
Abstract
Bacterial genomes are notable in that they contain large numbers of lineage-restricted (“ORFan”) genes, which have been postulated to originate from either horizontal transfer, rapid divergence from pre-existing genes, or de novo emergence from noncoding sequences. We assess the body of research that explores each of these hypotheses and demonstrate that the mystery of the origin of bacterial ORFans still remains unresolved. Nonetheless, bacteria offer several unique avenues for research into the process and mechanics of gene birth at a resolution not feasible in other organisms. Both their amenability to experimental evolutionary analysis and their strain-level variation in gene content foster investigations of how noncoding sequences acquire expression and transition into functionality—questions central to the origin of phenotypic novelty.
Most new genes are postulated to have formed through a process of duplication and divergence (Chen et al. 2013; Tautz 2014). But if genes arise only from pre-existing gene sequences, one would expect all genes to have homologs. Since the first sequencing projects, researchers have been struck by the occurrence in almost every genome of “ORFan” genes, those that lack homologs outside of the taxon in which they are found. Numerous explanations have been provided for the existence of ORFans (aka “orphans”), including rapid divergence from functional genes, inadequacies of search strategies, and de novo birth from noncoding sequences without a genic precursor (Khalturin et al. 2009; Tautz and Domazet-Lošo 2011; Van Oss and Carvunis 2019).
The mystery surrounding the origin of ORFans is fundamental to the evolution of bacteria, whose genomes contain numerous species- and strain-restricted genes (Siew and Fischer 2003; Daubin and Ochman 2004a; Siew et al. 2004) but exhibit very low frequencies of gene duplication (Treangen and Rocha 2011; Tria and Martin 2021). Despite wide acknowledgement of the presence of ORFans in bacteria, few studies have attempted to investigate their emergence (Table 1). Of particular note is the phenomenon of de novo gene birth from noncoding sequences, a mode of gene origin that has been investigated in depth in diverse eukaryotic taxa (Begun et al. 2007; Cai et al. 2008; Knowles and McLysaght 2009; McLysaght and Guerzoni 2015; Zhang et al. 2019; Zhuang et al. 2019) but has gone virtually unexplored in bacteria (Vakirlis and Kupczok 2024). Here, we analyze the efforts undertaken to discover the contribution of different evolutionary processes to the enormous ORFan gene pool in bacteria, with a special focus on de novo gene birth.
Studies investigating the origin of ORFans in bacteria
| Year of publication | Hypotheses investigated | Taxa investigated | Reference |
|---|---|---|---|
| 2024 | Emergence from noncoding sequences, frameshifting, phage origin | Various taxa in human gut microbiome | Vakirlis and Kupczok 2024 |
| 2023 | Emergence from noncoding sequences | Bacillus genus | Karlowski et al. 2023 |
| 2021 | Frameshifting | Escherichia coli | Watson et al. 2021 |
| 2015 | Phage origin | Various | Lobb et al. 2015 |
| 2010 | Emergence from intergenic sequences, frameshifting, phage origin | Various | Yomtovian et al. 2010 |
| 2009 | Phage origin | Various | Cortez et al. 2009 |
| 2006 | Phage origin | Various | Yin and Fischer 2006 |
| 2004 | Phage origin | Escherichia coli | Daubin and Ochman 2004a |
ORFans as artifacts
Our discussion of bacterial ORFans focuses only on protein-coding annotated genes, noting that not all annotated genes are functional (Ghatak et al. 2019) and that conventional annotations can miss functional genes (Armengaud 2009; de Souza et al. 2009). Identifying the ORFans in an organism's genome involves searching all of its annotated protein sequences against the proteins encoded by all other taxa (“outgroups”) and retaining only those that have no recognizable homolog among outgroups (McLysaght and Hurst 2016; Vakirlis and McLysaght 2019). Because this approach depends on the robustness of the search strategy, it admits the possibility that homologs to a putative ORFan might exist in the database but that the search has failed to identify them. Therefore, before addressing issues concerning ORFan origins, it is essential to rule out such false positives. A conventional protein BLAST search could fail to return hits owing to inadequate annotation, inappropriate application of an e-value cutoff, or failure to account for remote homology (Fig. 1A).
Workflow for detecting ORFans and de novo–emerged genes. (A) An augmented search strategy for detecting actual and excluding artifactual ORFan genes. (B) The gold standard of de novo gene detection. (Created with BioRender; https://www.biorender.com/.)
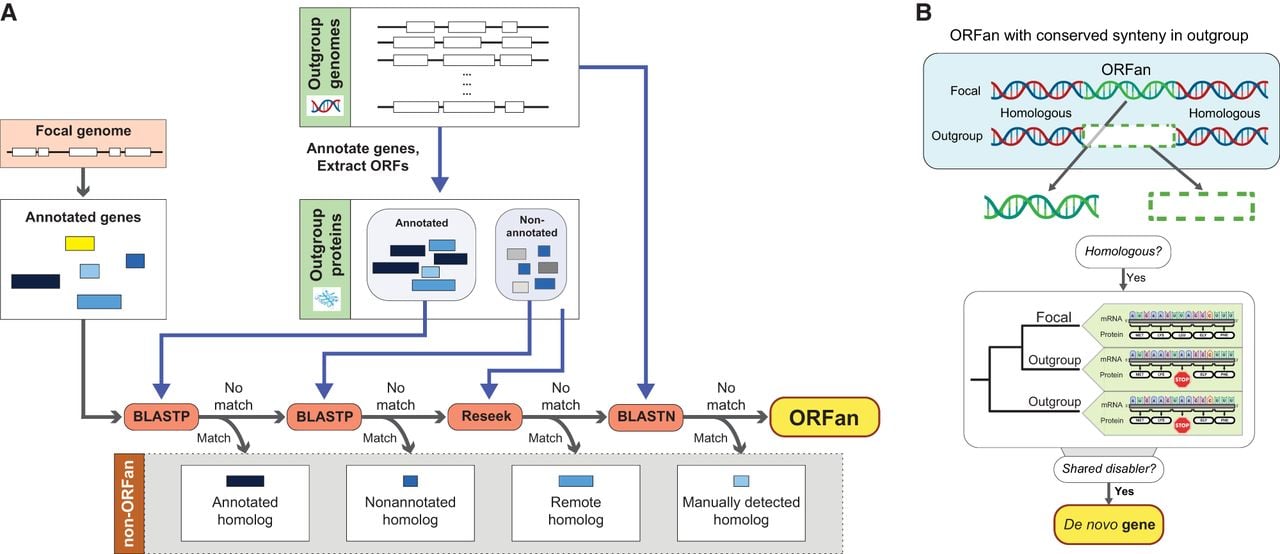
Annotation inadequacies
A protein homologous to a putative ORFan might possibly exist in the outgroup database but not be annotated as such in any of the surveyed genomes. Short proteins are particularly vulnerable to this concern because they often evade detection by conventional annotation programs (Storz et al. 2014; Tonkin-Hill et al. 2023). As a remedy, a protein BLAST search should be performed against all translated open reading frames, not just the annotated genes, in outgroup genomes (Fig. 1A). This procedure can be implemented either by searching all genome sequences with tFASTy and excluding frameshifted or truncated proteins from the results (Pearson et al. 1997; Karlowski et al. 2023) or by extracting all ORFs from outgroup genomes and conducting a protein BLAST search against their translated products.
Reliance on e-value thresholds
In a typical BLAST search, an e-value-based cutoff is implemented to distinguish genuine matches between the query and target sequences from those that are spurious (Vakirlis and McLysaght 2019). But because of the short length of some proteins, even a high-confidence hit might fail to return a low e-value owing to the inherently greater possibility of spurious short alignments. One remedy would be to manually curate protein alignments with higher e-values to rule out false-positive hits (Kuchibhatla et al. 2014). An alternative strategy is to search against the nucleotide sequences of outgroup genomic regions that are in conserved synteny with the putative ORFan. This restriction significantly reduces the size of the search database from entire genomes to, at most, several kilobases for each genome in which a hit is found, making it computationally manageable to incorporate a smaller “word size” parameter, thus leading to significant gains in sensitivity. The resulting alignments can then be manually curated to cull false positives.
Recognition of remote homology
Although sequence homology decays quickly between rapidly diverging homologs, structural homology can persist beyond the point at which there is virtually no sequence similarity (Weisman et al. 2020; Stern and Han 2022). Remote homology has been detected by searching hidden Markov model profiles derived from gene alignments against outgroup sequence or profile databases (Remmert et al. 2011; Lobb et al. 2015). The recent advent of protein structure prediction tools, such as ESMFold (Lin et al. 2023), coupled with structure-based search strategies (Edgar 2024; van Kempen et al. 2024) can potentially aid in the recovery of very distant homologs.
The origins of ORFan genes
A lack of homologs after these exhaustive search strategies leads to the more provocative issue of determining how such an ORFan gene actually emerges. There are three routes by which ORFans can arise in bacterial genomes: (1) de novo evolution from noncoding sequences (including noncoding alternative reading frames of functional genes), (2) extreme divergence from other functional genes, and (3) horizontal transfer from a source not present in the database (Daubin and Ochman 2004a; Yomtovian et al. 2010; Vakirlis and Kupczok 2024; Pereira et al. 2025).
De novo gene birth is the most tractable and traceable of these three routes because it is possible, in principle, to identify the ancestral noncoding sequence from which the new gene formed. Based on extensive work in eukaryotes, a “gold standard” for detection of such genes has been proposed (Vakirlis and McLysaght 2019; Zhang et al. 2019; Vakirlis et al. 2022).
The gold standard of de novo gene detection
To unambiguously establish that a gene arose de novo, the noncoding sequence that gave rise to the ORFan needs to be detected. This first requires identification of the template sequence that retains the ancestral noncoding status in outgroup genomes (Fig. 1B). To increase confidence in homology inference, the noncoding sequence in the outgroup genomes should be in conserved synteny with the ORFan, with its inert status confirmed by the lack of a start codon and/or interruption by stop codons or frameshift mutations.
Note that the mere presence of a homologous noncoding sequence in an outgroup genome is not sufficient to establish a de novo origin of the ORFan because it is possible that the outgroup sequence is noncoding as a result of gene inactivation after the ORFan emerged. To exclude this possibility, not only must the noncoding sequence display homology with the ORFan sequence but its “disabling” mutation (i.e., at least one start-codon disrupting mutation, stop-codon or frameshift mutation) must occur in two or more outgroup lineages (Fig. 1B). The rationale for this criterion is that when the same disabling mutation is present in at least two outgroup lineages, the most parsimonious reconstruction of events is that the noncoding status of the sequence is ancestral to the ORFan.
Reported cases of de novo gene birth in bacterial genomes
Only two studies have attempted to investigate de novo birth of bacterial ORFans by tracing their sequences to noncoding ancestors. In an analysis of the genus Bacillus, Karlowski et al. (2023) traced 331 ORFans to syntenic noncoding sequences in outgroup genomes. However, this study did not establish whether these noncoding sequences share a disabling mutation in more than one distinct lineage, thereby raising uncertainty about whether they are ancestral to the ORFans or simply represent cases in which a pre-existing gene has become pseudogenized in the outgroup genomes.
Recently, Vakirlis and Kupczok (2024) traced 1075 species-specific ORFans to their putative noncoding ancestral sequences. The authors were able to identify the corresponding syntenic regions in at least two outgroup genomes, one from the same species and one from a close outgroup, but because of the small number of outgroup genomes bearing the putative noncoding sequence, they could not investigate whether the identical disabling mutation was present in two outgroup lineages. As such, their methodology does not exclude the possibility that ORFan genes emerged via rapid divergence from pre-existing genes or that their status as ORFans resulted from loss in multiple lineages via repeated pseudogenization. To acknowledge this uncertainty, the authors refer to these as “de novo gene candidates,” but considering the massive size of their database (4.7 million protein families from 4644 species in the human gut microbiome), the fact that de novo gene candidacy was assigned to only 0.2% of their recognized ORFans underscores the technical limitations in detecting this mode of gene birth in bacterial genomes.
Difficulties intrinsic to detecting de novo gene birth
It is only practical to apply the gold standard of de novo gene detection when sequences evolve slowly and are distributed across multiple closely related lineages. Furthermore, bacterial genomes are buffeted by a pervasive deletional bias that removes noncoding regions (Mira et al. 2001; Kuo et al. 2009), thereby rendering it less likely that noncoding sequences would be conserved across multiple lineages. For bacteria that engage in frequent interspecific gene transfer, even the gold-standard criterion demanding the same debilitating mutation(s) in two outgroup lineages may, in fact, be too permissive and should instead require their presence in more than two outgroup lineages to establish the ancestral state. The complicated nature of phylogenetic inferences in bacteria, combined with the low retention of noncoding sequences, suggests that evidence of real de novo genes is uncommon in these genomes.
As an alternative to the stringent requirements of the gold standard, Vakirlis et al. (2024) have applied an ancestral sequence reconstruction method to detect noncoding ancestral sequence states. Although this might circumvent the need for identifying shared disabling mutation(s), it nonetheless requires the retention of noncoding sequences across multiple lineages, which is feasible for the yeast species investigated in the study but is less common in bacteria.
It is notable that many of the problems facing the inference that a gene emerged de novo apply equally to discerning whether these genes arose through rapid divergence from functional genes that retain no similarity to the ORFan sequence. Despite these challenges, two indirect methods of investigating the rapid divergence scenario have been proposed. First, if the rate of sequence evolution of a protein can be calculated, one can infer the likelihood that a homolog to the protein exists at a given evolutionary distance, but the sequence-level homology has decayed past the point of recognition (Weisman et al. 2020; Barrera-Redondo et al. 2023). However, such an approach requires the protein to be present in at least three species to calculate a rate of sequence evolution, which limits its applicability to species-specific ORFans. Furthermore, proteins may experience lineage-specific changes in their rates of evolution, compromising the utility of such an approach (Prabh and Tautz 2021).
Second, Vakirlis et al. (2020a) propose a synteny-informed method to calculate the overall rate of genes emerging via sequence divergence across a genome. Because bacterial ORFans can rarely be traced to a conserved syntenic region found in outgroups, it remains unclear whether this approach would be useful in case of bacteria.
Native versus foreign origins of ORFan genes
Although the competing hypotheses that account for bacterial ORFans—sequence divergence, de novo gene birth, and gene loss—can only rarely be resolved, broad questions relating to the source of ORFan genes can still be addressed. Specifically, do ORFans arise locally, from sequences already present in the genome, or do they arise via transfer from external sources not present in the database?
It has been posited that ORFans originate in and are acquired from bacteriophages (Daubin and Ochman 2004a,b), an hypothesis bearing some appeal because bacteriophages comprise the largest group of biological entities and are underrepresented in the databases, they serve as agents of gene transfer, and their high mutation rates can generate an extraordinary amount of genetic novelty (Hendrix et al. 1999; Sanjuán et al. 2010; Bar-On et al. 2018; Benler and Koonin 2021). Moreover, it has been known since the 1950s that “lysogenic conversion genes” introduced by temperate bacteriophages can confer beneficial traits to bacteria (Lwoff 1953; Canchaya et al. 2003). Remnants of phage infection are evident in most bacterial genomes and are implicated in a variety of cellular functions (Wang et al. 2010; Bobay et al. 2014; Bondy-Denomy and Davidson 2014), including in defense (Touchon et al. 2017) and in maintaining cell morphology (Randich et al. 2019).
Phages are unquestionably involved in bacterial gene transfer, but their role as a source of new bacterial genes is uncertain. The contribution of phages to the repertoire of bacterial ORFans has been investigated by two complementary methods. One method is to identify the fraction of ORFans that are traceable to existing phage genes, and in the first study of its kind, Yin and Fischer (2006) reported that ∼3% of all bacterial ORFans have a phage homolog. Although this value is certainly an underestimate on account of early database limitations, Vakirlis and Kupczok (2024) recently reported that only 5% of bacterial ORFans had phage homologs, even after they applied a low stringency threshold. In contrast to what might be expected if ORFans originated in phage, they found that the likelihood of phage homology increases with persistence of a gene, such that more conserved genes are more likely to have a phage homolog. Lobb et al. (2015) reported a similar estimate of phage-traceable ORFans based on remote homology searches but with a significant, albeit minor, enrichment of viral processes among ORFan functional classes.
Alternatively, similarities in the sequence characteristics of ORFans and phage-encoded genes seeded the idea that many ORFan genes originated in phage, even if they share no observable homology with phage proteins. Compared with the majority of genes residing in a bacterial genome, both ORFans and phage genes are short and AT-rich and have atypical dinucleotide signatures (Daubin and Ochman 2004a,b). Subsequently, Cortez et al. (2009) found that 60% of bacterial ORFans are situated within clusters of genes that display atypical sequence compositions, which led them to deduce that ORFans within such clusters stemmed from events of horizontal transfer, with about one-half derived from viral and plasmid sources. In contrast to their results, Yomtovian et al. (2010) observed no significant similarities in amino acid composition between ORFans and phage proteins. Drawing on a much larger data set, Vakirlis and Kupczok (2024) reported that ORFans do not differ from conserved genes in their average GC content or other sequence properties, thereby undercutting the phage origin hypothesis and reinforcing the view of a local origin of ORFans.
Because the vast majority of the phage sequences remains unsampled, as evidenced by the wealth of taxa and genes identified with each new metagenomic or metaproteomic survey (Nayfach et al. 2019, 2021; Sberro et al. 2019; Durrant and Bhatt 2021; Fremin et al. 2022), it is difficult to completely discount the hypothesis that a sizeable fraction of ORFans originate in phage even in light of the low proportion of ORFans with phage homologs. Although the association between gene age and their similarity to phage sequences has been taken as evidence that phages are an unlikely source of newly emerged ORFans (Yin and Fischer 2006; Vakirlis and Kupczok 2024), this finding is a predicted consequence of sampling bias because younger, rarer bacterial genes are expected to be rarer in phage sequence space as well.
Because of the contradictory interpretations drawn from these studies, it is difficult to know how further sequence comparisons will help in resolving this debate. Even if, as previous studies report, ORFans manifest a lower GC content than conserved genes, a feature considered to be a hallmark of phage origin, this compositional bias is also observed in rapidly diverging genes owing to the inherent pattern of mutations in bacterial genomes (Schaaper and Dunn 1991; Sargentini and Smith 1994; Yamamura et al. 2000). Conversely, acquired genes eventually take on the sequence properties as their new genomic host, obscuring their origins (Daubin and Ochman 2004a,b). Limitations of sampling notwithstanding, only the consistent discovery of clear homologs to ORFan genes in viral databases can lend support to the phage-origin hypothesis.
Alternative routes of de novo gene birth in bacteria
Although the exact contribution of the different pathways to gene birth remains unclear, it is likely that each of the processes discussed so far have contributed to the generation of ORFan genes in bacteria, with vestiges of some gene-birth events traceable in extant genomes. Owing to their amenability to experimental evolutionary analysis and their strain-level variation in gene contents, bacteria offer the opportunity to identify forms of de novo gene birth that are not readily captured by conventional detection methods.
De novo transcription or translation
Detecting de novo gene birth need not rely only on the identification of new ORFs originating from noncoding regions. New genes can also arise as a result of transcription and/or translation of pre-existing ORFs that were previously unexpressed, a process that represents one of the more frequently detected routes of new gene birth (Fig. 2A; Grandchamp et al. 2023). Because these ORFans have homologous ORFs in outgroup genomes, they would be ignored based on conventional criteria (Fig. 1).
Alternative forms of de novo gene birth. (A) De novo emerged transcription of a pre-existing ORF. (B) De novo gene emergence from a frameshifted gene sequence. (Created with BioRender; https://www.biorender.com/.)
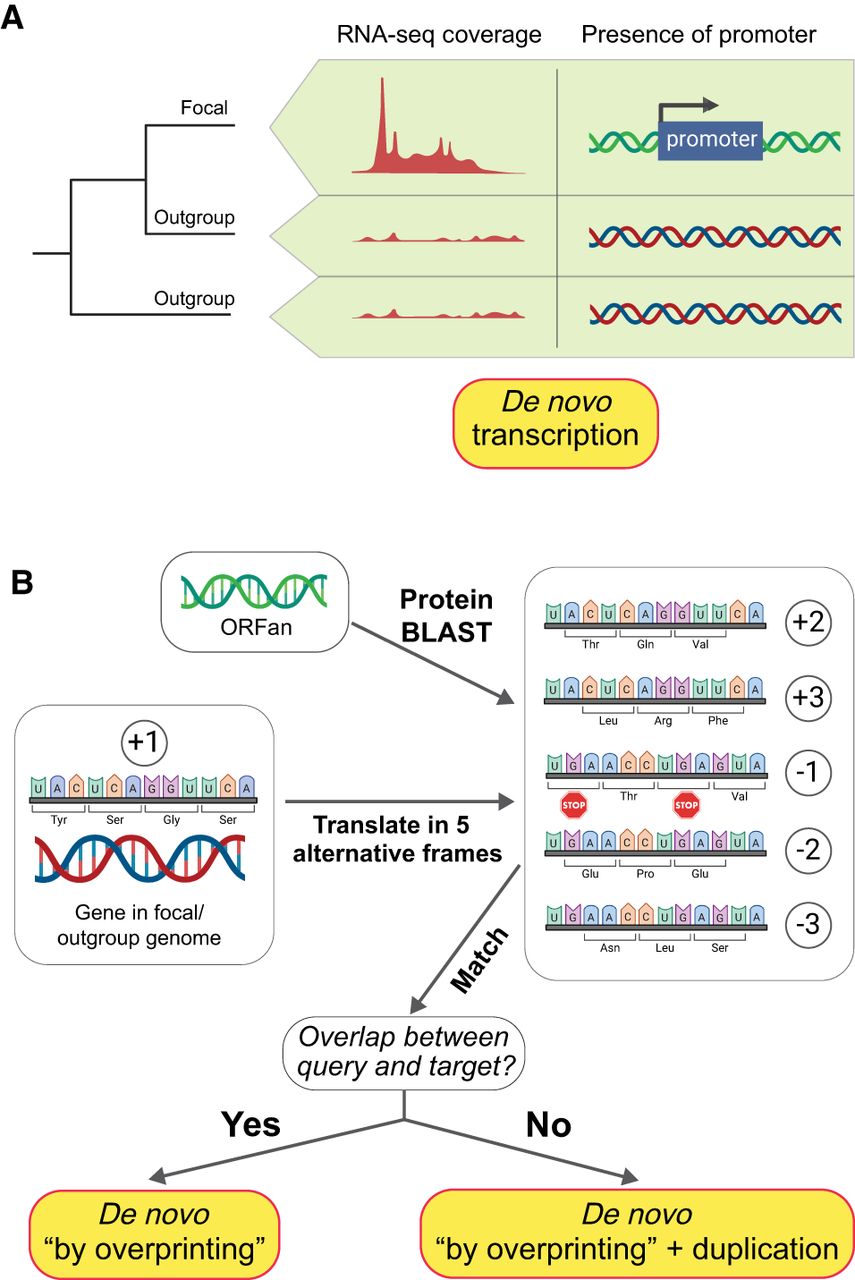
Merging comparative genomic and transcriptomic data allow detection of both the lineage-specific transcripts and the mutations responsible for the formation of new promoters (Blevins et al. 2021). Although bacterial promoters are often imprecise and difficult to detect (Coppens and Lavigne 2020; Lagator et al. 2022), and the absence of a canonical promoter or of transcription is not irrefutable evidence that a sequence is noncoding, the detection of novel expression and its causative regulatory sequences can be identified across short evolutionary timescales. Working with the Escherichia coli Long-Term Evolution Experiment (Lenski et al. 1991; Tenaillon et al. 2016; Good et al. 2017), a system in which the ancestral states of all genomes and each new mutation are known, we leveraged expression data assayed across a large number of growth conditions (Houser et al. 2015; Caglar et al. 2017; Tjaden 2023) to establish the de novo emergence of new transcripts and proteins (uz-Zaman et al. 2024). But because such idealized conditions are not available for natural populations, inferences about de novo gains in transcription or translation cannot be generalized across species.
Gene birth via overprinting
Paralleling the emergence of new transcription and translation of pre-existing open reading frames is the appearance of new genes from the noncoding reading frames of pre-existing genes (Fig. 2B). There is ample evidence that bacterial genes produce transcripts and proteins from alternative reading frames along both strands of DNA (Raghavan et al. 2012; Stringer et al. 2021; Smith et al. 2022) and that some of these proteins exhibit evidence of purifying selection, implying that they are functional (Ardern et al. 2020; Zehentner et al. 2020; Kreitmeier et al. 2022). That such products can serve as raw material for the formation of new genes (Ruiz-Orera et al. 2018) helps mitigate the idea that the lack of intergenic DNA in bacterial genomes limits the potential for new gene formation. Genes formed from within existing coding regions are apt to be easier to detect because their precursor sequences have a greater likelihood of being preserved over evolutionary timescales than do intergenic noncoding sequences. Also, because frameshifted proteins retain many properties of the protein encoded in the original coding frame (Bartonek et al. 2020), they may more easily transition to functionality compared with those originating from intergenic sequences.
The evolution of new genes via frameshifted overprinting has been widely investigated in viruses (Sabath et al. 2012; Pavesi 2021), and there are numerous cases of gene overlap in bacterial genomes (Rogozin et al. 2002; Wright et al. 2022). But owing to the very short length of most overlaps, often involving only the start and stop codons of adjacent genes, few represent cases of de novo gene birth (Wright et al. 2022). To date, only two studies have investigated the more extensive overlaps between annotated bacterial genes. In a study of chimeric genes in the E. coli pangenome, Watson et al. (2021) identified 767 gene families that contain at least one domain derived from the shifted frame of an annotated protein. Because this study focused solely on chimeric proteins, they described no cases in which a protein was derived exclusively from the shifted frame of another gene. However, this feature was considered in a survey of all species-specific genes in the gut microbiome in which 1.2% of ORFans (representing 7585 families) were derived from the frameshifting of other genes in the same genome (Vakirlis and Kupczok 2024). Although these genes represent unambiguous examples of de novo emergence from a previously noncoding sequence, their estimates suggest that this mode of de novo gene birth is a minor contributor to the pool of bacterial ORFans.
It is noteworthy that the frameshifted genes identified by Vakirlis and Kupczok (2024) did not overlap the reading frame from which they were derived, which is indicative of past gene duplication events, after which the two paralogs retain functionality in different frames. Similarly, a large fraction (31.5%) of the chimeric proteins reported by Watson et al. (2021) were nonoverlapping and could therefore be implicated in duplication events. Because this mode of gene origination requires paralogs to persist in the same genome, it is predictably rare in bacterial genomes owing to their very low retention of duplicated genes (Treangen and Rocha 2011).
Investigating the mechanics of de novo gene birth
The birth of a new gene from pre-existing noncoding sequences can be broadly conceptualized as having two phases: the acquisition of expression (and translation in the case of protein-coding genes) and the transition to functionality. Despite the complications accompanying the identification of fully formed de novo genes in bacteria, insights into the mechanisms of gene origin have been gained by studying these two phases separately.
Phase 1: the transition to expression
According to the proto-gene model of gene birth (Carvunis et al. 2012; Weisman and Eddy 2017), the functionality of gene sequences is preceded by their gain of expression and subsequent translation, such that the pool of expressed but nonfunctional proteins (“proto-genes”) harbored in each cell is the raw materials from which de novo genes arise. Recent genome-wide surveys of translation in a number of bacterial species have identified an abundance of novel proteins that cannot be detected by conventional gene annotation algorithms (Tables 2, 3; Baek et al. 2017; Hücker et al. 2017; Meydan et al. 2018, 2019; Weaver et al. 2019; Venturini et al. 2020; Stringer et al. 2021; Smith et al. 2022). Although translation of these proteins can be detected by ribosome profiling, their corresponding products mostly escape detection by mass spectrometry and western blots (VanOrsdel et al. 2018). For example, all but one of the studies listed in Table 2 failed to establish mass spectrometric evidence for 90% of the proteins detected by ribosome profiling, with three studies failing to find evidence for a single new protein resolved by ribosome profiling. Discrepancies in the detection of novel proteins have been attributed to their shorter lengths (which limits the generation of tryptic peptides), high hydrophobicity, and low stability in the cell (VanOrsdel et al. 2018; Fijalkowski et al. 2022), features that also explain why their detection suffers from poor reproducibility between studies (Weaver et al. 2019). Many of these novel sequences have been shown to be under purifying selection (Fesenko et al. 2025), but owing to their high rates of divergence (Stringer et al. 2021) and inconsistent translation, they are likely in the prefunctional, proto-gene phase of gene birth. Investigations of the properties and rate of emergence of these proto-genes can ultimately shed light on the first phases of gene birth: the de novo acquisition of open reading frames or expression.
Studies reporting the presence of nonannotated bacterial proteins using ribosome profiling
| Year of publication | Organisms studied | Number of nonannotated proteins identified | How many validated by mass spectrometry? | Reference |
|---|---|---|---|---|
| 2023 | Escherichia coli | 18 | N/A | Schumacher et al. 2023 |
| 2023 | Sinorhizobium meliloti | 37 | 0 | Hadjeras et al. 2023 |
| 2025 | Campylobacter jejuni | 42 | 0 | Froschauer et al. 2025 |
| 2022 | Streptococcus pneumoniae | 114 | N/A | Laczkovich et al. 2022 |
| 2022 | Samonella enterica | 49 | 12 (out of 36 tested) | Fijalkowski et al. 2022 |
| 2022 | Mycobacterium tuberculosis | 1689 | 44 | Smith et al. 2022 |
| 2021 | Escherichia coli | 283 | N/A | Stringer et al. 2021 |
| 2020 | Salmonella enterica | 42 | N/A | Venturini et al. 2020 |
| 2019 | Escherichia coli | 101 | N/A | Meydan et al. 2019 |
| 2019 | Escherichia coli | 68 | N/A | Weaver et al. 2019 |
| 2017 | Escherichia coli, Salmonella enterica, Bacillus subtilis | 49 (E. coli), 79 (S. enterica), 214 (B. subtilis) | 2 (E. coli), 8 (S. enterica) | Ndah et al. 2017 |
| 2017 | Escherichia coli | 465 | N/A | Hücker et al. 2017 |
| 2017 | Salmonella enterica | 61 | N/A | Giess et al. 2017 |
| 2017 | Salmonella enterica | 149 | N/A | Baek et al. 2017 |
| 2017 | Listeria monocytogenes | 6 | N/A | Impens et al. 2017 |
| 2016 | Mycobacterium abscessus | 130 | 0 | Miranda-CasoLuengo et al. 2016 |
| 2016 | Escherichia coli | 328 | N/A | Nakahigashi et al. 2016 |
| 2016 | Escherichia coli | 72 | 7 | Neuhaus et al. 2016 |
| 2015 | Mycobacterium smegmatis | 22 | N/A | Shell et al. 2015 |
[i] Only publications appearing since 2015 are listed.
Studies reporting the presence of nonannotated bacterial proteins using mass spectrometry
| Year of publication | Organisms studied | Number of nonannotated proteins identified | Reference |
|---|---|---|---|
| 2023 | Pseudomonas stutzeri | 29 | Meier-Credo et al. 2023 |
| 2021 | Nostoc sp. | 26 | Yu et al. 2021 |
| 2021 | Staphylococcus aureus | 24 | Fuchs et al. 2021 |
| 2021 | SIHUMIx (eight species in the simplified human intestinal microbiota) | 31 | Petruschke et al. 2021 |
| 2020 | Salmonella enterica, Deinococcus radiodurans | 18 (S. enterica), 33 (D. radiodurans) | Willems et al. 2020 |
| 2020 | Listeria monocytogenes | 4 | Varadarajan et al. 2020 |
| 2019 | Neomegalonema perideroedes | 38 | Herbst et al. 2019 |
| 2019 | Mycoplasma pneumoniae | 11 | Miravet-Verde et al. 2019 |
| 2018 | Methylobacterium extorquens | 39 | Bibi-Triki et al. 2018 |
| 2017 | Bartonella henselae | 10 | Omasits et al. 2017 |
| 2017 | Escherichia coli | 4 | D'Lima et al. 2017 |
| 2017 | Xanthomonas euvesicatoria | 30 | Abendroth et al. 2017 |
| 2017 | Brucella abortus | 6 | Zai et al. 2017 |
| 2016 | Mycoplasma pneumoniae, Mycoplasma genitalium | 38 (M. pneumoniae), 23 (M. genitalium) | Chen et al. 2016 |
[i] Only publications appearing since 2015 are listed.
Phase 2: the transition to functionality
Because of their rapid generation time and ease of propagation and genetic manipulation, bacteria provide excellent model systems to experimentally assay the functional potential of proteins encoded by nongenic sequences. Such evidence is usually achieved by expressing large pools of protein libraries in bacterial cells and testing for a functional phenotype. Using this approach, Knopp et al. (2019, 2021) have demonstrated the ability of random proteins to function as antibiotic-resistance peptides, either by modulating membrane potential or by engaging in specific interactions with transmembrane proteins. More recently, Frumkin and Laub (2023) have demonstrated the activity of a random peptide in inducing antitoxin resistance by interfering with the activity of protein chaperones in the cell. Using a rationally designed library of binary-patterned proteins, proteins that contain alternating polar and nonpolar residues, a wide range of auxotroph-rescue phenotypes could be demonstrated in bacteria (Kamtekar et al. 1993; Patel et al. 2009; Fisher et al. 2011; Donnelly et al. 2018). Also, in an approach that straddles the phage-transfer and de novo routes to gene origin, Warsi et al. (2020) constructed a gene fusion between bacterial and phage DNA that conferred a temperature-resistance phenotype.
Cumulatively, such studies not only demonstrate the ability of random proteins to confer beneficial phenotypes but allow direct tests of hypotheses about the transition to functionality during de novo gene birth. For example, the functional peptides identified by Knopp et al. (2019) were all membrane-associated, which coheres to the “transmembrane-first” model of gene birth, according to which new genes initially acquire functionality by acting as transmembrane domains (Vakirlis et al. 2020b).
State of the field and future prospects
Questions pertaining to the origin of ORFan genes in bacteria, and the degree to which de novo gene evolution contributes to their formation, remain almost as mysterious today as they were two decades ago. This is surprising, given that sequence information has resolved so many other aspects of gene and genome evolution in bacteria (Ochman et al. 2000; Gevers et al. 2004; Lerat et al. 2005; Bratlie et al. 2010; Treangen and Rocha 2011; Tria and Martin 2021). A key barrier to progress is that none of the three mechanisms proposed to explain the origin of ORFans—rapid divergence, de novo birth, and transfer from sources absent in the database—are expected to leave remnants in genomes, making it unusually difficult to reconstruct the origins of most taxon-specific genes. Rapid divergence from a pre-existing gene, by definition, leaves no homologous sequences in the outgroup; detecting de novo origin requires the improbable persistence of noncoding sequences across multiple bacterial lineages and concerns still remain about phage undersampling. Despite these hurdles, bacterial model systems can provide unique and unexplored research avenues. Because of the abundance of genomics and transcriptomics data sets, bacteria offer the opportunity to study the fine-grained stages in the emergence of genes within the history of a single species. Furthermore, bacterial experimental evolution presents an avenue by which the mechanisms of gene birth can be explored at a resolution otherwise not possible in more complex systems. In these ways, research on gene birth in bacteria can illuminate unanswered questions pertaining to the origin of novelty across all life-forms.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Kim Hammond for her help in preparing the figures. This work was supported by the National Institutes of Health (R35GM118038 to H.O.). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Notes
[3] Article published online before print. Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280157.124.
References
- ↵Abendroth U, Adlung N, Otto A, Grüneisen B, Becher D, Bonas U. 2017. Identification of new protein-coding genes with a potential role in the virulence of the plant pathogen Xanthomonas euvesicatoria. BMC Genomics 18: 625. 10.1186/s12864-017-4041-7
- ↵Ardern Z, Neuhaus K, Scherer S. 2020. Are antisense proteins in prokaryotes functional? Front Mol Biosci 7: 187. 10.3389/fmolb.2020.00187
- ↵Armengaud J. 2009. A perfect genome annotation is within reach with the proteomics and genomics alliance. Curr Opin Microbiol 12: 292–300. 10.1016/j.mib.2009.03.005
- ↵Baek J, Lee J, Yoon K, Lee H. 2017. Identification of unannotated small genes in Salmonella. G3 7: 983–989. 10.1534/g3.116.036939
- ↵Bar-On YM, Phillips R, Milo R. 2018. The biomass distribution on earth. Proc Natl Acad Sci 115: 6506–6511. 10.1073/pnas.1711842115
- ↵Barrera-Redondo J, Lotharukpong JS, Drost H-G, Coelho SM. 2023. Uncovering gene-family founder events during major evolutionary transitions in animals, plants and fungi using GenEra. Genome Biol 24: 54. 10.1186/s13059-023-02895-z
- ↵Bartonek L, Braun D, Zagrovic B. 2020. Frameshifting preserves key physicochemical properties of proteins. Proc Natl Acad Sci 117: 5907–5912. 10.1073/pnas.1911203117
- ↵Begun DJ, Lindfors HA, Kern AD, Jones CD. 2007. Evidence for de novo evolution of testis-expressed genes in the Drosophila yakuba/Drosophila erecta clade. Genetics 176: 1131–1137. 10.1534/genetics.106.069245
- ↵Benler S, Koonin EV. 2021. Fishing for phages in metagenomes: What do we catch, what do we miss? Curr Opin Virol 49: 142–150. 10.1016/j.coviro.2021.05.008
- ↵Bibi-Triki S, Husson G, Maucourt B, Vuilleumier S, Carapito C, Bringel F. 2018. N-terminome and proteogenomic analysis of the Methylobacterium extorquens DM4 reference strain for dichloromethane utilization. J Proteomics 179: 131–139. 10.1016/j.jprot.2018.03.012
- ↵Blevins WR, Ruiz-Orera J, Messeguer X, Blasco-Moreno B, Villanueva-Cañas JL, Espinar L, Díez J, Carey LB, Albà MM. 2021. Uncovering de novo gene birth in yeast using deep transcriptomics. Nat Commun 12: 604. 10.1038/s41467-021-20911-3
- ↵Bobay L-M, Touchon M, Rocha EPC. 2014. Pervasive domestication of defective prophages by bacteria. Proc Natl Acad Sci 111: 12127–12132. 10.1073/pnas.1405336111
- ↵Bondy-Denomy J, Davidson AR. 2014. When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J Microbiol 52: 235–242. 10.1007/s12275-014-4083-3
- ↵Bratlie MS, Johansen J, Sherman BT, Huang DW, Lempicki RA, Drabløs F. 2010. Gene duplications in prokaryotes can be associated with environmental adaptation. BMC Genomics 11: 588. 10.1186/1471-2164-11-588
- ↵Caglar MU, Houser JR, Barnhart CS, Boutz DR, Carroll SM, Dasgupta A, Lenoir WF, Smith BL, Sridhara V, Sydykova DK, 2017. The E. coli molecular phenotype under different growth conditions. Sci Rep 7: 45303. 10.1038/srep45303
- ↵Cai J, Zhao R, Jiang H, Wang W. 2008. De novo origination of a new protein-coding gene in Saccharomyces cerevisiae. Genetics 179: 487–496. 10.1534/genetics.107.084491
- ↵Canchaya C, Proux C, Fournous G, Bruttin A, Brüssow H. 2003. Prophage genomics. Microbiol Mol Biol Rev 67: 238–276. 10.1128/MMBR.67.2.238-276.2003
- ↵Carvunis A-R, Rolland T, Wapinski I, Calderwood MA, Yildirim MA, Simonis N, Charloteaux B, Hidalgo CA, Barbette J, Santhanam B, 2012. Proto-genes and de novo gene birth. Nature 487: 370–374. 10.1038/nature11184
- ↵Chen S, Krinsky BH, Long M. 2013. New genes as drivers of phenotypic evolution. Nat Rev Genet 14: 645–660. 10.1038/nrg3521
- ↵Chen W-H, van Noort V, Lluch-Senar M, Hennrich ML, Wodke JAH, Yus E, Alibés A, Roma G, Mende DR, Pesavento C, 2016. Integration of multi-omics data of a genome-reduced bacterium: prevalence of post-transcriptional regulation and its correlation with protein abundances. Nucleic Acids Res 44: 1192–1202. 10.1093/nar/gkw004
- ↵Coppens L, Lavigne R. 2020. SAPPHIRE: a neural network based classifier for σ70 promoter prediction in Pseudomonas. BMC Bioinformatics 21: 415. 10.1186/s12859-020-03730-z
- ↵Cortez D, Forterre P, Gribaldo S. 2009. A hidden reservoir of integrative elements is the major source of recently acquired foreign genes and ORFans in archaeal and bacterial genomes. Genome Biol 10: R65. 10.1186/gb-2009-10-6-r65
- ↵Daubin V, Ochman H. 2004a. Bacterial genomes as new gene homes: the genealogy of ORFans in E. coli. Genome Res 14: 1036–1042. 10.1101/gr.2231904
- ↵Daubin V, Ochman H. 2004b. Start-up entities in the origin of new genes. Curr Opin Genet Dev 14: 616–619. 10.1016/j.gde.2004.09.004
- ↵de Souza GA, Søfteland T, Koehler CJ, Thiede B, Wiker HG. 2009. Validating divergent ORF annotation of the Mycobacterium leprae genome through a full translation data set and peptide identification by tandem mass spectrometry. Proteomics 9: 3233–3243. 10.1002/pmic.200800955
- ↵D'Lima NG, Khitun A, Rosenbloom AD, Yuan P, Gassaway BM, Barber KW, Rinehart J, Slavoff SA. 2017. Comparative proteomics enables identification of nonannotated cold shock proteins in E. coli. J Proteome Res 16: 3722–3731. 10.1021/acs.jproteome.7b00419
- ↵Donnelly AE, Murphy GS, Digianantonio KM, Hecht MH. 2018. A de novo enzyme catalyzes a life-sustaining reaction in Escherichia coli. Nat Chem Biol 14: 253–255. 10.1038/nchembio.2550
- ↵Durrant MG, Bhatt AS. 2021. Automated prediction and annotation of small open reading frames in microbial genomes. Cell Host Microbe 29: 121–131.e4. 10.1016/j.chom.2020.11.002
- ↵Edgar RC. 2024. Protein structure alignment by Reseek improves sensitivity to remote homologs. Bioinformatics 40: btae687. 10.1093/bioinformatics/btae687
- ↵Fesenko I, Sahakyan H, Dhyani R, Shabalina SA, Storz G, Koonin EV. 2025. The hidden bacterial microproteome. Mol Cell 85: 1025–1041. 10.1016/j.molcel.2025.01.025
- ↵Fijalkowski I, Willems P, Jonckheere V, Simoens L, Van Damme P. 2022. Hidden in plain sight: challenges in proteomics detection of small ORF-encoded polypeptides. MicroLife 3: uqac005. 10.1093/femsml/uqac005
- ↵Fisher MA, McKinley KL, Bradley LH, Viola SR, Hecht MH. 2011. De novo designed proteins from a library of artificial sequences function in Escherichia coli and enable cell growth. PLoS One 6: e15364. 10.1371/journal.pone.0015364
- ↵Fremin BJ, Bhatt AS, Kyrpides NC, Global Phage Small Open Reading Frame (GP-SmORF) Consortium. 2022. Thousands of small, novel genes predicted in global phage genomes. Cell Rep 39: 110984. 10.1016/j.celrep.2022.110984
- ↵Froschauer K, Svensson SL, Gelhausen R, Fiore E, Kible P, Klaude A, Kucklick M, Fuchs S, Eggenhofer F, Yang C, 2025. Complementary Ribo-seq approaches map the translatome and provide a small protein census in the foodborne pathogen Campylobacter jejuni. Nat Commun 16: 3078. 10.1038/s41467-025-58329-w
- ↵Frumkin I, Laub MT. 2023. Selection of a de novo gene that can promote survival of Escherichia coli by modulating protein homeostasis pathways. Nat Ecol Evol 7: 2067–2079. 10.1038/s41559-023-02224-4
- ↵Fuchs S, Kucklick M, Lehmann E, Beckmann A, Wilkens M, Kolte B, Mustafayeva A, Ludwig T, Diwo M, Wissing J, 2021. Towards the characterization of the hidden world of small proteins in Staphylococcus aureus, a proteogenomics approach. PLoS Genet 17: e1009585. 10.1371/journal.pgen.1009585
- ↵Gevers D, Vandepoele K, Simillon C, Van de Peer Y. 2004. Gene duplication and biased functional retention of paralogs in bacterial genomes. Trends Microbiol 12: 148–154. 10.1016/j.tim.2004.02.007
- ↵Ghatak S, King ZA, Sastry A, Palsson BO. 2019. The y-ome defines the 35% of Escherichia coli genes that lack experimental evidence of function. Nucleic Acids Res 47: 2446–2454. 10.1093/nar/gkz030
- ↵Giess A, Jonckheere V, Ndah E, Chyżyńska K, Van Damme P, Valen E. 2017. Ribosome signatures aid bacterial translation initiation site identification. BMC Biol 15: 76. 10.1186/s12915-017-0416-0
- ↵Good BH, McDonald MJ, Barrick JE, Lenski RE, Desai MM. 2017. The dynamics of molecular evolution over 60,000 generations. Nature 551: 45–50. 10.1038/nature24287
- ↵Grandchamp A, Kühl L, Lebherz M, Brüggemann K, Parsch J, Bornberg-Bauer E. 2023. Population genomics reveals mechanisms and dynamics of de novo expressed open reading frame emergence in Drosophila melanogaster. Genome Res 33: 872–890. 10.1101/gr.277482.122
- ↵Hadjeras L, Heiniger B, Maaß S, Scheuer R, Gelhausen R, Azarderakhsh S, Barth-Weber S, Backofen R, Becher D, Ahrens CH, 2023. Unraveling the small proteome of the plant symbiont Sinorhizobium meliloti by ribosome profiling and proteogenomics. MicroLife 4: uqad012. 10.1093/femsml/uqad012
- ↵Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF. 1999. Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage. Proc Natl Acad Sci 96: 2192–2197. 10.1073/pnas.96.5.2192
- ↵Herbst F-A, Gonçalves SCL, Behr T, McIlroy SJ, Nielsen PH. 2019. Proteogenomic refinement of the Neomegalonema perideroedesT genome annotation. Proteomics 19: e1800330. 10.1002/pmic.201800330
- ↵Houser JR, Barnhart C, Boutz DR, Carroll SM, Dasgupta A, Michener JK, Needham BD, Papoulas O, Sridhara V, Sydykova DK, 2015. Controlled measurement and comparative analysis of cellular components in E. coli reveals broad regulatory changes in response to glucose starvation. PLoS Comput Biol 11: e1004400. 10.1371/journal.pcbi.1004400
- ↵Hücker SM, Ardern Z, Goldberg T, Schafferhans A, Bernhofer M, Vestergaard G, Nelson CW, Schloter M, Rost B, Scherer S, 2017. Discovery of numerous novel small genes in the intergenic regions of the Escherichia coli O157:H7 Sakai genome. PLoS One 12: e0184119. 10.1371/journal.pone.0184119
- ↵Impens F, Rolhion N, Radoshevich L, Bécavin C, Duval M, Mellin J, García Del Portillo F, Pucciarelli MG, Williams AH, Cossart P. 2017. N-terminomics identifies Prli42 as a membrane miniprotein conserved in Firmicutes and critical for stressosome activation in Listeria monocytogenes. Nat Microbiol 2: 17005. 10.1038/nmicrobiol.2017.5
- ↵Kamtekar S, Schiffer JM, Xiong H, Babik JM, Hecht MH. 1993. Protein design by binary patterning of polar and nonpolar amino acids. Science 262: 1680–1685. 10.1126/science.8259512
- ↵Karlowski WM, Varshney D, Zielezinski A. 2023. Taxonomically restricted genes in Bacillus may form clusters of homologs and can be traced to a large reservoir of noncoding sequences. Genome Biol Evol 15: evad023. 10.1093/gbe/evad023
- ↵Khalturin K, Hemmrich G, Fraune S, Augustin R, Bosch TCG. 2009. More than just orphans: Are taxonomically-restricted genes important in evolution? Trends Genet 25: 404–413. 10.1016/j.tig.2009.07.006
- ↵Knopp M, Gudmundsdottir JS, Nilsson T, König F, Warsi O, Rajer F, Ädelroth P, Andersson DI. 2019. De novo emergence of peptides that confer antibiotic resistance. mBio 10: e00837-19. 10.1128/mBio.00837-19
- ↵Knopp M, Babina AM, Gudmundsdóttir JS, Douglass MV, Trent MS, Andersson DI. 2021. A novel type of colistin resistance genes selected from random sequence space. PLoS Genet 17: e1009227. 10.1371/journal.pgen.1009227
- ↵Knowles DG, McLysaght A. 2009. Recent de novo origin of human protein-coding genes. Genome Res 19: 1752–1759. 10.1101/gr.095026.109
- ↵Kreitmeier M, Ardern Z, Abele M, Ludwig C, Scherer S, Neuhaus K. 2022. Spotlight on alternative frame coding: Two long overlapping genes in Pseudomonas aeruginosa are translated and under purifying selection. iScience 25: 103844. 10.1016/j.isci.2022.103844
- ↵Kuchibhatla BD, Sherman AW, Chung YWB, Cook S, Schneider G, Eisenhaber B, Karlin DG. 2014. Powerful sequence similarity search methods and in-depth manual analyses can identify remote homologs in many apparently “orphan” viral proteins. J Virol 88: 10–20. 10.1128/JVI.02595-13
- ↵Kuo C-H, Moran NA, Ochman H. 2009. The consequences of genetic drift for bacterial genome complexity. Genome Res 19: 1450–1454. 10.1101/gr.091785.109
- ↵Laczkovich I, Mangano K, Shao X, Hockenberry AJ, Gao Y, Mankin A, Vázquez-Laslop N, Federle MJ. 2022. Discovery of unannotated small open reading frames in Streptococcus pneumoniae D39 involved in quorum sensing and virulence using ribosome profiling. mBio 13: e0124722. 10.1128/mbio.01247-22
- ↵Lagator M, Sarikas S, Steinrueck M, Toledo-Aparicio D, Bollback JP, Guet CC, Tkačik G. 2022. Predicting bacterial promoter function and evolution from random sequences. eLife 11: e64543. 10.7554/eLife.64543
- ↵Lenski RE, Rose MR, Simpson SC, Tadler SC. 1991. Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2,000 generations. Am Nat 138: 1315–1341. 10.1086/285289
- ↵Lerat E, Daubin V, Ochman H, Moran NA. 2005. Evolutionary origins of genomic repertoires in bacteria. PLoS Biol 3: e130. 10.1371/journal.pbio.0030130
- ↵Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, Smetanin N, Verkuil R, Kabeli O, Shmueli Y, 2023. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379: 1123–1130. 10.1126/science.ade2574
- ↵Lobb B, Kurtz DA, Moreno-Hagelsieb G, Doxey AC. 2015. Remote homology and the functions of metagenomic dark matter. Front Genet 6: 234. 10.3389/fgene.2015.00234
- ↵Lwoff A. 1953. Lysogeny. Bacteriol Rev 17: 269–337. 10.1128/br.17.4.269-337.1953
- ↵McLysaght A, Guerzoni D. 2015. New genes from non-coding sequence: the role of de novo protein-coding genes in eukaryotic evolutionary innovation. Philos Trans R Soc Lond B Biol Sci 370: 20140332. 10.1098/rstb.2014.0332
- ↵McLysaght A, Hurst LD. 2016. Open questions in the study of de novo genes: what, how and why. Nat Rev Genet 17: 567–578. 10.1038/nrg.2016.78
- ↵Meier-Credo J, Heiniger B, Schori C, Rupprecht F, Michel H, Ahrens CH, Langer JD. 2023. Detection of known and novel small proteins in Pseudomonas stutzeri using a combination of bottom-up and digest-free proteomics and proteogenomics. Anal Chem 95: 11892–11900. 10.1021/acs.analchem.3c00676
- ↵Meydan S, Vázquez-Laslop N, Mankin AS. 2018. Genes within genes in bacterial genomes. Microbiol Spect 6: 10.1128/microbiolspec.rwr-0020-2018. 10.1128/9781683670247.ch9
- ↵Meydan S, Marks J, Klepacki D, Sharma V, Baranov PV, Firth AE, Margus T, Kefi A, Vázquez-Laslop N, Mankin AS. 2019. Retapamulin-assisted ribosome profiling reveals the alternative bacterial proteome. Mol Cell 74: 481–493.e6. 10.1016/j.molcel.2019.02.017
- ↵Mira A, Ochman H, Moran NA. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet 17: 589–596. 10.1016/S0168-9525(01)02447-7
- ↵Miranda-CasoLuengo AA, Staunton PM, Dinan AM, Lohan AJ, Loftus BJ. 2016. Functional characterization of the Mycobacterium abscessus genome coupled with condition specific transcriptomics reveals conserved molecular strategies for host adaptation and persistence. BMC Genomics 17: 553. 10.1186/s12864-016-2868-y
- ↵Miravet-Verde S, Ferrar T, Espadas-García G, Mazzolini R, Gharrab A, Sabido E, Serrano L, Lluch-Senar M. 2019. Unraveling the hidden universe of small proteins in bacterial genomes. Mol Syst Biol 15: e8290. 10.15252/msb.20188290
- ↵Nakahigashi K, Takai Y, Kimura M, Abe N, Nakayashiki T, Shiwa Y, Yoshikawa H, Wanner BL, Ishihama Y, Mori H. 2016. Comprehensive identification of translation start sites by tetracycline-inhibited ribosome profiling. DNA Res 23: 193–201. 10.1093/dnares/dsw008
- ↵Nayfach S, Shi ZJ, Seshadri R, Pollard KS, Kyrpides NC. 2019. New insights from uncultivated genomes of the global human gut microbiome. Nature 568: 505–510. 10.1038/s41586-019-1058-x
- ↵Nayfach S, Páez-Espino D, Call L, Low SJ, Sberro H, Ivanova NN, Proal AD, Fischbach MA, Bhatt AS, Hugenholtz P, 2021. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat Microbiol 6: 960–970. 10.1038/s41564-021-00928-6
- ↵Ndah E, Jonckheere V, Giess A, Valen E, Menschaert G, Van Damme P. 2017. REPARATION: ribosome profiling assisted (re-)annotation of bacterial genomes. Nucleic Acids Res 45: e168. 10.1093/nar/gkx758
- ↵Neuhaus K, Landstorfer R, Fellner L, Simon S, Schafferhans A, Goldberg T, Marx H, Ozoline ON, Rost B, Kuster B, 2016. Translatomics combined with transcriptomics and proteomics reveals novel functional, recently evolved orphan genes in Escherichia coli O157:H7 (EHEC). BMC Genomics 17: 133. 10.1186/s12864-016-2456-1
- ↵Ochman H, Lawrence JG, Groisman EA. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405: 299–304. 10.1038/35012500
- ↵Omasits U, Varadarajan AR, Schmid M, Goetze S, Melidis D, Bourqui M, Nikolayeva O, Québatte M, Patrignani A, Dehio C, 2017. An integrative strategy to identify the entire protein coding potential of prokaryotic genomes by proteogenomics. Genome Res 27: 2083–2095. 10.1101/gr.218255.116
- ↵Patel SC, Bradley LH, Jinadasa SP, Hecht MH. 2009. Cofactor binding and enzymatic activity in an unevolved superfamily of de novo designed 4-helix bundle proteins. Protein Sci 18: 1388–1400. 10.1002/pro.147
- ↵Pavesi A. 2021. Origin, evolution and stability of overlapping genes in viruses: a systematic review. Genes (Basel) 12: 809. 10.3390/genes12060809
- ↵Pearson WR, Wood T, Zhang Z, Miller W. 1997. Comparison of DNA sequences with protein sequences. Genomics 46: 24–36. 10.1006/geno.1997.4995
- ↵Pereira AB, Marano M, Bathala R, Zaragoza RA, Neira A, Samano A, Owoyemi A, Casola C. 2025. Orphan genes are not a distinct biological entity. Bioessays 47: e2400146. 10.1002/bies.202400146
- ↵Petruschke H, Schori C, Canzler S, Riesbeck S, Poehlein A, Daniel R, Frei D, Segessemann T, Zimmerman J, Marinos G, 2021. Discovery of novel community-relevant small proteins in a simplified human intestinal microbiome. Microbiome 9: 55. 10.1186/s40168-020-00981-z
- ↵Prabh N, Tautz D. 2021. Frequent lineage-specific substitution rate changes support an episodic model for protein evolution. G3 11: jkab333. 10.1093/g3journal/jkab333
- ↵Raghavan R, Sloan DB, Ochman H. 2012. Antisense transcription is pervasive but rarely conserved in enteric bacteria. mBio 3: e00156-12. 10.1128/mBio.00156-12
- ↵Randich AM, Kysela DT, Morlot C, Brun YV. 2019. Origin of a core bacterial gene via co-option and detoxification of a phage lysin. Curr Biol 29: 1634–1646. 10.1016/j.cub.2019.04.032
- ↵Remmert M, Biegert A, Hauser A, Söding J. 2011. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 9: 173–175. 10.1038/nmeth.1818
- ↵Rogozin IB, Spiridonov AN, Sorokin AV, Wolf YI, Jordan IK, Tatusov RL, Koonin EV. 2002. Purifying and directional selection in overlapping prokaryotic genes. Trends Genet 18: 228–232. 10.1016/S0168-9525(02)02649-5
- ↵Ruiz-Orera J, Verdaguer-Grau P, Villanueva-Cañas JL, Messeguer X, Albà MM. 2018. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nat Ecol Evol 2: 890–896. 10.1038/s41559-018-0506-6
- ↵Sabath N, Wagner A, Karlin D. 2012. Evolution of viral proteins originated de novo by overprinting. Mol Biol Evol 29: 3767–3780. 10.1093/molbev/mss179
- ↵Sanjuán R, Nebot MR, Chirico N, Mansky LM, Belshaw R. 2010. Viral mutation rates. J Virol 84: 9733–9748. 10.1128/JVI.00694-10
- ↵Sargentini NJ, Smith KC. 1994. DNA sequence analysis of gamma-radiation (anoxic)-induced and spontaneous lacId mutations in Escherichia coli K-12. Mutat Res 309: 147–163. 10.1016/0027-5107(94)90088-4
- ↵Sberro H, Fremin BJ, Zlitni S, Edfors F, Greenfield N, Snyder MP, Pavlopoulos GA, Kyrpides NC, Bhatt AS. 2019. Large-scale analyses of human microbiomes reveal thousands of small, novel genes. Cell 178: 1245–1259.e14. 10.1016/j.cell.2019.07.016
- ↵Schaaper RM, Dunn RL. 1991. Spontaneous mutation in the Escherichia coli lacI gene. Genetics 129: 317–326. 10.1093/genetics/129.2.317
- ↵Schumacher K, Gelhausen R, Kion-Crosby W, Barquist L, Backofen R, Jung K. 2023. Ribosome profiling reveals the fine-tuned response of Escherichia coli to mild and severe acid stress. mSystems 8: e0103723. 10.1128/msystems.01037-23
- ↵Shell SS, Wang J, Lapierre P, Mir M, Chase MR, Pyle MM, Gawande R, Ahmad R, Sarracino DA, Ioerger TR, 2015. Leaderless transcripts and small proteins are common features of the mycobacterial translational landscape. PLoS Genet 11: e1005641. 10.1371/journal.pgen.1005641
- ↵Siew N, Fischer D. 2003. Twenty thousand ORFan microbial protein families for the biologist? Structure 11: 7–9. 10.1016/S0969-2126(02)00938-3
- ↵Siew N, Azaria Y, Fischer D. 2004. The ORFanage: an ORFan database. Nucleic Acids Res 32: D281–D283. 10.1093/nar/gkh116
- ↵Smith C, Canestrari JG, Wang AJ, Champion MM, Derbyshire KM, Gray TA, Wade JT. 2022. Pervasive translation in Mycobacterium tuberculosis. eLife 11: e73980. 10.7554/eLife.73980
- ↵Stern DL, Han C. 2022. Gene structure-based homology search identifies highly divergent putative effector gene family. Genome Biol Evol 14: evac069. 10.1093/gbe/evac069
- ↵Storz G, Wolf YI, Ramamurthi KS. 2014. Small proteins can no longer be ignored. Annu Rev Biochem 83: 753–777. 10.1146/annurev-biochem-070611-102400
- ↵Stringer A, Smith C, Mangano K, Wade JT. 2021. Identification of novel translated small ORFs in Escherichia coli using complementary ribosome profiling approaches. J Bacteriol 204: e00352-21. 10.1101/2021.07.02.450978
- ↵Tautz D. 2014. The discovery of de novo gene evolution. Perspect Biol Med 57: 149–161. 10.1353/pbm.2014.0006
- ↵Tautz D, Domazet-Lošo T. 2011. The evolutionary origin of orphan genes. Nat Rev Genet 12: 692–702. 10.1038/nrg3053
- ↵Tenaillon O, Barrick JE, Ribeck N, Deatherage DE, Blanchard JL, Dasgupta A, Wu GC, Wielgoss S, Cruveiller S, Médigue C, 2016. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature 536: 165–170. 10.1038/nature18959
- ↵Tjaden B. 2023. Escherichia coli transcriptome assembly from a compendium of RNA-seq data sets. RNA Biol 20: 77–84. 10.1080/15476286.2023.2189331
- ↵Tonkin-Hill G, Corander J, Parkhill J. 2023. Challenges in prokaryote pangenomics. Microb Genom 9: 001021. 10.1099/mgen.0.001021
- ↵Touchon M, Moura de Sousa JA, Rocha EP. 2017. Embracing the enemy: the diversification of microbial gene repertoires by phage-mediated horizontal gene transfer. Curr Opin Microbiol 38: 66–73. 10.1016/j.mib.2017.04.010
- ↵Treangen TJ, Rocha EPC. 2011. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet 7: e1001284. 10.1371/journal.pgen.1001284
- ↵Tria FDK, Martin WF. 2021. Gene duplications are at least 50 times less frequent than gene transfers in prokaryotic genomes. Genome Biol Evol 13: evab224. 10.1093/gbe/evab224
- ↵uz-Zaman MH, D'Alton S, Barrick JE, Ochman H. 2024. Promoter recruitment drives the emergence of proto-genes in a long-term evolution experiment with Escherichia coli. PLoS Biol 22: e3002418. 10.1371/journal.pbio.3002418
- ↵Vakirlis N, Kupczok A. 2024. Large-scale investigation of species-specific orphan genes in the human gut microbiome elucidates their evolutionary origins. Genome Res 34: 888–903. 10.1101/gr.278977.124
- ↵Vakirlis N, McLysaght A. 2019. Computational prediction of de novo emerged protein-coding genes. Methods Mol Biol 1851: 63–81. 10.1007/978-1-4939-8736-8_4
- ↵Vakirlis N, Carvunis A-R, McLysaght A. 2020a. Synteny-based analyses indicate that sequence divergence is not the main source of orphan genes. eLife 9: e53500. 10.7554/eLife.53500
- ↵Vakirlis N, Acar O, Hsu B, Castilho Coelho N, Van Oss SB, Wacholder A, Medetgul-Ernar K, Bowman RW, Hines CP, Iannotta J, 2020b. De novo emergence of adaptive membrane proteins from thymine-rich genomic sequences. Nat Commun 11: 781. 10.1038/s41467-020-14500-z
- ↵Vakirlis N, Vance Z, Duggan KM, McLysaght A. 2022. De novo birth of functional microproteins in the human lineage. Cell Rep 41: 111808. 10.1016/j.celrep.2022.111808
- ↵Vakirlis N, Acar O, Cherupally V, Carvunis A-R. 2024. Ancestral sequence reconstruction as a tool to detect and study de novo gene emergence. Genome Biol Evol 16: evae151. 10.1093/gbe/evae151
- ↵van Kempen M, Kim SS, Tumescheit C, Mirdita M, Lee J, Gilchrist CLM, Söding J, Steinegger M. 2024. Fast and accurate protein structure search with Foldseek. Nat Biotechnol 42: 243–246. 10.1038/s41587-023-01773-0
- ↵VanOrsdel CE, Kelly JP, Burke BN, Lein CD, Oufiero CE, Sanchez JF, Wimmers LE, Hearn DJ, Abuikhdair FJ, Barnhart KR, 2018. Identifying new small proteins in Escherichia coli. Proteomics 18: e1700064. 10.1002/pmic.201700064
- ↵Van Oss SB, Carvunis A-R. 2019. De novo gene birth. PLoS Genet 15: e1008160. 10.1371/journal.pgen.1008160
- ↵Varadarajan AR, Goetze S, Pavlou MP, Grosboillot V, Shen Y, Loessner MJ, Ahrens CH, Wollscheid B. 2020. A proteogenomic resource enabling integrated analysis of Listeria genotype–proteotype–phenotype relationships. J Proteome Res 19: 1647–1662. 10.1021/acs.jproteome.9b00842
- ↵Venturini E, Svensson SL, Maaß S, Gelhausen R, Eggenhofer F, Li L, Cain AK, Parkhill J, Becher D, Backofen R, 2020. A global data-driven census of Salmonella small proteins and their potential functions in bacterial virulence. MicroLife 1: uqaa002. 10.1093/femsml/uqaa002
- ↵Wang X, Kim Y, Ma Q, Hong SH, Pokusaeva K, Sturino JM, Wood TK. 2010. Cryptic prophages help bacteria cope with adverse environments. Nat Commun 1: 147. 10.1038/ncomms1146
- ↵Warsi O, Knopp M, Surkov S, Jerlström Hultqvist J, Andersson DI. 2020. Evolution of a new function by fusion between phage DNA and a bacterial gene. Mol Biol Evol 37: 1329–1341. 10.1093/molbev/msaa007
- ↵Watson AK, Lopez P, Bapteste E. 2021. Hundreds of out-of-frame remodeled gene families in the Escherichia coli pangenome. Mol Biol Evol 39: msab329. 10.1093/molbev/msab329
- ↵Weaver J, Mohammad F, Buskirk AR, Storz G. 2019. Identifying small proteins by ribosome profiling with stalled initiation complexes. mBio 10: e02819-18. 10.1128/mBio.02819-18
- ↵Weisman CM, Eddy SR. 2017. Gene evolution: getting something from nothing. Curr Biol 27: R661–R663. 10.1016/j.cub.2017.05.056
- ↵Weisman CM, Murray AW, Eddy SR. 2020. Many, but not all, lineage-specific genes can be explained by homology detection failure. PLoS Biol 18: e3000862. 10.1371/journal.pbio.3000862
- ↵Willems P, Fijalkowski I, Van Damme P. 2020. Lost and found: re-searching and re-scoring proteomics data aids genome annotation and improves proteome coverage. mSystems 5: e00833-20. 10.1128/mSystems.00833-20
- ↵Wright BW, Molloy MP, Jaschke PR. 2022. Overlapping genes in natural and engineered genomes. Nat Rev Genet 23: 154–168. 10.1038/s41576-021-00417-w
- ↵Yamamura E, Nunoshiba T, Kawata M, Yamamoto K. 2000. Characterization of spontaneous mutation in the oxyR strain of Escherichia coli. Biochem Biophys Res Commun 279: 427–432. 10.1006/bbrc.2000.3961
- ↵Yin Y, Fischer D. 2006. On the origin of microbial ORFans: quantifying the strength of the evidence for viral lateral transfer. BMC Evol Biol 6: 63. 10.1186/1471-2148-6-63
- ↵Yomtovian I, Teerakulkittipong N, Lee B, Moult J, Unger R. 2010. Composition bias and the origin of ORFan genes. Bioinformatics 26: 996–999. 10.1093/bioinformatics/btq093
- ↵Yu S, Yang M, Xiong J, Zhang Q, Gao X, Miao W, Ge F. 2021. Proteogenomic analysis provides novel insight into genome annotation and nitrogen metabolism in Nostoc sp. PCC 7120. Microbiol Spect 9: e00490-21. 10.1128/Spectrum.00490-21
- ↵Zai X, Yang Q, Liu K, Li R, Qian M, Zhao T, Li Y, Yin Y, Dong D, Fu L, 2017. A comprehensive proteogenomic study of the human Brucella vaccine strain 104 M. BMC Genomics 18: 402. 10.1186/s12864-017-3800-9
- ↵Zehentner B, Ardern Z, Kreitmeier M, Scherer S, Neuhaus K. 2020. A novel pH-regulated, unusual 603 bp overlapping protein coding gene pop Is encoded antisense to ompA in Escherichia coli O157:H7 (EHEC). Front Microbiol 11: 377. 10.3389/fmicb.2020.00377
- ↵Zhang L, Ren Y, Yang T, Li G, Chen J, Gschwend AR, Yu Y, Hou G, Zi J, Zhou R, 2019. Rapid evolution of protein diversity by de novo origination in Oryza. Nat Ecol Evol 3: 679–690. 10.1038/s41559-019-0822-5
- ↵Zhuang X, Yang C, Murphy KR, Cheng C-HC. 2019. Molecular mechanism and history of non-sense to sense evolution of antifreeze glycoprotein gene in northern gadids. Proc Natl Acad Sci 116: 4400–4405. 10.1073/pnas.1817138116