal ERC interaction network compared with the yeast ERC network to determine the types of functional interactions best captured in each. We first subsetted each data set to the top 1% of FtERC values to capture the strongest set of gene–gene interactions. We then used a Markov chain clustering (MCL) algorithm in Cytoscape (Shannon et al. 2003) to create clusters of genes based on similar ERC profiles.
The resulting yeast network consisted of 257 clusters, with cluster 1 containing 3346 of the 4181 genes (Supplemental Fig. S5; Supplemental File 1). We performed GO-BP enrichment tests (Thomas et al. 2022) on the 12 clusters with greater than 10 members (Fig. 5A). This allows us to determine what types of processes within the cell/organism are shared by the genes within a cluster. Given the density of cluster 1, we performed another round of MCL clustering on only that cluster which broke it into seven subgroups. These subgroups were largely defined by cytoplasmic translation, ascospore wall assembly, mitochondrial transcription/translation, cytogamy, RNA polymerase I initiation, protein import into the peroxisome, and DNA double-stranded break repair (Fig. 5B; for full lists, see Supplemental File 2). This indicates that the strongest signal coming from the yeast data set is related to fundamental cellular processes like transcription, translation, and growth. Select clusters outside of cluster 1 are also enriched for processes such as mitochondrial double-stranded break repair, galactose catabolic processes, and attachment of mitotic spindle (Fig. 5C). Once again indicating that these fundamental cellular processes are strongly linked in the ERC network.
Yeast ERC is largely dominated by transcription/translation related processes. Cytoscape MCL clusters using the top 500,000 yeast FtERC values as the weights. (A) MCL clusters with more than six members; clusters highlighted in B and C are displayed in orange. A full network of all clusters is pictured in Supplemental Figure S5. (B) Enlarged image of MCL cluster 1. The large cluster is colored by subclusters with the highest enriched GO-BP term listed next to the subcluster in the corresponding color. (C) Select clusters with the highest enriched GO-BP term is shown on top of the cluster. (DSB) Double-stranded break. The full enrichment lists are in Supplemental File 2.
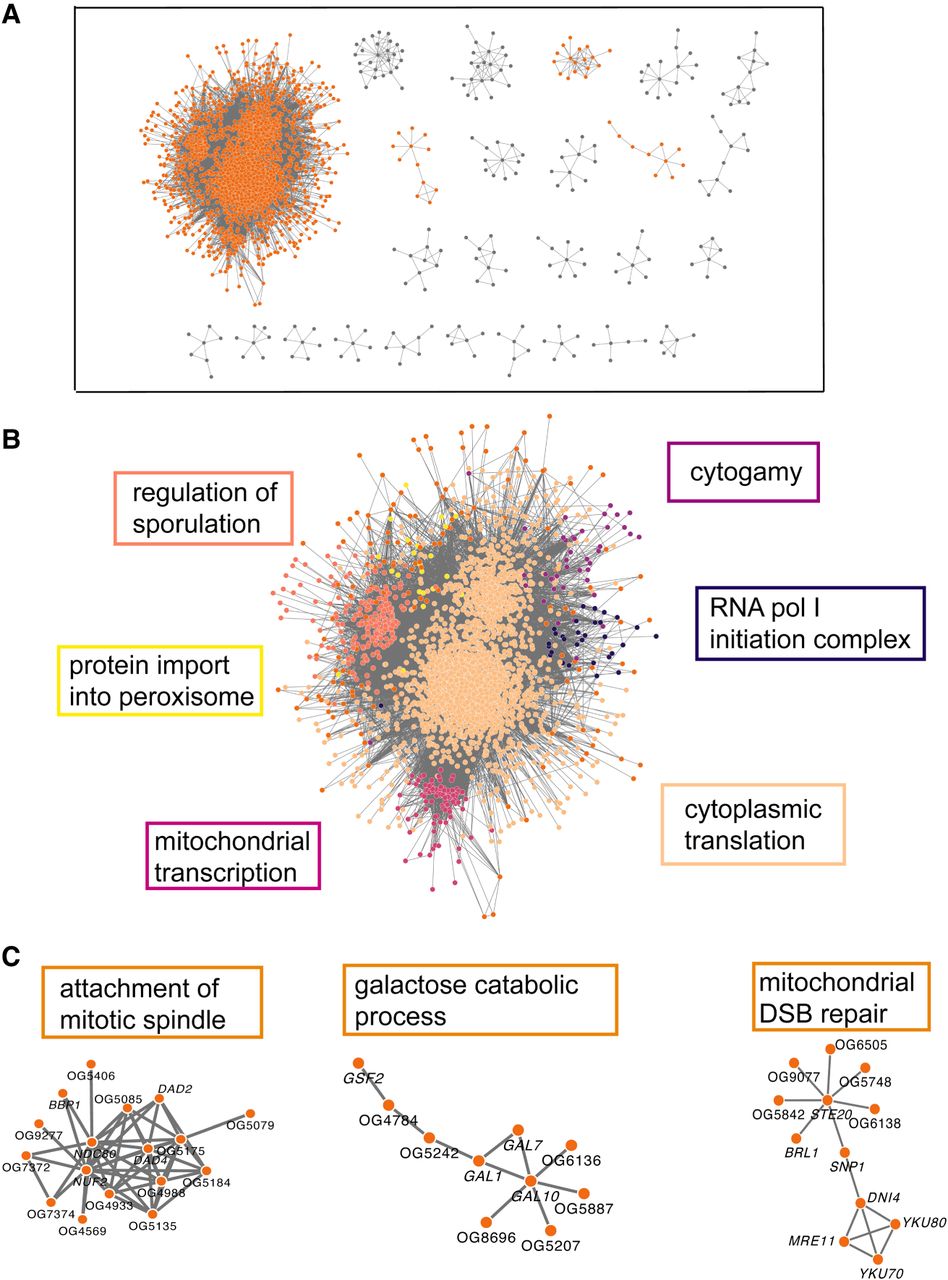
The yeast networks also make specific predictions of new genes in each of these functional categories, because each network contained some genes not previously appreciated to be in the enriched function (Supplemental Table S2). Some of these novel genes were completely uncharacterized, whereas others were already annotated to unrelated functions, which suggests an additional, pleiotropic function. Other novel genes are not in the Saccharomyces cerevisiae genome at all, in which most functional annotation has occurred. They were instead present in many non-cerevisiae yeast species. Those novelties show the potential to assign functions to genes in nonmodel species, including in core processes shared more broadly across taxonomic groups. For example, we observed several non-cerevisiae genes that had high ERC values with recognized mitochondrial genes; those predictions were upheld in databases from non-cerevisiae yeast species (Supplemental File 7).
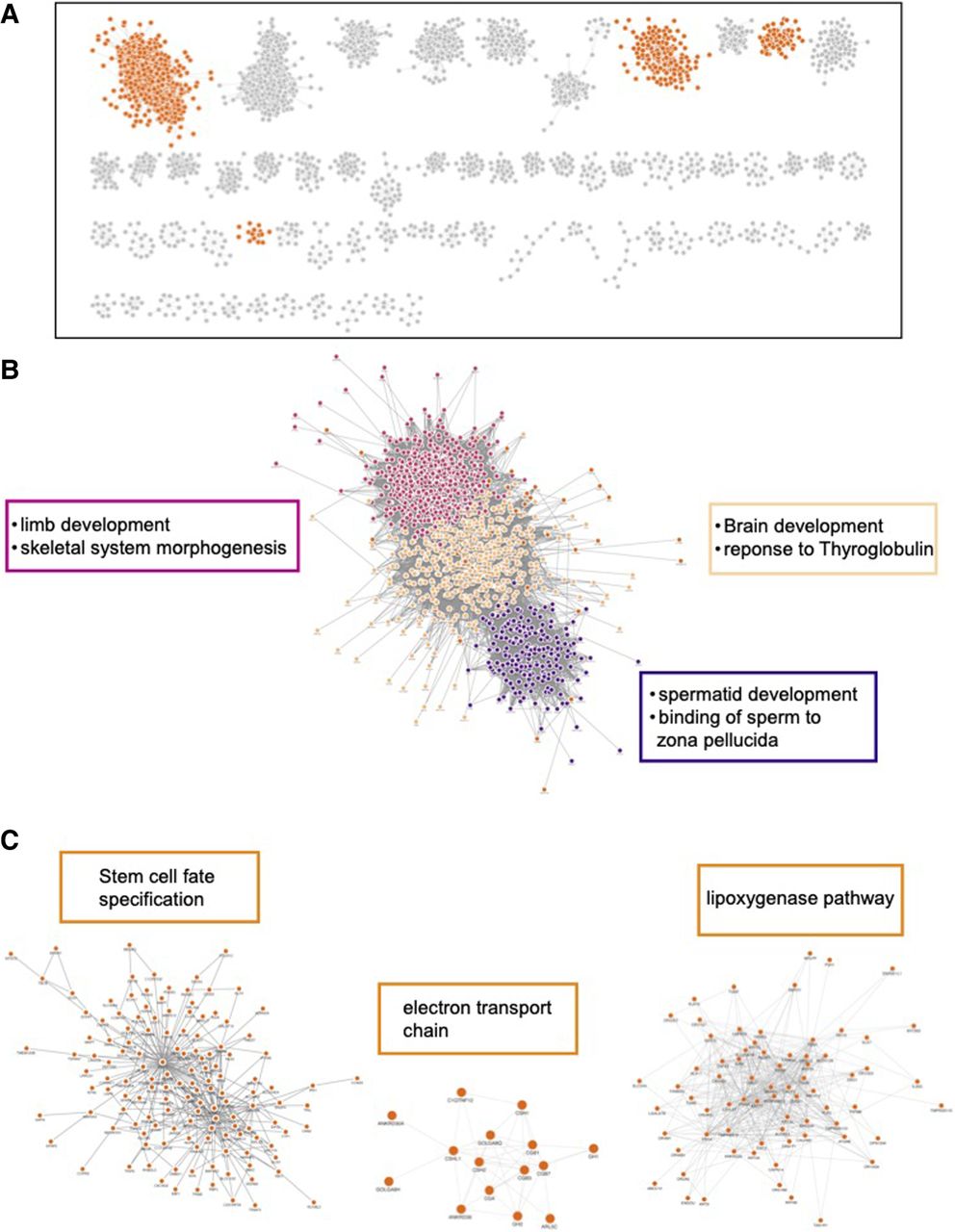
In contrast, the mammal data set formed networks capturing more organismal level biological processes. The mammalian network, similar to the yeast network, has one large cluster containing 1504 of all 4805 nodes. (Fig. 6A; Supplemental Fig. S6; Supplemental File 3). After subclustering, there were three major subgroups of cluster 1 that were enriched for a variety of GO-BP terms such as brain development, limb development, and spermatid development (Fig. 6B; Supplemental File 4). Among the clusters with 10 or more members, there are many organism-level enrichment terms such as stem cell fate specification, animal organ formation, and detection of chemical stimuli involved in sensory perception of smell (Fig. 6C). Although there are clusters that capture transcription/translation (2, 13, and 24), overall, the mammalian network shows that ERC is capturing more organismal-level biological processes compared with the yeast network, which is heavily dominated by processes involved in transcription and translation.
Mammal ERC captures organismal processes. Cytoscape MCL clusters using the top 500,000 mammal FtERC values as the weights. (A) Clusters with 10 or more members; all network clusters can be found in Supplemental Figure S6. Clusters highlighted in B and C are highlighted in orange. (B) Enlarged image of MCL cluster 1. The large cluster is colored by subclusters with the highest enriched GO-BP term listed next to the subcluster in the corresponding color. (C) Select clusters with the highest enriched GO-BP term shown on top of the cluster. The full enrichment lists are in Supplemental File 3.
Case study: ERC2.0 identifies known and novel functional interactions with histone chaperones
To demonstrate that ERC2.0 analysis is sufficiently informative to identify both known and novel relationships between genes, we examined transcription-associated histone chaperones as a test case using the 343 yeast species data set. Histone chaperones interact with histones and modulate the disassembly and assembly of nucleosomes in an ATP-independent manner during DNA-templated processes (Hammond et al. 2017). In S. cerevisiae, Spt6p, Spn1p, and the FAcilitates Chromatin Transcription (FACT) complex, containing Spt16p and Pob3p, function as histone chaperones during transcription by RNA polymerase II (RNAPII) (Robert and Jeronimo 2023; Miller et al. 2023). All four of these proteins are conserved in mammalian cells and are essential for the viability of yeast cells, indicating that they perform critical independent functions. Recent genetic and genomic work in yeast suggests that despite being essential individually, these factors cooperate to maintain proper chromatin architecture in the wake of RNAPII transcription (Viktorovskaya et al. 2021; López-Rivera et al. 2022; Warner et al. 2024).
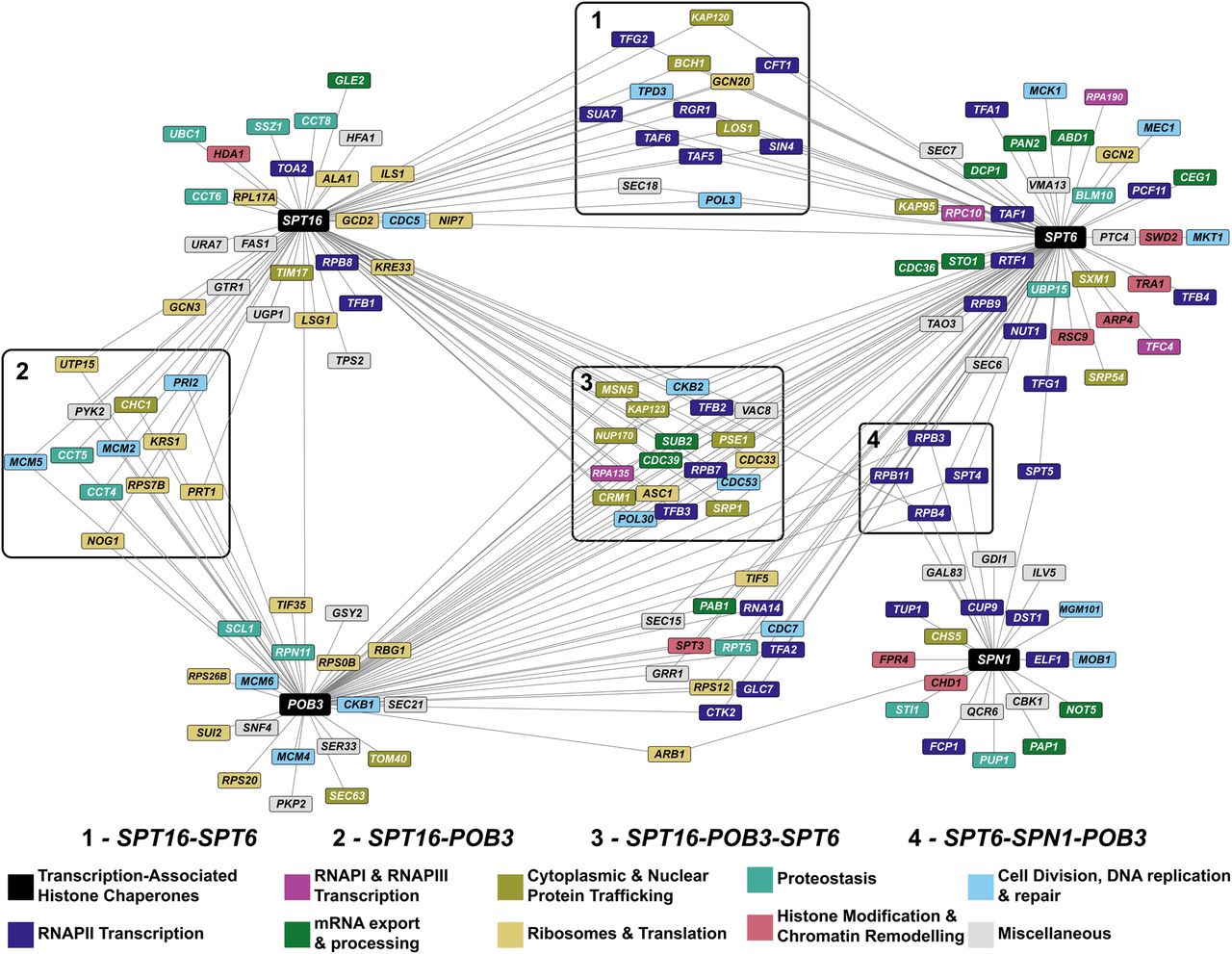
To understand the functional coordination among these histone chaperones, we asked which genes have a high ERC with SPT16, POB3, SPT6, and SPN1. Because the distribution of ERC values for each gene was different (Supplemental Fig. S7A), we standardized the results by calculating Z-scores for ERC values in each distribution (Supplemental Fig. S7B). In this section, we define a gene pair to have a high ERC value if it passes a Z-score cutoff of greater than or equal to 3.00, which selects roughly the top 1% of genes with each histone chaperone. We visualized genes that have a high ERC value with the histone chaperone genes as a network (Fig. 7; Supplemental File 5).
ERC network of transcription-associated histone chaperones identifies both known and putative functional interactions. Each node in the network is a gene, and an edge between two nodes indicates that the genes share an ERC value above a Z-score cutoff of three (SPT16, N = 72; POB3, N = 67; SPT6, N = 86; SPN1, N = 25). The ERC values in this network range from 6.94 to 16.01. Clusters in boxes represent nodes that are connected to two or more of the queried histone chaperones. The colors of the nodes represent the biological process associated with the gene. Biological process associations were manually curated from the Saccharomyces Genome Database. The miscellaneous category consists of genes involved in autophagy (GTR1), budding (CBK1 and TAO3), metabolism (FAS1, GAL83, GRR1, GSY2, HFA1, ILV5, PKP2, PYK2, QCR6, SER33, SNF4, TPS2, UGP1, and URA7), vesicular transport (GDI1, SEC15, SEC18, SEC21, SEC6, and SEC7), vacuole acidification (VMA13), and vacuole fusion (VAC8).
A high degree of interconnectivity within the histone chaperone network would indicate that many of the same genes share a high ERC with SPT16, POB3, SPT6, and SPN1. To test whether this network was more interconnected than would be expected by chance, we compared its global clustering coefficient to that of 10,000 randomly generated networks. Each sampled network was created by selecting one random gene corresponding to each of the four histone chaperone genes and collecting the top N ERC hits to match the number (N) passing the Z-score cutoff for that chaperone gene. The result is a network with the same number of edges. We found that the histone chaperone network had a higher global clustering coefficient than all 10,000 sampled networks, indicating a significant degree of connectivity between the genes that shared a high ERC value with the histone chaperones (Supplemental Fig. S7C). Because high ERC values suggest potentially shared functional roles, the strong interconnectivity observed in the histone chaperone network implies that these genes might participate in common biological processes or molecular pathways.
Several genes in the network are connected to two or more of the queried histone chaperones (Fig. 7, clusters 1–4). These shared genes support existing literature showing that transcription-associated histone chaperones functionally coordinate with one another (Viktorovskaya et al. 2021; López-Rivera et al. 2022; Warner et al. 2024). Genes involved in the nuclear import of proteins, RNAPII subunits, and transcription elongation factors have a high ERC value with SPT16, POB3, and SPT6 (Fig. 7, cluster 3). In addition to their roles in transcription elongation, SPT16 and SPT6 have been implicated in transcription initiation, and expectedly, several genes involved in the process (Fig. 7, blue nodes in the cluster 1) have a high ERC value with both histone chaperones (Biswas et al. 2005; Doris et al. 2018). Genes encoding the DNA replication factors Pol3p and Pol30p have a high ERC value with SPT16, POB3, and SPT6, and genes encoding subunits in the Mcm2-7 complex have a high ERC value with FACT. This is consistent with Spt6p, Pob3p, and Spt16p having functions in DNA replication (Formosa and Winston 2020; Miller and Winston 2023; Wang et al. 2023). Thus, the ERC network indicates functional interactions between the histone chaperones and proteins central to nuclear processes impacted by chromatin structure. However, the absence of an edge between a histone chaperone and another node in this network does not imply a lack of functional connection. For example, although Spt6p physically interacts with Mcm2p and Mcm4p and has been implicated in DNA replication, an edge was not drawn between the two as the ERC values were just under the Z-score cutoff (Z-scoreSPT6-MCM2 = 2.88 and Z-scoreSPT6-MCM4 = 2.95) (Supplemental File 6; Miller and Winston 2023). We do note that Spn1p, which physically interacts with Spt6p in the transcription elongation complex (Farnung 2025), has a narrower ERC value distribution (Supplemental Fig. S7A) and is less connected to other nodes in the network.
We also asked if the network suggests previously unexpected connections between transcription-associated histone chaperones and other biological processes. Some translation and ribosome-associated genes have a high ERC value with SPT16 and POB3 (Fig. 7, yellow nodes in cluster 2). There are even broader relationships captured by the edges between the black and gray nodes. These edges suggest relationships between proteins involved in transcription-associated histone chaperoning with functions involved in metabolism and vesicular transport, among others. This may be because of the direct relationships between the histone chaperones and these other functions, or individual proteins on either side of these edges may play a noncanonical role elsewhere that is contributing to the high ERC score. These observations highlight the power of ERC analysis to generate hypotheses to examine underappreciated relationships between factors in pathways thought to be disparate.
Discussion
ERC2.0 allows for faster and more accurate computation of ERC for large species data sets. We show that for a data set of 343 yeast species or 120 mammalian species, there is an improvement in predictive power over using the S. cerevisiae and Homo sapiens STRING databases alone. The new methodology provides increased power over older methods owing to improvements to normalization of rates (removing heteroskedasticity) and correlation measures (Fisher transformation), which also allow better comparisons between ERC data sets calculated in different taxonomic groups. Most gained power, however, comes from using more species. As more species are added, there seems to be a plateau of power above 147 species in yeast. We attribute some of this to the fact that we are limited to annotations in S. cerevisiae, and some of the high-scoring pairs in the 196–343 species bins are specific to clades outside of S. cerevisiae and, therefore, would not be recorded as a true positive. There could also be a plateau because approximately 100 species are enough to capture the majority of variation in selective environments encountered in that taxonomic group.
ERC can be used to rank genome-wide interactions with a gene/complex/pathway of interest to identify candidate interactors and has had great success predicting previously unknown functional relationships valuable to experimental biologists (Clark et al. 2013; Findlay et al. 2014; Priedigkeit et al. 2015; Raza et al. 2019; Kowalczyk et al. 2021). We also built ERC-based functional gene networks using MCL clustering, which suggest new cofunctional interactions and discovered a discrepancy in the types of processes and functions captured by the highest-scoring edges for yeast and mammals. In the yeast networks, we saw a very strong cluster of transcription/translation-related genes, whereas the mammalian network captured more macrolevel terms, such as sperm motility. These differences could indicate a difference in the variation of evolutionary pressures experienced by single-celled yeast versus multicellular mammals. This pattern could also be an artifact of the much greater evolutionary distance captured in the yeast species, in which the majority of overlap between orthologous genes is captured by essential processes such as transcription. The latter hypothesis will require further exploration of individual clades within the 343-yeast data set by splitting the phylogeny into different subgroups of species, running ERC on each individually, and assessing the differences in FtERC scores for a given pair.
We introduced the application of a logistic regression model to make functional predictions that could be applied for any set of annotations either from a database or from a custom annotation specific to a given study. We showed that for the 343-yeast species data set, there is strong predictive power for two different annotation sets. There was less predictive power for the mammal ERC data set. However, across all three data sets, 60% of the terms still performed with an AUC greater than 0.8, which indicates useful predictive power, and when employed in a predictive capacity, the user would know how well ERC performed on known genes (using ROC-AUC), before deciding whether to pursue new candidates suggested by the model.
We also showed the potential for ERC to aid researchers who work in nonmodel systems. First, the yeast data set makes many strong functional predictions for non–S. cerevisiae genes, many of which were validated after consulting annotations from non-cerevisiae species (Supplemental File 7). Second, the combination of ERC with logistic regression predictions allows for a reassessment of already annotated genes. In some cases, an experimental assay shows a gene/protein as a part of a specific function, which creates a confirmation bias whenever that gene/protein shows up in other contexts. By looking at candidate interactions in an unbiased way, such as ERC, we may uncover novel functions of proteins beyond their current functional annotations. Third, even more potential for nonmodel systems is demonstrated by the fact that ERC alone has predictive power approaching that of the entire STRING database (Supplemental Fig. S4A). Considering that STRING draws from experimental and computational predictions amassed over decades by thousands of researchers, having ERC as a rapid and low-cost alternative to making functional predictions will be a huge boost to nonmodel systems, especially because ERC only requires a collection of genomes from related species.
We also present a specific example in transcription-associated histone chaperone genes SPT16, POB3, SPT6, and SPN1 to illustrate the potential of ERC2.0-derived networks as a screen for novel interactions with genes/functions of interest. The network showed edges between genes involved in related functional categories as well as suggested relationships between functions such as translation and histone chaperoning that have not yet been explored.
In general, the studies presented here demonstrate how ERC2.0 could be applied in the systems biology field as a complement to other functional and interaction prediction methods. Although we have demonstrated the improvements in the ERC methodology and power to predict from ERC1.0 to ERC2.0, there are still limitations. These include concepts that challenge the field of comparative biology, such as how to deal with paralogous genes and gene trees that are incongruous with the species tree. The current study was also limited to yeast and mammal species; however, there are increasingly larger collections of related species genomes and more refined phylogenies, both of which promise greater power in those systems. Some more neglected taxonomic groups, such as bacteria and parasites, are of great interest to the health community, and ERC will allow researchers to rapidly identify functions and interactions within their genomes. These predictions would be especially useful in species that are more difficult to study in the laboratory.
Methods
Generating sequence alignments and gene trees
To calculate ERC, we first need to align and generate gene trees for each of the orthologous genes. For the data set of 343 yeast, the orthologous groups (OGs) were sourced from Shen et al. (2018). Each OG was then aligned using the MUSCLE alignment algorithm (Edgar 2004).
For the 120-mammal data set, the coding sequence alignments were generated from the whole-genome alignment found in the work of Hecker and Hiller (2020). First the protein-coding portions were extracted from the whole-genome alignment based on the canonical hg38 UCSC human gene models. The exons were then extracted from the MAF file using sub.msa from the RPHAST package (Hubisz et al. 2011). Next, the human reading frame was enforced, and stop codons were masked as gaps using codon.clean.msa in the RPHAST package. The sequences were translated into amino acid sequences using translate.msa in RPHAST.
Finally, gene trees for both sets of alignments were calculated using the estimatePhangornTreeAll wrapper for phangorn (Schliep 2011) wrapper included with RERconverge (Kowalczyk et al. 2021). To ensure congruent topologies, we supplied estimatePhangornTreeAll with a species master tree whose topology was used to generate the individual gene trees.
Efficient computation of correlations between features
The complexity of ERC combinations stems from the fact that each feature (e.g., orthologous gene set) exhibits a unique presence–absence pattern across species. These patterns are largely driven by technical artifacts and evolutionary gain/loss and are effectively random, resulting in each pair of features typically sharing a unique set of species. To compare tree-encoded data between features, they must first be standardized onto a common species set by pruning the original trees. Because this operation needs to be performed separately for every pair of features, it is critical to minimize the computational cost of tree operations.
Our implementation addresses this challenge in two ways. First, we exhaustively record every unidirectional path between nodes within the phylogenetic tree and store this information in a matrix. This allows us to store the information in a format that is conducive to efficient computations. This is essential for quick pruning because we are now able to precompute all possible paths resulting from any pruning operation for each tree and store them in a feature-by-path matrix. Second, we construct an indexing structure that, given a common species set, quickly retrieves the subset of paths corresponding to the tree pruned to match that set. This approach allows for a matrix indexing lookup, returning the results as a vector for fast correlation computations.
These optimizations leverage highly efficient C-based tree operations provided as helper functions in the R (R Core Team 2024) TreeTools package (Smith 2019). As a result, our approach ensures that ∼80% of the running time is dedicated to actual correlation computations. The code implementing ERC2.0 is available at its repository on GitHub (see Software availability).
Creating clustered ERC networks
To create clustered networks from ERC scores, we first subset the genome-by-genome matrices, both mammalian and yeast, to only Fisher-transformed values of 10 or greater. We then imported the data into Cytoscape (Shannon et al. 2003) and clustered using the clusterMaker2 (Morris et al. 2011) MCL clustering algorithm. We kept the inflation parameter at 2.5 and removed edges between clusters for visualization purposes. The clusters were then colored by MCL cluster number along a continuous scale.
We performed GO biological process enrichment analyses on all clusters with at least 10 members for both data sets using the Gene Ontology PANTHER enrichment analysis tool (Supplemental Files 2, 4; The Gene Ontology Consortium 2000; Thomas et al. 2022).
Generating glmnet models and predictions
We used the glmnet (Tay et al. 2023) R package to perform 10-fold cross-validation across two different annotation data sets collected from Enrichr (Kuleshov et al. 2016) for each of the ERC matrices. We used the GO-BP and KEGG pathways annotation data sets.
We performed two iterations of cv.glmnet for the yeast ERC matrix. The first was run with the entirety of the 12,552 orthologous genes as the input for the model. In the second matrix, only genes that are in both the ERC matrix and the STRING database were used, leaving the matrix with 4423 genes. The STRING-matched ERC matrix was also used to predict annotations for the non–S. cerevisiae orthologous genes (Supplemental Table S2), which was run using the command
Software availability
The source code for ERC2.0 can be found in Supplemental Code, as well as at GitHub (https://github.com/nclark-lab/erc).
Data access
The ERC data generated in this study have been submitted to Dryad (https://datadryad.org/) under the identifier dryad.6m905qg8q. All output data can also be found in output_data.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
The support and resources from the Center for High-Performance Computing at the University of Utah are gratefully acknowledged. We acknowledge key funding from the National Institutes of Health as R01 HG009299 to J.H.L. and N.C. and R35 GM141964 to K.M.A.
Author contributions: Conceptualization, data curation, formal analysis, software, validation, visualization, writing were by J.H.L. Software and reviewing and editing were by G.H.M. Data curation, formal analysis, investigation, visualization, and writing were by A.G. Data curation, formal analysis, investigation, visualization, and writing were by A.M.F. Methodology and software were by R.P. Formal analysis, funding acquisition, supervision, and writing were by K.M.A. Software and reviewing and editing were by M.S. Conceptualization, funding acquisition, methodology, project administration, resources, software, supervision, and writing were by N.C. Conceptualization, formal analysis, funding acquisition, methodology, resources, software, supervision, validation, and writing were by M.C.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280586.125.
References
- ↵Biswas D, Yu Y, Prall M, Formosa T, Stillman DJ. 2005. The yeast FACT complex has a role in transcriptional initiation. Mol Cell Biol 25: 5812–5822. 10.1128/MCB.25.14.5812-5822.2005
- ↵Brunette GJ, Jamalruddin MA, Baldock RA, Clark NL, Bernstein KA. 2019. Evolution-based screening enables genome-wide prioritization and discovery of DNA repair genes. Proc Natl Acad Sci 116: 19593–19599. 10.1073/pnas.1906559116
- ↵Chen J, Aronow BJ, Jegga AG. 2009. Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinformatics 10: 73. 10.1186/1471-2105-10-73
- ↵Clark NL, Alani E, Aquadro CF. 2012. Evolutionary rate covariation reveals shared functionality and coexpression of genes. Genome Res 22: 714–720. 10.1101/gr.132647.111
- ↵Clark NL, Alani E, Aquadro CF. 2013. Evolutionary rate covariation in meiotic proteins results from fluctuating evolutionary pressure in yeasts and mammals. Genetics 193: 529–538. 10.1534/genetics.112.145979
- ↵Doris SM, Chuang J, Viktorovskaya O, Murawska M, Spatt D, Churchman LS, Winston F. 2018. Spt6 is required for the fidelity of promoter selection. Mol Cell 72: 687–699.e6. 10.1016/j.molcel.2018.09.005
- ↵Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. 10.1093/nar/gkh340
- ↵Farnung L. 2025. Chromatin transcription elongation: a structural perspective. J Mol Biol 437: 168845. 10.1016/j.jmb.2024.168845
- ↵Findlay GD, Sitnik JL, Wang W, Aquadro CF, Clark NL, Wolfner MF. 2014. Evolutionary rate covariation identifies new members of a protein network required for Drosophila melanogaster female post-mating responses. PLoS Genet 10: e1004108. 10.1371/journal.pgen.1004108
- ↵Fisher RA. 1915. Frequency distribution of the values of the correlation coefficient in samples from an indefinitely large population. Biometrika 10: 507–521. 10.2307/2331838
- ↵Formosa T, Winston F. 2020. The role of FACT in managing chromatin: disruption, assembly, or repair? Nucleic Acids Res 48: 11929–11941. 10.1093/nar/gkaa912
- ↵Forsythe ES, Williams AM, Sloan DB. 2021. Genome-wide signatures of plastid-nuclear coevolution point to repeated perturbations of plastid proteostasis systems across angiosperms. Plant Cell 33: 980–997. 10.1093/plcell/koab021
- ↵The Gene Ontology Consortium. 2000. Gene Ontology: tool for the unification of biology. Nat Genet 25: 25–29. 10.1038/75556
- ↵Goh CS, Bogan AA, Joachimiak M, Walther D, Cohen FE. 2000. Co-evolution of proteins with their interaction partners. J Mol Biol 299: 283–293. 10.1006/jmbi.2000.3732
- ↵Hammond CM, Strømme CB, Huang H, Patel DJ, Groth A. 2017. Histone chaperone networks shaping chromatin function. Nat Rev Mol Cell Biol 18: 141–158. 10.1038/nrm.2016.159
- ↵Hecker N, Hiller M. 2020. A genome alignment of 120 mammals highlights ultraconserved element variability and placenta-associated enhancers. GigaScience 9: giz159. 10.1093/gigascience/giz159
- ↵Hotaling S, Kelley JL, Frandsen PB. 2021. Toward a genome sequence for every animal: Where are we now? Proc Natl Acad Sci 118: e2109019118. 10.1073/pnas.2109019118
- ↵Hubisz MJ, Pollard KS, Siepel A. 2011. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief Bioinform 12: 41–51. 10.1093/bib/bbq072
- ↵Kanehisa M. 2019. Toward understanding the origin and evolution of cellular organisms. Protein Sci 28: 1947–1951. 10.1002/pro.3715
- ↵Kowalczyk A, Gbadamosi O, Kolor K, Sosa J, Andrzejczuk L, Gibson G, St Croix C, Chikina M, Aizenman E, Clark N, 2021. Evolutionary rate covariation identifies SLC30A9 (ZnT9) as a mitochondrial zinc transporter. Biochem J 478: 3205–3220. 10.1042/BCJ20210342
- ↵Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97. 10.1093/nar/gkw377
- ↵Little J, Chikina M, Clark NL. 2024. Evolutionary rate covariation is a reliable predictor of co-functional interactions but not necessarily physical interactions. eLife 12: RP93333. 10.7554/eLife.93333.3
- ↵López-Rivera F, Chuang J, Spatt D, Gopalakrishnan R, Winston F. 2022. Suppressor mutations that make the essential transcription factor Spn1/Iws1 dispensable in Saccharomyces cerevisiae. Genetics 222: iyac125. 10.1093/genetics/iyac125
- ↵Lovell SC, Robertson DL. 2010. An integrated view of molecular coevolution in protein–protein interactions. Mol Biol Evol 27: 2567–2575. 10.1093/molbev/msq144
- ↵Miller CLW, Winston F. 2023. The conserved histone chaperone Spt6 is strongly required for DNA replication and genome stability. Cell Rep 42: 112264. 10.1016/j.celrep.2023.112264
- ↵Miller CLW, Warner JL, Winston F. 2023. Insights into Spt6: a histone chaperone that functions in transcription, DNA replication, and genome stability. Trends Genet 39: 858–872. 10.1016/j.tig.2023.06.008
- ↵Morris JH, Apeltsin L, Newman AM, Baumbach J, Wittkop T, Su G, Bader GD, Ferrin TE. 2011. clusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC Bioinformatics 12: 436. 10.1186/1471-2105-12-436
- ↵Paredes-Sánchez FA, Sifuentes-Rincón AM, Segura Cabrera A, García Pérez CA, Parra Bracamonte GM, Ambriz Morales P. 2015. Associations of SNPs located at candidate genes to bovine growth traits, prioritized with an interaction networks construction approach. BMC Genet 16: 91. 10.1186/s12863-015-0247-3
- ↵Partha R, Kowalczyk A, Clark NL, Chikina M. 2019. Robust method for detecting convergent shifts in evolutionary rates. Mol Biol Evol 36: 1817–1830. 10.1093/molbev/msz107
- ↵Pazos F, Valencia A. 2001. Similarity of phylogenetic trees as indicator of protein–protein interaction. Protein Eng 14: 609–614. 10.1093/protein/14.9.609
- ↵Priedigkeit N, Wolfe N, Clark NL. 2015. Evolutionary signatures amongst disease genes permit novel methods for gene prioritization and construction of informative gene-based networks. PLoS Genet 11: e1004967. 10.1371/journal.pgen.1004967
- ↵Ramani AK, Marcotte EM. 2003. Exploiting the co-evolution of interacting proteins to discover interaction specificity. J Mol Biol 327: 273–284. 10.1016/S0022-2836(03)00114-1
- ↵Raza Q, Choi JY, Li Y, O'Dowd RM, Watkins SC, Chikina M, Hong Y, Clark NL, Kwiatkowski AV. 2019. Evolutionary rate covariation analysis of E-cadherin identifies Raskol as a regulator of cell adhesion and actin dynamics in Drosophila. PLoS Genet 15: e1007720. 10.1371/journal.pgen.1007720
- ↵R Core Team. 2024. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/.
- ↵Robert F, Jeronimo C. 2023. Transcription-coupled nucleosome assembly. Trends Biochem Sci 48: 978–992. 10.1016/j.tibs.2023.08.003
- ↵Saha S, Prasad A, Chatterjee P, Basu S, Nasipuri M. 2019. Protein function prediction from dynamic protein interaction network using gene expression data. J Bioinform Comput Biol 17: 1950025. 10.1142/S0219720019500252
- ↵Schliep KP. 2011. phangorn: phylogenetic analysis in R. Bioinformatics 27: 592–593. 10.1093/bioinformatics/btq706
- ↵Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. 10.1101/gr.1239303
- ↵Sharan R, Ulitsky I, Shamir R. 2007. Network-based prediction of protein function. Mol Syst Biol 3: 88. 10.1038/msb4100129
- ↵Shen X-X, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh KV, Haase MAB, Wisecaver JH, Wang M, Doering DT, 2018. Tempo and mode of genome evolution in the budding yeast subphylum. Cell 175: 1533–1545.e20. 10.1016/j.cell.2018.10.023
- ↵Smith MR. 2019. TreeTools: create, modify and analyse phylogenetic trees. Comprehensive R Archive Network. 10.32614/CRAN.package.TreeTools
- ↵Steenwyk JL, Phillips MA, Yang F, Date SS, Graham TR, Berman J, Hittinger CT, Rokas A. 2022. An orthologous gene coevolution network provides insight into eukaryotic cellular and genomic structure and function. Sci Adv 8: eabn0105. 10.1126/sciadv.abn0105
- ↵Sun J, Jia P, Fanous AH, van den Oord E, Chen X, Riley BP, Amdur RL, Kendler KS, Zhao Z. 2010. Schizophrenia gene networks and pathways and their applications for novel candidate gene selection. PLoS One 5: e11351. 10.1371/journal.pone.0011351
- ↵Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, 2019. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47: D607–D613. 10.1093/nar/gky1131
- ↵Tay JK, Narasimhan B, Hastie T. 2023. Elastic net regularization paths for all generalized linear models. J Stat Softw 106: 1–31. 10.18637/jss.v106.i01
- ↵Thomas PD, Ebert D, Muruganujan A, Mushayahama T, Albou L-P, Mi H. 2022. PANTHER: making genome-scale phylogenetics accessible to all. Protein Sci 31: 8–22. 10.1002/pro.4218
- ↵Viktorovskaya O, Chuang J, Jain D, Reim NI, López-Rivera F, Murawska M, Spatt D, Churchman LS, Park PJ, Winston F. 2021. Essential histone chaperones collaborate to regulate transcription and chromatin integrity. Genes Dev 35: 698–712. 10.1101/gad.348431.121
- ↵Wang Y, Robinson PS, Coorens THH, Moore L, Lee-Six H, Noorani A, Sanders MA, Jung H, Katainen R, Heuschkel R, 2023. APOBEC mutagenesis is a common process in normal human small intestine. Nat Genet 55: 246–254. 10.1038/s41588-022-01296-5
- ↵Warner JL, Lux V, Veverka V, Winston F. 2024. The histone chaperone Spt6 controls chromatin structure through its conserved N-terminal domain. bioRxiv 10.1101/2024.11.25.625227
- ↵Xiong W, Xie L, Zhou S, Guan J. 2014. Active learning for protein function prediction in protein–protein interaction networks. Neurocomputing 145: 44–52. 10.1016/j.neucom.2014.05.075
- ↵Yosef N, Kupiec M, Ruppin E, Sharan R. 2009. A complex-centric view of protein network evolution. Nucleic Acids Res 37: e88. 10.1093/nar/gkp414
- ↵Zhu W, Hou J, Phoebe Chen Y-P. 2010. Semantic and layered protein function prediction from PPI networks. J Theor Biol 267: 129–136. 10.1016/j.jtbi.2010.08.005