
Developmental transcriptomics in Pristionchus reveals the environmental responsiveness of a plasticity gene-regulatory network
Abstract
Developmental plasticity enables the production of alternative phenotypes in response to different environmental conditions. Although significant advances in understanding the ecological and evolutionary implications of plasticity have been made, understanding its genetic basis has lagged. However, a decade of genetic screens in the model nematode Pristionchus pacificus has culminated in the identification of more than 30 genes that affect mouth form. We also recently reported the critical window of environmental sensitivity and therefore have clear expectations for when differential gene expression should matter. Here, we collated existing data into a gene-regulatory network (GRN) and performed developmental transcriptomics across different environmental conditions, genetic backgrounds, and mutants to assess the regulatory logic of mouth-form plasticity. We find that only two genes in the GRN (eud-1 and seud-1/sult-1) are sensitive to the environment during the critical window. The time points of their sensitivity differ, suggesting that they act as sequential checkpoints. Additionally, seud-1/sult-1 is differentially expressed across strains and species with different mouth-form biases, highlighting the potential role of switch-gene regulation in the evolution of plasticity. We also observe temporal constraint upon the transcriptional effects of mutating the GRN and reveal unexpected feedback between mouth-form genes. Finally, a comprehensive analysis of all samples identifies metabolism as a shared pathway for regulating mouth-form plasticity. These data are presented in a Shiny app to facilitate gene expression comparisons across development in up to 14 different conditions. Collectively, our results divide the GRN for mouth-form plasticity into environmentally sensitive switch genes and downstream genes that execute the decision.
Phenotypic plasticity is a widespread phenomenon by which exposure to different environments elicits different phenotypes from the same genotype (Pigliucci 2001; West-Eberhard 2003; DeWitt and Scheiner 2004). In sexually mature adults, plasticity is typically limited to physiological and behavioral traits, but when plasticity is channeled through development, often during an environmentally sensitive period or “window,” large differences in morphology can be achieved (Nijhout 2003). Several nontraditional model organisms that exhibit qualitative differences in morphology and behavior (i.e., polyphenism) have been developed to study plasticity, including social insects (Wilson 1971; Wheeler 1986; Simpson et al. 2011), dung beetles (Emlen 1994; Moczek 1998), and spadefoot toads (Pfennig 1990). These systems have revealed important roles for plasticity in evolution through its provision of environment-matched phenotypes and masking of genetic variants (West-Eberhard 1989; Pfennig et al. 2010; Moczek et al. 2011; Sommer 2020). However, to fully incorporate plasticity into the modern synthesis, a genetic framework is needed. Although possible (Kucharski et al. 2008; Kijimoto et al. 2012; Xu et al. 2015; Klein et al. 2016; Mazo-Vargas et al. 2017; Trible et al. 2017; Yan et al. 2017; Zhang et al. 2017; Parker and Brisson 2019), genetic analysis remains challenging in non-model organisms owing to long reproductive cycles and difficulties in laboratory rearing.
The nematode Pristionchus pacificus was introduced as a model system for studying developmental plasticity in 2010 (Bento et al. 2010). Similar to Caenorhabditis elegans, P. pacificus has four larval stages (J1–J4), although the first stage remains enclosed in the egg. The laboratory strain PS312 reaches sexual maturity within 72 h, at which point adults exhibit either a narrow, deep stenostomatous (St) mouth form with a single dorsal tooth, or a wide, shallow eurystomatous (Eu) mouth form containing two hooked teeth (Fig. 1A,C). This morphological decision has ecological consequences as the St morph is limited to a bacterial diet, whereas the Eu morph is a facultative predator on other nematodes. Mouth-form development is sensitive to many environmental factors including pheromones, crowding, salt concentration, temperature, and culturing substrate (Bento et al. 2010; Bose et al. 2012; Ragsdale et al. 2013; Werner et al. 2017, 2018; Lenuzzi et al. 2023). Reciprocal transplantation experiments recently identified 36–60 h (J3–J4) as the critical window for mouth-form plasticity (Werner et al. 2023). Additionally, more than a decade of sampling for Pristionchus has revealed a world-wide distribution with at least 48 species and hundreds of natural isolates (McGaughran et al. 2016; Kanzaki et al. 2021). Importantly, different natural isolates and species vary in the percentage of animals that are Eu or St, even when grown under the same environmental conditions (Ragsdale et al. 2013; Werner et al. 2017), demonstrating a genetic basis, and possible selection, for mouth-form ratios in the wild.
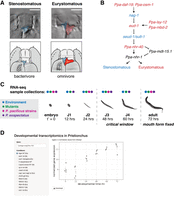
Developmental transcriptomics of mouth-form plasticity in Pristionchus pacificus. (A) The two mouth forms developed by adult P. pacificus with teeth colored for ease of visualization. (B) A gene-regulatory network for mouth-form development with St-promoting genes colored in blue and Eu-promoting genes in red. Genes in black may regulate the development of both morphs. (C) Experimental design for developmental transcriptomics. The timing of different stages in the life cycle of P. pacificus is shown. Different conditions are indicated by color per the legend on the left. Colored circles above time points represent when the different conditions were sampled. (D) A screenshot of the Shiny app that can be used to visualize transcriptomic data for any gene in any condition.
P. pacificus is easy to maintain in the laboratory and possesses many of the same traits that have made C. elegans a powerful model organism: hermaphroditism, short generation time, small genome, and amenability to genetic analysis. Over the past 15 years, forward genetic screens and targeted mutagenesis in P. pacificus have identified many genes that affect mouth-form development (Bento et al. 2010; Ragsdale et al. 2013; Kieninger et al. 2016; Markov et al. 2016; Serobyan et al. 2016; Sieriebriennikov et al. 2017, 2018, 2020; Bui et al. 2018; Moreno et al. 2018, 2019; Namdeo et al. 2018; Lo et al. 2022; Sun et al. 2022a,b; Casasa et al. 2023; Ishita et al. 2023; Lenuzzi et al. 2023; Levis and Ragsdale 2023; Piskobulu et al. 2025). Although there are undoubtedly more components involved, these experiments represent an important milestone as all phases of mouth-form development are now represented from environmental sensation to morphological execution of the decision. Furthermore, the identification of these genes enables mechanistic investigations into how plasticity is regulated.
Previous studies have examined transcriptional differences between a few mouth-form mutants from mixed-staged populations. These data helped to identify downstream targets of mouth-form genes (Bui and Ragsdale 2019; Sieriebriennikov et al. 2020) and gene coexpression modules (Casasa et al. 2021). Here, we expand upon these studies by assembling a gene-regulatory network (GRN) of mouth-form plasticity and use developmental transcriptomics in different Pristionchus strains, species, and environmental conditions to identify (1) which genes in the network are environmentally responsive, (2) when they are responsive during development, and (3) how these patterns change over evolutionary time.
Results
A GRN for mouth-form plasticity
First, we performed a comprehensive literature analysis to compile all genes that have been shown to affect mouth form in P. pacificus. We identified 39 genes across 20 publications from 2010 to 2025. In Table 1 we provide a list of these genes with their identifiers, chromosomal position, Gene Ontology, mouth-form phenotype, and relevant citations. For the rest of this paper, we chose to focus on genes that have a ≥90% penetrant mouth-form phenotype when mutated. We ordered these genes into a GRN for mouth-form plasticity using annotated protein function and previous epistatic experiments (Fig. 1B). At the start of the pathway are the chemosensory genes daf-19 and osm-1, which control cilia development and function. The core of the network consists of a multigene locus containing the sulfatase eud-1 and an antagonistic gene nag-1, two chromatin modifiers (lsy-12, mbd-2), and the sulfotransferase seud-1/sult-1. Downstream are two nuclear hormone receptors (nhr-40, nhr-1) and a Mediator subunit (mdt-15.1). This pathway, based on more than a decade of forward and reverse genetic experiments, represents a comprehensive GRN of developmental plasticity.
Genes that affect mouth-form development in Pristionchus pacificus
Environmental responsiveness of the mouth-form GRN
Developmental plasticity is mediated by environmental exposure during a sensitive period of development. Establishing the temporal action of molecular factors relative to this critical window is required to interpret the regulatory logic of developmental plasticity. We also sought to investigate potential feedback loops and to understand how the responsiveness of the GRN evolves. To address these questions, we generated a developmental transcriptomic data set that incorporates two environmental conditions, seven mouth-form mutant strains, four natural isolates of P. pacificus, and its sister species, Pristionchus exspectatus (Supplemental Fig. S1A). For each condition, we collected worms at multiple time points across development from 0 h post egg-synchronization (embryos) to 72 h (young adults) (Fig. 1C). We extracted and sequenced mRNA to a minimum depth of 3 million reads and with a minimum quality score of 30, ultimately resulting in more than 3.5 billion high-quality reads that we anticipate will be a valuable resource for the P. pacificus and phenotypic plasticity communities (Supplemental Figs. S1–S7; Supplemental Tables S1, S4). To facilitate visualization of these data, we made a Shiny app in R that renders downloadable plots of gene expression for any P. pacificus gene in each of the tested conditions (Fig. 1D; https://CRAN.R-project.org/package=shiny). This app can be found at https://pristionchus-transcriptomics.shinyapps.io/GRN-shiny/.
First, we investigated the transcriptome-wide response to different environmental conditions. We cultured the P. pacificus laboratory strain PS312 on NGM-agar plates, which yielded ∼95% Eu animals, and in S-Medium liquid culture, which yielded 5%–10% Eu animals (Werner et al. 2017), and collected samples at six developmental time points (0 h, 12 h, 24 h, 48 h, 60 h, and 72 h) (Supplemental Fig. S8). We then performed poly(A)-tail-based bulk RNA-seq and used DESeq2 (Love et al. 2014) to assess differential gene expression (adjusted P-value < 0.05) at each time point. A principal component analysis of normalized expression showed that PC1, which separated samples by developmental time, explained 88% of the variance in gene expression (Supplemental Fig. S9A). This suggests that developmental time is the single largest factor that explains differences in gene expression and highlights the importance of using stage-synchronized samples for transcriptomic analysis.
We found a total of 7648 differentially expressed genes (DEGs) between agar and liquid culture samples across all time points. In general, the number of DEGs between the two conditions increased with developmental time. The 12 h time point, which contains a mix of eggs and J1s, had only 81 DEGs (1% of all DEGs) (Fig. 2A). At 24 and 48 h, there was a notable increase to 1255 and 842 DEGs, respectively (Fig. 2B,C). Yet, at the 60 and 72 h time points, there were far more DEGs (4937 and 4640, respectively) (Fig. 2D,E; Supplemental Fig. S9B,C). Slightly fewer than half of the DEGs genes were differentially expressed at multiple developmental time points, with the last two developmental time points sharing >60% (2995) of their DEGs (Supplemental Fig. S9C). Altogether, these data show a large transcriptomic response to the different environmental conditions starting between 48 h and 60 h that persists through to adulthood. This transcriptomic response coincides with the closing of the critical window for mouth-form plasticity (Werner et al. 2023). We conclude that the early stages of development are relatively canalized with only minor changes in gene expression occurring in response to the environment. Leading up to the end of the critical window, large-scale transcriptional rewiring occurs in response to the environment, which bifurcates development into different trajectories affecting multiple physiological and morphological traits including, but not limited to, mouth-form development. Presumably, the mouth-form genes that are differentially expressed early in the critical window represent environmental switch genes, which if expressed above a threshold, initiate downstream transcriptional changes observed at the end of the critical window (i.e., ≥60 h).
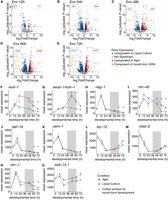
A subset of the mouth-form GRN is environmentally responsive. (A–E) Volcano plots of genes differentially expressed between worms grown on agar and in liquid culture at five developmental time points. Significant genes are colored by the condition in which they were upregulated: blue for liquid culture and red for agar. Significant mouth-form genes are labeled on the plot in gold. A dashed line represents the significance cutoff (adjusted P-value < 0.05). (F–O) Mean expression of normalized counts from DESeq2 with SEM for mouth-form genes. (*) Time points with significant differences in expression (adjusted P-value < 0.05).
Next, we investigated whether the environment induces changes in expression of the mouth-form GRN and whether any members are differentially expressed during the critical window. We found that even though each of these genes has a ∼100% penetrant phenotype when mutated (Ragsdale et al. 2013; Kieninger et al. 2016; Serobyan et al. 2016; Namdeo et al. 2018; Sieriebriennikov et al. 2018, 2020; Moreno et al. 2019; Casasa et al. 2023), only six of the 10 genes were significantly differentially expressed (adjusted P-value < 0.05) at any point during development (Fig. 2F–O). Four of these genes exhibited a change in expression that is consistent with their loss-of-function phenotype (eud-1, seud-1/sult-1, nag-1, and nhr-1). However, nhr-40 and daf-19, which are thought to promote the Eu mouth form based on their loss-of-function phenotypes (Table 1; Moreno et al. 2019; Sieriebriennikov et al. 2020), were upregulated in St-promoting liquid culture during late development (Fig. 2I,J). Although the basis of the discrepancy is not currently clear, nhr-40’s expression may be untethered to its role in mouth form as many nuclear hormone receptors are post-transcriptionally regulated (Aranda and Pascual 2001).
All the differentially expressed mouth-form genes showed differences in expression at the 60 h time point, and four of these genes continued to show differences in expression into adulthood. At 60 h, animals are almost exclusively in juvenile stage 4 (J4) in both conditions, which means they go through one more molt before adulthood. Hence, it is possible that these differences contribute to the mouth-form decision. However, as the critical window has largely closed by 60 h, we suspect that genes that are not differentially expressed before this time point do not contribute to the switch mechanism per se. Instead, either their function is unrelated to their role in mouth form or late differential expression pertains to their role in executing the mouth-form decision. For example, nhr-1 exhibits late differential expression (Fig. 2N) and yields an intermediate mouth form (an intermediate width between Eu and St and an underdeveloped second tooth) when mutated (Sieriebriennikov et al. 2020). These results are consistent with nhr-1 regulating the expression of additional genes required for the formation of the adult mouth.
In contrast, the genes eud-1 and seud-1/sult-1 showed significant differences in expression as early as 24 and 48 h, respectively (Fig. 2F,G). These data support the role of eud-1 and seud-1/sult-1 as opposing genetic switches for mouth-form development, as well as the downstream placement of the nuclear hormone receptors. Moreover, the early differential expression of eud-1 at 24 h, the only mouth-form gene to be differentially expressed prior to the critical window, suggests that it is the pivotal environmental switch gene.
Transcriptional effects of mutations in the mouth-form GRN are temporally restricted
To explore the relationship between mouth-form genes, we performed RNA-seq on seven mutant lines of P. pacificus (Supplemental Fig. S1A) and quantified changes in gene expression relative to wild-type PS312 grown on agar. These mutants exhibit fixed mouth-form phenotypes (Eu: Ppa-Ex[eud-1], Ppa-nhr-40, Ppa-sult-1, Ppa-nag-1/nag-2; St: Ppa-eud-1, Ppa-mbd-2, Ppa-lsy-12) (Table 1) and were also grown on NGM agar plates. We reasoned that keeping the environment consistent across samples should yield fewer changes in gene expression than between-environment comparisons. However, we found that mutating or overexpressing individual mouth-form genes still generated substantial transcriptomic differences across development, especially at 60 and 72 h time points (Supplemental Fig. S10A). Notably, the eud-1 loss-of function mutant had the fewest DEGs and appeared to be an outlier in this regard (Z = −1.9) (Supplemental Fig. S10B). Collectively, a meta-analysis of GRN mutants across development suggests that eud-1 is the most specific component of mouth-form regulation.
We next investigated the transcriptional effects of mutating individual mouth-form genes on other components of the GRN. First, we observed that mutations in mouth-form genes supported the topology of the GRN (Fig. 1B). eud-1, which promotes the Eu morph, is part of a multigene locus and is bookended by two genes, nag-1 and nag-2, which promote the St morph. The nag-1/nag-2 double mutant showed upregulation of eud-1 across development that was significantly higher than wild-type expression at 12 h and 48 h (Fig. 3H), supporting nag-1’s placement upstream of eud-1. Overexpression of eud-1 led to upregulation of the two downstream nuclear hormone receptors (Fig. 3B,C), whereas a loss-of-function mutation in eud-1 resulted in an upregulation of seud-1/sult-1 (Fig. 3D). Conversely, mutating seud-1/sult-1 had no effect on eud-1 expression (Fig. 3G), supporting its downstream placement in the GRN relative to eud-1. Differential expression of seud-1/sult-1 in the eud-1 mutant was notable because neither of their protein products is a transcription factor, and they are believed to be expressed in different cells (Bui et al. 2018). EUD-1, SEUD-1/SULT-1, and NAG-1 are all cytosolic enzymes, so these regulatory connections are presumably mediated indirectly via their enzymatic products functioning as signaling molecules.
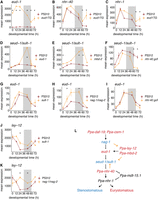
Transcriptional effects of mutations in the mouth-form GRN are temporally restricted. (A–K) Mean expression from DESeq2 normalized counts of mouth-form genes in wild-type PS312 and the indicated mutant line. (*) Time point with significant differences in expression (adjusted P-value < 0.05). (L) Mouth-form GRN with St-promoting genes colored in blue and Eu-promoting genes colored in red. Genes in black may regulate the development of both morphs. Solid lines represent gene ordering resulting from epistatic analysis, whereas dashed lines represent untested gene interactions. Gold lines represent new regulatory links inferred from the results of this study.
Second, we found evidence for additional regulation between components of the mouth-form GRN. The mbd-2 mutant displayed significant upregulation of seud-1/sult-1 at 48 and 60 h (Fig. 3E). MBD-2 may recruit chromatin modifiers to regulate seud-1/sult-1’s expression (Gutierrez and Sommer 2007). Alternatively, it may indirectly upregulate seud-1/sult-1 by decreasing eud-1’s expression in J2 larvae (Serobyan et al. 2016). We also noted upregulation of lsy-12 at 12 h in the sult-1 mutant (Fig. 3J) and at 24 and 48 h in the nag-1/nag-2 double mutant (Fig. 3K). Finally, our data set indicated a potential feedback mechanism from a downstream nuclear hormone receptor to an upstream switch gene. The gain-of-function mutation in nhr-40 caused a significant downregulation of seud-1/sult-1, but not eud-1, at 60 and 72 h compared with the wild type (Fig. 3F,I). Whether direct or indirect, these results enabled us to add new regulatory connections to the mouth-form GRN (Fig. 3L).
Third, we were struck by the temporal restriction of mouth-form mutants on other mouth-form genes. For example, even though eud-1 was overexpressed at each sampled time point in the transgenic line (Fig. 3A), its upregulation of the downstream nuclear hormone receptors was limited to the last two time points (60 h and 72 h) (Fig. 3B,C). Similarly, the upregulation of seud-1/sult-1 in the eud-1 mutant was limited to the 48 h time point (Fig. 3D), and the gain-of-function mutation in nhr-40 resulted in upregulation of both nuclear hormone receptors, but only at 60 and 72 h (Supplemental Fig. S11A,G). We found that nhr-40 was upregulated at 60 h and/or 72 h in all mutant lines relative to the wild type regardless of whether the phenotype was Eu or St (Supplemental Fig. S11B–F). These results indicate that nhr-40’s expression is particularly sensitive to perturbations in the GRN. However, because the activity of nuclear receptors can be governed by their ligand-binding domains or their dimerization partners, increased expression of nhr-40 may not equate to increased activity of the transcription factor. Overall, our transcriptomic analyses of mouth-form mutants show strong connections between multiple nodes in the GRN. This regulation is largely restricted to the time points at which individual genes are environmentally responsive. This constraint is consistent with transcriptional regulation being confined to discrete developmental checkpoints.
Genetic background has different effects on the GRN than the environment
Natural isolates of P. pacificus vary in the ratio of Eu to St animals even when grown under the same environmental conditions, indicating a strong effect of genetic background on mouth-form development (Ragsdale et al. 2013). A recent study employed recombinant inbred lines and QTL mapping with two strains from clade B (Dardiry et al. 2023) and found copy-number variation of cis-regulatory elements underlying differences in eud-1 expression and mouth form. However, outside of this case study, it is unknown which nodes of the network are being acted on by evolution. To expand upon these findings, we compared gene expression between two Eu-biased strains (RSA100 and RS5427) and two St-biased strains (RSC017 and RS5410) of P. pacificus from clade C (Supplemental Fig. S12A). These strains were all grown on NGM-agar plates, and we isolated mRNA from samples at 0 h, 24 h, 48 h, and 72 h.
We observed that both St strains exhibited elevated seud-1/sult-1 expression relative to Eu strains, whereas the reciprocal pattern for eud-1 was less apparent (Supplemental Fig. S12B,C). To increase our statistical power, we pooled the two Eu-biased strains and two St-biased strains. Overall, we found relatively few DEGs compared with the number of genes that were differentially expressed between environmentally induced Eu and St worms (Fig. 4A–D). The greatest number of DEGs between Eu- and St-biased strains was at the 0 h time point (Fig. 4A). We wondered if this could be because of differences in developmental rate or in egg retention. However, staging of worms from a subset of each sampled time point revealed development to be tightly synced between the four different strains (Supplemental Fig. S13A–C). Furthermore, clustering of the samples in the PCA was independent of strain identity and even mouth-form bias (Supplemental Fig. S13D–F). A Gene Ontology revealed a broad distribution of cellular functions, making it challenging to interpret the underlying cause or consequence of this difference (Supplemental Table S2). Nevertheless, these results suggest that Eu- and St-biased strains are primed for different trajectories from the earliest stages of development.
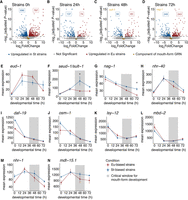
Natural variation in mouth-form transcriptomics. (A–D) Volcano plots of differentially expressed genes between pooled St- and Eu-biased strains of P. pacificus at four developmental time points. Significant genes (adjusted P-values < 0.05) upregulated in St are colored blue, and those upregulated in Eu are colored red. Gold points represent components of the mouth-form GRN, and significant mouth-form genes are labeled in gold on the plot. (E–N) Mean expression of normalized counts from DESeq2 for mouth-form genes in pooled St- and Eu-biased strains. (*) Time points with significant expression differences (adjusted P-value < 0.05).
Consistent with the environmental comparisons, the 72 h time point exhibited substantially more DEGs than 48 h or 24 h (Fig. 4C,D), suggesting that distinct transcriptional changes accompany the formation of the two different adult mouth forms (although we note that some of these differences may be unrelated to mouth form). In this comparison only two of the genes from the mouth-form GRN showed significant differences in expression: seud-1/sult-1 and nag-1 (Fig. 4E–N). The significant difference in seud-1/sult-1’s expression was at 48 h (Fig. 4F), essentially in the middle of the critical window, whereas nag-1 was differentially expressed at 72 h (Fig. 4G). This temporal pattern, as well as the absence of other differentially expressed mouth-form genes, suggests that seud-1/sult-1 is the key GRN component responsible for the differences in mouth form between the four natural isolates in our analysis.
Pristionchus sister species exhibit large transcriptomic differences
To further investigate the effect of genetic background on the mouth-form GRN, we compared gene expression between two sister species of Pristionchus with opposite biases in mouth form. In contrast to the main laboratory strain of P. pacificus (PS312), which is >95% Eu on agar plates, the laboratory strain of P. exspectatus (RS5522B) is <1% Eu (Kanzaki et al. 2012; Ragsdale et al. 2013). Evaluating gene expression between species is notoriously challenging (Munro et al. 2022) owing to both evolutionary divergence and the need to normalize RNA-seq reads between different gene annotations. We took a conservative approach and mapped the reads of each species to its own transcriptome and then performed a differential expression analysis exclusively on the 1:1 species orthologs using DESeq2 (see Methods). We found that P. pacificus and P. exspectatus have large differences in gene expression across all developmental time points (Supplemental Fig. S14A–F). Principal component analysis of the normalized expression values separated samples first by developmental time along PC1 and second by species along PC2 (Supplemental Fig. S15). The mbd-2 gene in P. exspectatus seems to be incorrectly annotated and was excluded from this analysis. Consistent with the large-scale transcriptomic changes, the GRN also exhibited significant differences in expression (Supplemental Fig. S14G–O). Notably, seud-1/sult-1 and nag-1 were significantly upregulated in St-biased P. exspectatus during the critical window (Supplemental Fig. S14H,I). eud-1 was differentially expressed only at 12 h and was unexpectedly upregulated in St-biased P. exspectatus along with two paralogs (see Discussion) (Supplemental Fig. S14G,P,Q). In summary, comparing gene expression between sister species shows a massive rewiring of transcription. Our PCA suggests that most of these differences are species specific and not related to mouth-form plasticity. Nonetheless, our results argue that the transcriptional regulation of seud-1/sult-1 is a conserved feature of mouth-form plasticity across environments, natural isolates, and sister species. We note, however, that the other switch gene (eud-1) was elevated in Eu-biased strains, and promoters and enhancers of both genes are likely targets for selection (Dardiry et al. 2023).
Differential expression analysis across diverse conditions reveals shared pathways
After examining the developmental regulation of genes known to affect mouth-form (i.e., the GRN), we leveraged our data set to identify additional genes and pathways involved. We combined all previously analyzed samples (PS312 in agar and liquid culture, seven mouth-form mutant strains, four P. pacificus strains, and P. exspectatus samples) (Supplemental Fig. S1A) to build a comprehensive transcriptomic data set that should filter out all but a core set of mouth-form DEGs. We used k-medoid clustering to separate samples into four developmental clusters (Fig. 5A; Supplemental Fig. S16) based solely on gene expression (agnostic to sampling time point) to accommodate any discrepancy in developmental rate between different conditions. Cluster 1 contained samples from 0 and 12 h. Cluster 2 contained mostly 24 h samples, as well as a few samples from 12 h and 48 h. Cluster 3 was almost entirely 48 h samples, and cluster 4 contained exclusively 60 h and 72 h samples (Fig. 5A).
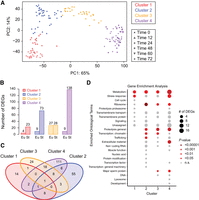
Differential expression analysis across diverse conditions reveals shared pathways. (A) PCA and k-medoid clustering of all samples based on gene expression. Color indicates cluster. Shape indicates time point. (B) The number of differentially expressed genes (DEGs) between Eu and St samples within each cluster (adjusted P-value < 0.05, FC > 1.5). (C) Venn diagram showing the overlap in DEGs between each cluster. (D) Gene enrichment analysis of DEGs within each cluster using WormCat. The size of the bubble corresponds to the number of DEGs in that category, and the color corresponds to the P-value.
We then performed a differential expression analysis on all samples, with specific comparisons made between St and Eu samples within each cluster (adjusted P-value < 0.05, log2FC > 1.5). In general, we found that more genes were upregulated in St worms (i.e., downregulated in Eu worms). Cluster 1 had only 27 DEGs (Fig. 5B). Cluster 2 had 82 DEGs, 90% of which were upregulated in St worms. Only cluster 3 had a roughly equal number of genes that were upregulated in Eu and St worms with a total number of 55 DEGs. Cluster 3 contained almost entirely 48 h samples and represents the middle of the critical window for mouth-form development. This reduction in the number of DEGs at 48 h relative to both earlier and later time points is consistent with our observations in the environmental comparison (Fig. 2C), strain comparison (Fig. 4C), and, to a lesser extent, species comparison (Supplemental Fig. S14C). Also consistent with the environmental comparison, we found the largest transcriptomic differences during late larval development and early adulthood, with cluster 4 having 147 DEGs. Again, most of these (138 genes) were upregulated in St worms. Clusters 3 and 4 shared the greatest number of DEGs; however, the majority of DEGs were unique to a single cluster (Fig. 5C). Only one DEG was found in all four clusters (PPA36778) and is most likely a ribosomal protein. Importantly, only two mouth-form genes were significantly differentially expressed in this analysis. The sulfatase eud-1 showed significant differences in expression between Eu and St worms in clusters 1 and 3, and the sulfotransferase seud-1/sult-1 showed significant differences in cluster 3. Thus, even when comparing across multiple inputs to mouth-form development, which effectively filters the total number of DEGs, sulfation comes out as the key switch mechanism for plasticity.
We performed a gene enrichment analysis using WormCat 2.0 (Holdorf et al. 2020) on the C. elegans homologs of the DEGs from each cluster to find common pathways affecting morph development (Supplemental Table S3). We found significant enrichment of metabolic genes in every cluster (Fig. 5D). These metabolic genes included both switch genes (eud-1 and seud-1/sult-1) in cluster 3 and paralogs of each switch gene in cluster 4. Within cluster 4, there was a specific enrichment of genes involved in lipid metabolism. This is consistent with prior comparisons of gene expression between different mouth-form GRN mutants that implicated genes involved in lipid metabolism (Bui and Ragsdale 2019) and starvation response (Casasa et al. 2021) and with the recently identified role of a Mediator homolog in regulating mouth form (Casasa et al. 2023). We also found an enrichment of genes pertaining to extracellular material in cluster 4, which includes the samples in which the adult mouth is being formed. The tooth-like denticles of P. pacificus are cuticular structures composed of chitin (Sun et al. 2022b); therefore, the differential expression of collagens and matrix-secreting genes between Eu and St worms suggests that distinct extracellular matrix components contribute to the formation of each mouth form. This compositional difference could be owing to either the presence/absence of certain components or their relative quantities. In summary, comparing gene expression across multiple conditions highlighted metabolism, stress response, ribosomal function, and chromatin regulation as molecular processes that likely contribute to the regulation of mouth-form development.
Discussion
The environmental triggers and resulting phenotypes for several model systems of plasticity have been described over the past two decades, contributing to the appreciation of plasticity in development as a potential source of evolutionary novelty (Moczek et al. 2011; Simpson et al. 2011; Levis et al. 2018; Sommer 2020). However, the molecular mechanisms that mediate plasticity remain unknown beyond a few isolated genetic factors, limiting the incorporation of plasticity into standard evolutionary theory (Laland et al. 2015). Recently, genetic studies in P. pacificus have laid the groundwork for a GRN for alternative mouth forms. Here, we describe the temporal regulation of mouth-form genes in response to environmental induction and in different genetic backgrounds. When interpreted in the context of the critical window (Werner et al. 2023), these data allow us to evaluate the regulatory logic of mouth-form plasticity.
Our results highlight four tenets of genetic regulation of mouth-form plasticity. First, we found that only a subset of the GRN is differentially expressed by environmental conditions. This indicates that not all genes that have dominant effects on a plastic trait are involved in its environmental induction. However, it is possible that other conditions could reveal different patterns of environmental induction or that these genes are post-transcriptionally regulated by the environment. Moreover, only two mouth-form switch genes were differentially expressed during the critical window for mouth-form development. This finding argues that of the many genes that comprise a GRN, there may be only one to two major switch genes controlling the developmental decision of a plastic trait.
Second, transcriptional regulation of the GRN is largely limited to discrete time points. When upstream genes are mutated or overexpressed, the effect on downstream genes is (with some exceptions) confined to their wild-type time points of regulation, indicating that nodes in plasticity networks are under temporal regulatory constraint.
Third, regulatory circuits exist between genes that code for enzymes with different functions (e.g., nag-1/nag-2, eud-1, seud-1/sult-1, and lsy-12). Some of these links, like nag-1/nag-2 and eud-1, were known through genetic epistasis (Sieriebriennikov et al. 2018). However, other apparent regulatory connections, such as between nag-1/2 and lsy-12, were unexpected. The molecular mechanisms through which these enzymes affect the mouth-form decision are not well understood. One hypothesis is that this circuit is connected by signaling molecules that are either directly or indirectly linked with the abundance of their substrates and products. For instance, our results underscore the importance of sulfation as a key switch mechanism for mouth-form plasticity. Sulfation is an important biochemical mechanism for regulating the activity of signaling molecules, metabolizing xenobiotics, and generating diverse extracellular ligands (Strott 2002). In our data, the sulfatase eud-1 and sulfotransferase seud-1/sult-1 were the only mouth-form genes to be differentially expressed during the critical window in both environmental and strain comparisons and were the only mouth-form genes to be differentially expressed in the combined analysis. Human homologs of these enzymes modify a variety of molecules, including hormones, glycosaminoglycans, cerebrosides, and catecholamines (Chapman et al. 2004; Hanson et al. 2004; Gamage et al. 2006). The molecules that are modified by these two enzymes in Pristionchus are currently unknown (Igreja and Sommer 2022). Identification of these substrates is necessary to determine whether EUD-1 and SEUD-1/SULT-1 act antagonistically on the same substrate, or on separate substrates, and to clarify their mode of action in mouth form. Going forward, it will also be important to see if any of the substrates used by enzymes in the GRN are affected by the environment (i.e., through diet, temperature, or pheromones).
Fourth, the two major switch genes, eud-1 and seud-1/sult-1, appear to act sequentially. This observed pattern of differential expression leads to a model in which elevated expression of eud-1 before and during the critical window sets an initial trajectory for mouth-form development. seud-1/sult-1’s expression in J3 and J4 larvae provides a second checkpoint closer to the end of the critical window, with binary mouth-form outcomes depending on the balance between eud-1 and seud-1/sult-1’s expression. This conceptual model is reminiscent of a binary decision diagram (Akers 1978), ultimately yielding a Boolean outcome: omnivore (Eu) or bacterivore (St) (Fig. 6). Environmentally sensitive, sequential checkpoints enable the juvenile worm to alter its developmental trajectory up until the end of the critical window. Identification of sequential regulation in other systems of plasticity would support its role as a general mechanism for integrating environmental information across development.
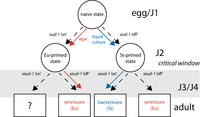
Conceptual model for how switch genes act sequentially across development to regulate mouth-form plasticity. Mouth-form outcomes given in boxes are based on phenotypes of mutants except for the outcome when both eud-1 and seud-1/sult-1 are “on,” as that result is unknown.
Beyond the known GRN, we also found that metabolic genes were differentially expressed between Eu and St worms throughout development, corroborating previous results that mouth form is linked with metabolism in Pristionchus (Bui and Ragsdale 2019; Casasa et al. 2021, 2023). Notably, this induction begins prior to the critical window and may therefore feed into the regulation of switch gene transcription. Diet and metabolic state are known effectors of plasticity in many established systems (Pfennig 1990; Emlen 1994; Moczek 1998; Simpson et al. 2011). Thus, deciphering the molecular mechanisms connecting diet and metabolism to the Pristionchus mouth form may provide broad insight into a common biological phenomenon.
Finally, we also assessed how patterns in expression of the GRN have changed across evolutionary time. These comparisons yielded a couple results that merit further discussion. First, we found large transcriptomic differences between Eu- and St-biased strains at the t′ = 0 time point. Given that these samples include pooled strains from the same species with tightly synced development that were grown under the same conditions, our best interpretation for these results is that Eu- and St-biased strains are primed for different developmental trajectories from early development. One possible mechanism for such priming could be differences in maternally deposited mRNAs. Although little is known about maternal mRNAs in P. pacificus, it is known that maternal mRNAs and proteins direct early development in C. elegans prior to the maternal-to-zygotic transition (Robertson and Lin 2015). A maternal effect on mouth-form development in P. pacificus males has been documented (Serobyan et al. 2013). Males are generally St biased (70% St), but males from St hermaphrodites are ∼100% St. It is unknown how St hermaphrodites bias mouth-form development in their male progeny, but possible mechanisms include deposition of maternal mRNAs.
Second, we observed unexpected differences in switch gene expression across the strains and species used in our study. Although seud-1/sult-1 was upregulated at 48 h in St worms across all conditions tested, eud-1 was not differentially expressed between the Eu- and St-biased strains and sister species during the critical window. This is consistent with prior work showing that inbred lines of P. exspectatus, which had been selected for different mouth-form bias, showed differential expression of seud-1/sult-1 but not eud-1 (Bui et al. 2018). In contrast, however, Ragsdale et al. (2013) found eud-1 to be upregulated in P. pacificus relative to P. exspectatus. This discrepancy regarding eud-1 expression could be because of differences in the annotations or pipelines used for the different experiments. Ragsdale et al. used FPKM to quantify gene expression relative to all genes and did not test for significance between the two species. A recent quantitative trait locus (QTL) study between two strains of P. pacificus found differences in cis-regulatory elements of eud-1 that contributed to different mouth-form biases (Dardiry et al. 2023). Thus, our results, together with prior work, suggest that evolution has acted on the regulation of both switch genes to affect mouth-form bias. Identifying how these genes are regulated and how their enzymatic functions enact the cascade of events leading to each morph are the next key questions in this field.
Methods
Nematode strains and maintenance
The reference P. pacificus strain PS312 (RRID:WB-STRAIN:WBStrain00047433) was used for the environmental samples (NGM agar plates and S-medium liquid culture). All other strains were grown only on NGM agar plates. The following mutants were used: Ppa-eud-1 (tu1069), Ppa-Ex[eud-1] (RS2561), Ppa-lys-12 (tu319), Ppa-sult-1 (tu1061), Ppa-nag1/2 (RS3195/tu1142/tu1143), Ppa-nhr40 (tu505), and Ppa-mbd-2 (tu365). The eud-1 mutant is a novel CRISPR-Cas9-induced 10 bp deletion in exon 3, whereas all other mutants were previously published ethyl methanesulfonate (EMS)–induced or CRISPR-Cas9-induced mutations (Table 1). However, an inspection of the sequencing reads from the Ppa-sult-1 mutant line revealed that despite having a mutant phenotype, this line does not contain the reported frameshift mutation: It has a 9 bp deletion rather than the previously reported 10 bp deletion in the ninth exon (Supplemental Fig. S17, Namdeo et al. 2018). Four strains of P. pacificus (RS5410, RS5427, RSC017, and RSA100) and a strain of P. exspectatus (RS5522B) were used to assess the role of genetic background on the mouth-form GRN. The mouth-form ratios of three strains were empirically determined by us and that of RS5410 was adapted from Figure 4 of Ragsdale et al. (2013) (Supplemental Fig. S12A). For general maintenance of the nematode cultures, five young adults were passed every 4–6 days on 60 mm NGM-agar plates at 20°C seeded with 300 µL of overnight cultures of Escherichia coli OP50 (grown in LB medium at 37°C) and covered with parafilm. Majority hermaphrodite (>95%) cultures were used for all experiments.
Phenotyping of daf-12 mutants
We phenotyped worms from two different mutant lines of the nuclear hormone receptor daf-12 (Supplemental Fig. S18). RS2209 (tu381) has a mutation in the splice acceptor of the hinge region, and RS2272 (tu390) has a premature stop codon in the ligand binding domain (Ogawa et al. 2009). We grew worms on NGM agar plates and determined the mouth-form ratio (% Eu) of each strain and wild-type PS312. Error bars represent SEM for n = 3 independent biological replicates, with more than 20 worms phenotyped per replicate.
RNA sequencing
Developmental RNA-seq was performed on worms from different strains and grown in different environmental conditions (NGM agar plates and S-medium liquid culture) at the time points described in Figure 1C and Supplemental Figure S1A. Details regarding the culture state, sampled time points, number of replicates, and strain IDs for each condition are given in Supplemental Table S1. To synchronize worms, we performed cuticular disruption of gravid adults with bleach/NaOH (Stiernagle 2006) and aliquoted eggs to J1s to NGM-agar plates or S-medium liquid culture OP50 E. coli (Werner et al. 2017) or, for t′ = 0, directly resuspended in 500 µL TRIzol (Ambion 15596026). Thus, the agar t′ = 0 time point represents the start of both the agar and the liquid culture experiments for the environmental comparison and is composed of both embryos and some J1 larvae. Liquid cultures were incubated at 180 rpm, 20°C–22°C. Worms at different stages were then collected at corresponding time points by washing from plates with M9, or simply decanting from liquid culture, and filtering through 5 µM filters. Subsets of adult worms (10–20) were confirmed visually to have the expected mouth form for that condition. Washed worms were then resuspended in 500 µL TRIzol and freeze-thawed 3× between 37°C and a small liquid nitrogen container to disrupt the cuticle. RNA was then extracted following the manufacturer's protocol and purified using RNA Clean & Concentrator-25 columns (Zymo R1017). RNA was then eluted in 50 µL water and quantified by NanoDrop (A260/280). Five hundred nanograms to 1 µg of total RNA was converted to Illumina sequencing libraries using the NEB Ultra II directional RNA library prep kit with sample purification beads (NEB E7765), with 10–14 PCR cycles (determined empirically to achieve the minimum number of cycles per library). Libraries were then sequenced on an Illumina HiSeq 2000 in single-end mode, except for the 12 h time points, which were sequenced in paired-end mode. We kept samples with a minimum of 3 million mapped reads and average quality scores greater than 30.
Read mapping
We used Salmon 1.3.0 (Patro et al. 2017) to map P. pacificus sequencing reads to the P. pacificus transcriptome (El Paco annotation, version 2) and P. exspectatus reads to the reference transcriptome (Rödelsperger et al. 2014). We first indexed the El Paco transcriptome and then quantified the sequencing reads against this index, correcting for GC bias.
Differential expression analysis
We imported the quantified reads to RStudio (version 2022.2.3.492) (RStudio Team 2020) using tximport and performed differential gene-expression analysis using DESeq2 (Love et al. 2014) and R (version 4.2.0) (R Core Team 2024). We first did a differential expression analysis on each condition (agar, liquid culture, individual strains, individual mutant lines, P. exspectatus samples) to check that the replicates from the same time points clustered together within each condition (Supplemental Figs. S2–S7). We then moved forward with the analysis and compared gene expression across different conditions.
We performed separate analyses for each of the different conditions and compared samples at the same time point using DESeq2 (design = ∼MF_time) for environmental and genetic background comparisons and DESeq2 (design = ∼condition) for mutant comparisons. We did not impose a fold change threshold but considered all genes with an adjusted P-value < 0.05 to be significantly differentially expressed. Plots were made using base R or ggplot2 (Wickham 2016).
For the species comparison, we identified genes that were 1:1 orthologs through reciprocal protein BLASTing with an e-value of 1 × 10−3. This resulted in 11,556 1:1 orthologs between the two species. We found more orthologs when querying P. exspectatus against P. pacificus, but the number was greatly reduced when we queried P. pacificus proteins against P. exspectatus. However, this pipeline excluded both mbd-2 and eud-1, two genes that should be 1:1 orthologs. The problem appeared to be the poor P. exspectatus annotation. We were able to identify the P. exspectatus annotation for eud-1 and add it back into the data set, but we could not identify the P. exspectatus annotation for mbd-2, so it was left out of the species comparison. We then gave the P. exspectatus reads the P. pacificus ortholog's gene ID and performed a differential expression analysis between species by time point using DESeq2 (design = ∼MF_time). This resulted in a comparison of 11,504 transcripts with a nonzero total read count compared with 22,631 transcripts in the initial species comparison.
To identify a core list of genes that are differentially regulated in St and Eu worms, we performed a differential expression analysis on all samples from the different conditions together. We first ran DESeq2 on all samples with replicates grouped such that the comparison was between all unique sample types (i.e., agar samples from the zero time point were compared against all other samples, including other agar samples). We performed a principal component analysis on the log2-transformed results and noted that the first principal component explained 65% of the variance in gene expression (Fig. 5A). The samples separated along this principal component according to time point. We plotted the dissimilarity between samples against the number of clusters and noted diminishing returns when dividing the samples between more than four clusters (Supplemental Fig. S18). We then performed k-medoid clustering to subset the samples into four clusters. This code was adapted from Theska et al. (2020). Finally, we ran DESeq2 on all samples, with comparisons being made between Eu and St samples within each cluster (design = ∼MF_clust). This produced a list of DEGs for each cluster with fold change threshold of 1.5 (lfcthreshold = 0.585) and >95% confidence (adjusted P-value < 0.05). All code used to generate these data is included in the Supplemental Code file.
Gene enrichment analysis
We used BLASTP to identify C. elegans orthologs and WormBase IDs of the P. pacificus genes that were differentially expressed between Eu and St worms (see Supplemental Code). Only 53% (134 genes) of the P. pacificus DEGs from the combined analysis had a BLAST hit. Gene enrichment analysis was completed using WormCat 2.0 (Holdorf et al. 2020) on the WormBase IDs.
Shiny App
We made a shiny app in R (version 4.3.3) (R Core Team 2024) that plots the normalized expression of any Pristionchus gene in our different conditions across development. The app extracts the values from a spreadsheet that corresponds to the All_samples_ntd tab in Supplemental Table S4. The custom code used to generate the app is available at GitHub (https://github.com/drjuliejung/GRN-shiny) and as Supplemental Code.
Data access
The RNA sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA628502.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Thomas King and Audrey Brown for thoughtful critiques of the manuscript. This study was funded by the Max Planck Society and National Institutes of Health (National Institute of General Medical Sciences) grant R35GM150720-01 to M.S.W.
Author contributions: M.S.W. and R.J.S. conceived and designed experiments for sampling and sequencing RNA. M.S.W. conducted RNA-seq with assistance from T.L. S.R. performed all bioinformatic analyses with assistance from M.S.W. J.J. produced the Shiny app from the data. S.N. phenotyped daf-12 mutants. S.R. wrote the manuscript with assistance from M.S.W.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279783.124.
-
Freely available online through the Genome Research Open Access option.
- Received July 14, 2024.
- Accepted April 30, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








