
Abstract
Individuals living with rare diseases often undergo a frustrating and expensive diagnostic odyssey. Clinical geneticists who analyze exome or genome sequencing data from rare disease patients often encounter a list of variants of uncertain significance (VUS) in known disease-causing genes or rare variants in genes of uncertain significance (GUS) that are difficult to interpret, even with the integration of the latest bioinformatic tools. In this Perspective, we review how studies using the fruit fly Drosophila melanogaster have facilitated rare disease diagnosis by uncovering the clinical relevance of GUS and classifying rare variants into specific allelic categories (loss-of-function or gain-of-function, Muller's morphs). We showcase how fly researchers have been collaboratively studying the loss-of-function of orthologous fly genes, assessing the ability of the human genes to rescue the fly mutant phenotypes, determining the effect of overexpressing human proteins, and testing functional consequences of rare variants of interest by generating analogous fly mutants to contribute to rare disease diagnosis. We argue that data obtained using Drosophila can be leveraged to design effective multiplexed assays for variant effects (MAVEs) to decipher the vast human variome.
The burden of undiagnosed diseases and the importance of functional studies
The United States classifies a disease as rare if it affects less than 200,000 people (Orphan Drug Act of 1983, Public Law 97-414, 96 Stat. 2049). Even with this stringent criterion, there are more than 10,000 rare diseases documented, and it is estimated that one in 10 people within the United States is affected by a rare disease (Haendel et al. 2020). The process to receive a diagnosis for people living with rare diseases, on average, takes 6 years (Benito-Lozano et al. 2022). This takes a toll on, not only the individual, but also their family, friends, and society (Graungaard and Skov 2007; EveryLife Foundation for Rare Diseases 2023). It is estimated that over 70% of rare diseases are caused by genetic variants (Wakap et al. 2020). With the improvements in accuracy, advances in speed, and reduction in the cost of DNA sequencing, it has become possible to rapidly identify most variants in the genome of a single individual (Gorzynski et al. 2022; http://www.genome.gov/sequencingcostsdata). Most clinical geneticists follow the American College Medical Genetics/Association for Molecular Pathology (ACMG/AMP) guidelines to classify genetic variants into five categories: pathogenic, likely pathogenic, benign, likely benign, and variant(s) of uncertain significance (VUS) (Richards et al. 2015). Whereas the first four classifications could be useful in interpreting rare variants that are found in genes that have been previously linked to human diseases, many individuals (including people without rare diseases) also carry many rare variants in gene(s) of uncertain significance (GUS) that cannot be classified into these categories due to the uncertainty at the gene level. Currently (as of August 2024), even though the Human Genome Organization Gene Nomenclature Committee (HGNC) has annotated 19,261 protein coding genes in the human genome (Seal et al. 2023), only 4923 genes have been associated with human phenotypes according to the Online Mendelian Inheritance in Man (OMIM) (Hamosh et al. 2021). In addition, there are 9103 genes that encode non-coding RNAs, the majority of which have yet to be linked to Mendelian disorders or traits. Therefore, most human genes are GUS, especially in the context of rare diseases, and many are part of the “ignorome,” which consists of functionally enigmatic genes on which no one has performed functional studies in any biological context (Pandey et al. 2014). One effective way to resolve VUS or GUS is to identify other individuals with similar genotypes and phenotypes. Matchmaking databases such as GeneMatcher (Hamosh et al. 2022) and PhenomeCentral (Osmond et al. 2022) that are interconnected through the Matchmaker Exchange (Boycott et al. 2022) platform can be used to facilitate this process. However, because current matchmaking efforts are quite labor-intensive and largely depend on the luck of matching cases being submitted to the same database or other nodes of Matchmaker Exchange, complementary approaches are required to fill the gap. Although a number of bioinformatic tools have been developed to predict the likely pathogenicity of genetic variants, there is still a lot of room for improvement (Ghosh et al. 2017).
Experimental studies are particularly useful to determine whether the variant of interest affects the function or expression of the gene of interest. For some genes, well-established functional assays can be utilized (e.g., enzymatic assay, transcriptional reporter assay, cell survival/proliferation assay). However, for most genes, there are no standardized tests that have been established and implemented. Recently, high-throughput approaches collectively referred to as multiplexed assays for variant effects (MAVEs) have been explored (Gasperini et al. 2016; Starita et al. 2017). In principle, MAVEs have the power to reveal the functional consequences of every possible genetic variant in a given gene for a specific molecular function that can be screened in vitro or in cultured cells. According to MaveDB (Esposito et al. 2019) and MaveRegistery (Kuang et al. 2021), MAVE data sets have been published for ∼600 human genes. To design an effective MAVE that can classify a VUS into pathogenic or benign, several data sets need to be collected. First, it is important to have some idea regarding the mechanism by which previously known pathogenic variants cause the disease. For example, if the disease of interest is caused by a loss-of-function (LOF) mechanism, the level of RNA or protein of interest can be used as a functional readout. However, if the disease is caused by a gain-of-function (GOF) mechanism, a readout that reflects the specific activity of the RNA/protein will be required. Second, a set of well-defined pathogenic and benign variants that can be used as positive and negative controls are necessary to calibrate the sensitivity and specificity of the assay. Whereas such information is often readily available for well-established disease-causing genes, it is very difficult to develop an effective MAVE for diseases that are ultra-rare and for GUS that have yet to be clearly linked to a clinical condition. In addition, although approaches that utilize specific cell types derived from human stem cells such as induced pluripotent stem cells (iPSC) or harness the power of organoids that mimic organ systems are being actively explored to establish MAVEs that reflect in vivo conditions, most MAVEs are performed in vitro or using limited cell lines. This makes the evaluation of the impact of a VUS on non-cell-autonomous or noncanonical functions very challenging, especially for diseases that are caused by defects in cell type–specific functions of genes.
Over the last decade, in vivo functional studies using genetic model organisms have made significant contributions to resolve VUS and to reveal the function of GUS that have been associated with rare diseases (Yamamoto et al. 2024). In this Perspective, we will introduce how the fruit fly, Drosophila melanogaster, can be used to characterize the functional consequences of novel variants that are potential causes of rare diseases and highlight a number of rare disease gene discovery papers in which fly geneticists have played a critical role. We will further discuss how this organism can be used to further classify pathogenic variants into different allelic categories to begin to reveal the underlying pathogenic mechanism of newly discovered clinical conditions. In this Perspective, we will guide clinical researchers to understand how experimental data obtained from flies can support the discovery and characterization of novel clinical conditions. In the Supplemental Text, we provide additional practical tips and technical insights to help support Drosophila scientists to begin to engage in these types of research projects. Such information can be further leveraged to design effective MAVEs to decipher the vast human “variome” (Cotton et al. 2008) to facilitate the diagnosis and therapeutic research on rare and ultra-rare diseases.
Approaches and strategies to study VUS and GUS in Drosophila
Research using fruit flies has played a fundamental role in establishing the field of genetics. In the early 1900s, pioneering studies performed by Thomas H. Morgan and his students demonstrated the principles of how genes are organized on chromosomes and how various genetic traits can be linked together using Drosophila. In the 1930s, Herman Muller, who was the founding president of the American Society of Human Genetics, discovered that genes can be mutated in different ways using Drosophila and coined specific terminologies (amorph, hypomorph, hypermorph, antimorph, neomorph, isomorph) to classify them, which are now known as “Muller's morphs.” The discovery of Hox genes and developmental signaling pathways by Ed Lewis, Christiane Nüsslein-Volhard, Eric Wieschaus, and many other fly researchers in the late twentieth century has increased the understanding of embryonic patterning in both flies and humans. Research led by Jules A. Hoffmann and colleagues identified conserved pathways that govern innate immunity, whereas research pioneered by Seymour Benzer indicated that complex traits such as behavior are also under the tight regulation of genetic factors. The sequencing of the euchromatic portion of the Drosophila melanogaster genome (Adams et al. 2000) accelerated the utilization of fruit flies in human disease studies because 75% of human disease genes that were known at the time were found to be conserved in the fly genome (Fortini et al. 2000; Reiter et al. 2001). More recently, the high degree of conservation of both genes and genetic pathways between flies and humans has been utilized for studies of VUS and GUS (Bellen and Yamamoto 2015). For example, Drosophila is one of the model organisms that has been selected to perform functional studies by the Rare Diseases Models and Mechanisms (RDMM) network in Canada, a matchmaking platform to connect clinicians and scientists with shared interests in specific genes related to rare diseases (Boycott et al. 2020). Similar initiatives have been launched in Europe (Ellwanger et al. 2024), Japan (Takahashi et al. 2022), Australia, and Singapore, which also integrate Drosophila scientists as key members of the network (Boycott et al. 2020). In the United States, the NIH supports the Model Organisms Screening Center (MOSC) of the Undiagnosed Diseases Network (UDN) and the Center for Precision Medicine Model (CPMM) at Baylor College of Medicine, both of which use Drosophila as a key model organism to study the function of GUS and effects of VUS that are clinically relevant (Baldridge et al. 2021). Nonprofit organizations such as the Simons Foundation have also funded fly researchers who can provide functional information for VUS found in individuals with autism spectrum disorders (Post et al. 2020; Marcogliese et al. 2022). Whereas other model organisms such as yeast (e.g., Saccharomyces cerevisiae), worms (Caenorhabditis elegans), zebrafish (Danio rerio), and mice (Mus musculus) have also been used to study GUS and VUS with unique advantages (Boycott et al. 2020; Yamamoto et al. 2024), the versatility of the technology and large number of publicly available reagents available to Drosophila researchers make this model organism unique.
In the following subsections, we discuss several factors that need to be taken into account when considering the use of Drosophila in VUS resolution and outline different technical strategies that could be implemented, providing specific examples to which each strategy has previously been applied. We have generated a list of over 130 publications between 2020 and early 2024 that have used Drosophila to assess the functions of VUS and GUS in vivo (Fig. 1; Supplemental Table 1). Although this is not meant to be an exhaustive list, we hope this provides further support that Drosophila has been utilized as an effective model system to support novel disease gene discovery, phenotype expansions, and diagnosis of rare diseases in a research setting.
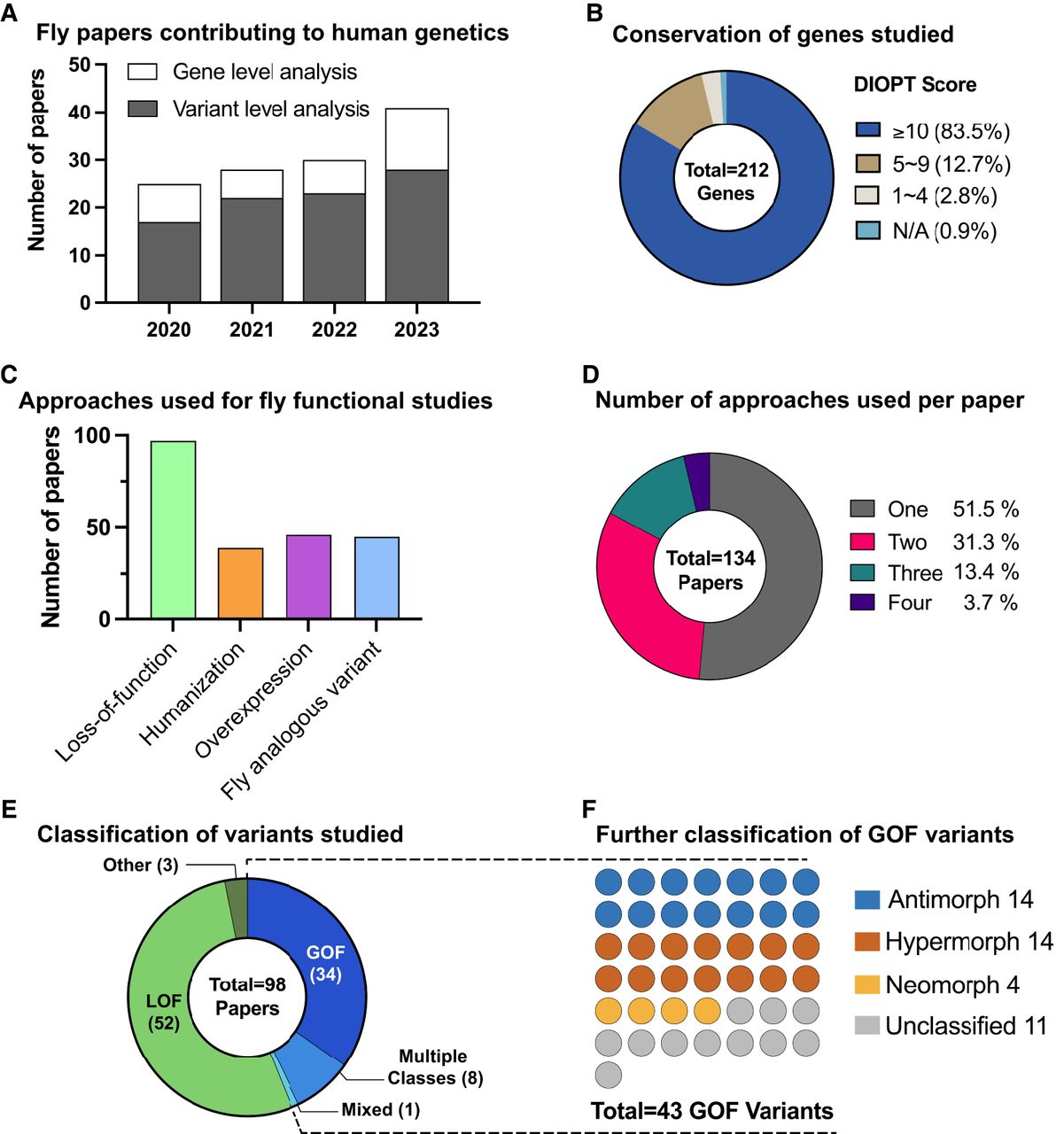
Utilization of Drosophila in rare disease gene discovery and functional classification of VUS. To compile a list of papers published using Drosophila melanogaster as a model organism to study rare disease genes and variants between January 2020 and April 2024, we first performed manual inspections of all abstracts published in the following journals where many new human disease gene discovery and phenotypic expansion papers are published: American Journal of Human Genetics, European Journal of Human Genetics, Genetics in Medicine, Genome Research, Human Molecular Genetics, Nature Genetics. We also utilized the advanced search option on PubMed by performing the search with the keywords [“Drosophila” + “Variant”, “Human Mutation”, “de novo”, “biallelic”, or “missense allele”] in the title or abstract and manually filtered the results to identify relevant papers that were published in other journals. Note that this search strategy may have omitted some relevant papers that were not captured by the selected keywords. (A) The number of the papers that studied GUS and VUS utilizing Drosophila models has continuously increased between 2020 and 2023 based on our search methods. The majority of the studies explored the function of specific variants in addition to assessing the function of the orthologous fly gene. (B) The DIOPT scores of fly-human ortholog candidate pairs used in each study. A great majority of papers studied ortholog candidate pairs with a DIOPT score of 10 or more, but some papers conducted functional studies of genes with lower DIOPT scores. (C) The percentages of papers that used each Drosophila-based functional approach in human genetic studies. Approximately 70% of papers assessed the loss-of-function of the orthologous genes as part of the functional study of the gene of interest, which are often GUS. Approximately 30% of manuscripts performed humanization of orthologous fly gene, and ∼35% of papers performed functional assessment of the VUS based on overexpression and/or fly analogous variant studies. (D) Breakdown of functional studies of rare variants by the number of Drosophila approaches employed. Whereas approximately half of the papers used a single method (Loss-of-function, Overexpression, Humanization, or Fly analogous variant investigation), the other half integrated multiple strategies. (E) Out of 98 papers that assessed the function of variants, 54 papers classified the variants of interest as LOF and 34 papers designated the variants as GOF alleles. Some studies found both LOF and GOF variants that contribute to clinical phenotypes and one study reported variants that have properties of both GOF and LOF (referred to here as “mixed” due to their mixed properties). (F) Of the 43 papers that identified GOF variants, ∼75% provided data to further classify them as specific Muller's morphs. See Supplemental Table 1 for details.
Identifying orthologous genes between humans and flies
Although studies using Drosophila have proven to be effective and successful, there are many variables that must be considered when designing, conducting, and interpreting functional studies using this model organism (Bellen et al. 2019). The first thing to check is whether the Drosophila genome possesses an orthologous gene (or genes) to the human gene of interest (Fig. 2). Whereas homologous genes can be identified relatively easily using simple sequence alignment tools, orthologous genes between different species are much more difficult to determine because one needs to take multiple factors into consideration such as phylogeny, whole genome duplication events during evolution, and gene duplication/deletion events that happen before and after speciation (Kuzniar et al. 2008; Linard et al. 2021). Among the variety of methodological approaches, Drosophila RNAi Screening Center Integrative Ortholog Prediction Tool (DIOPT) is widely used to identify ortholog candidates across multiple species, including flies and humans (see Supplemental Text 1; Hu et al. 2011). It is notable that 177 (83%) human genes among 212 that were studied in Drosophila over the past few years (Fig. 1B; Supplemental Table 1) have relatively high DIOPT scores (10 or more out of 19, DIOPT version 9.1). However, the DIOPT score alone should not be used as a sole conclusive method to judge whether the case could be successfully studied in Drosophila or not because there are some studies (∼16%) that successfully modeled specific VUS or supported new disease gene discovery using Drosophila with moderate to low DIOPT scores (1–9 out of 19). A few studies used Drosophila to evaluate the function of genes and variants that lack any predicted orthologs, which we will discuss further below. Often, one fly gene may correspond to multiple human genes due to two rounds of whole genome duplication events that likely happened in the vertebrate lineage (Holland 2003). In addition, there are scenarios in which a gene family has been amplified in the invertebrate lineage and cases in which multiple fly genes are orthologous to multiple human genes.
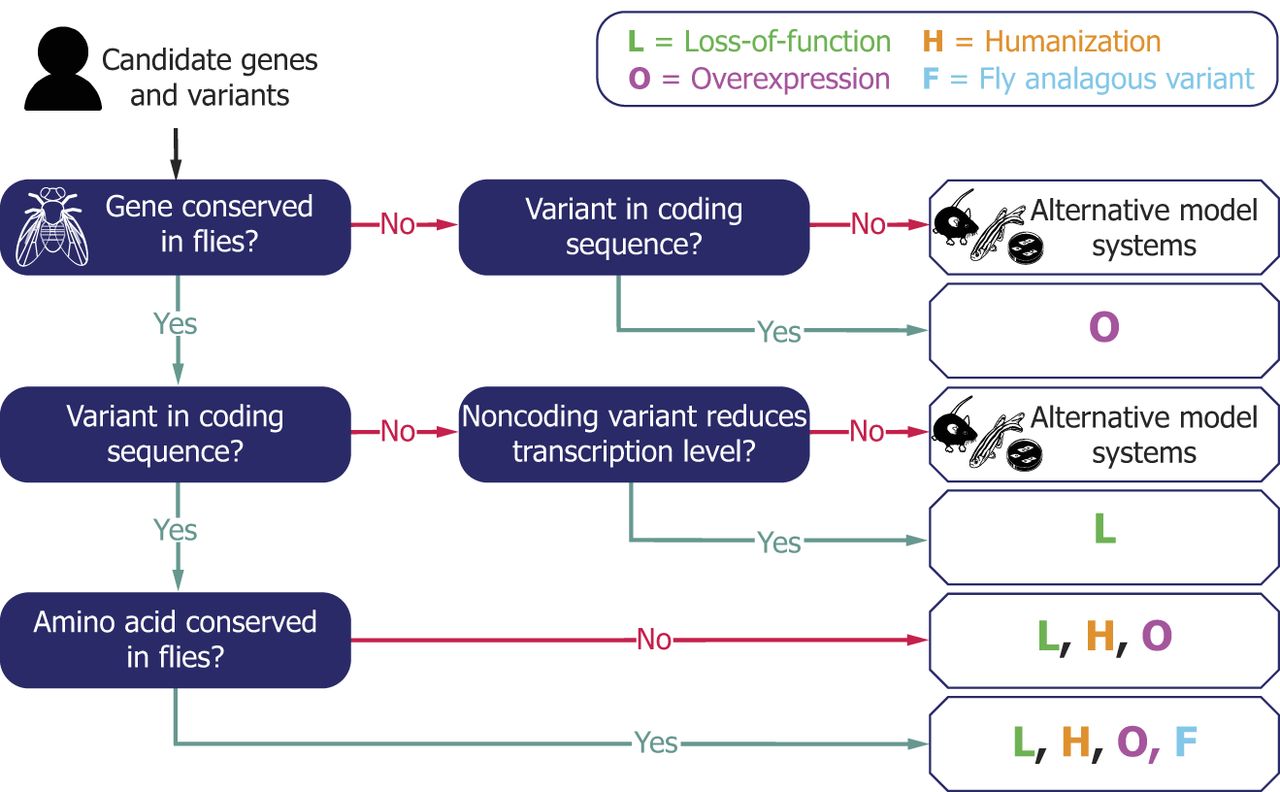
A workflow of utilizing Drosophila models for functional studies of GUS and VUS. An example of decision-making framework for employing Drosophila models in GUS and VUS interpretation. Four different strategies (L: Loss-of-function, H: Humanization, O: Overexpression, F: Fly analogous variant) can be selected depending on the characteristics of candidate genes and alleles. Multiple strategies could be utilized together to further support the findings.
Understanding the mutant phenotypes and expression pattern of orthologous fly genes
Before studying the function of a specific human genetic variant in vivo, it is useful to know what kind of phenotypes can be caused by manipulating the model organism gene. Amorphic (null) alleles that completely lack gene expression or function serve as an important reference point to interpret other types of alleles, such as hypomorphs (reduced activity or abundance), hypermorphs (increased activity or abundance), antimorphs (often referred to as “dominant negative” [Herskowitz 1987]), and neomorphs (gain of toxic/altered function or ectopic expression) (Muller 1932). In this Perspective, we group amorphic and hypomorphic alleles as loss-of-function alleles, and hypermorphic, antimorphic and neomorphic alleles as gain-of-function alleles. Although we acknowledge that there are some scientists who refer to antimorphic alleles as a type of LOF mutation (because the key function of the protein is often lost and their phenotype often resembles that of amorphic and strong hypomorphic alleles), the fact that these variants “gained a function to inhibit the endogenous copy of itself or paralogous genes” justifies the classification of these variants as a GOF allele.
Since the identification of the first mutant allele (amorphic allele of the white gene [Morgan 1910]), fly researchers have generated a great number of mutant strains (see Supplemental Text 2). Although it can be highly encouraging if the symptoms seen in patients have links to previously identified fly mutant phenotypes (i.e., in this case, mutant flies can be considered as a “disease model”), one should not be discouraged from pursuing functional studies of GUS and VUS in this model organism even if the previously reported phenotypes appear unrelated. Indeed, some clinical phenotypes are very difficult or impossible to model in flies as insects lack certain organ systems (e.g., endoskeleton, adaptive immune system), have significantly different anatomical features (e.g., cardiovascular system, nervous system), and do not seem to carry out sophisticated functions (e.g., higher cognition). Even when studying clinical cases that relate to these conditions, various phenotypes in flies can be used as readouts of gene and variant function. For example, bone morphogenetic protein (BMP) signaling is utilized in diverse developmental contexts in flies although this model organism lacks bones. In some cases, nonobvious phenotypic links based on shared molecular pathways may be present between orthologous genes (i.e., phenologs) (McGary et al. 2010). In other cases, flies can be considered as “living test tubes” that can provide qualitative or quantitative readouts of variant function. It is, of course, important not to overinterpret the data obtained in Drosophila because some genes may play species-specific roles. Also, when conducting an experiment in flies (and in other species), it is also essential to attempt to validate an observation reported in the literature because some information annotated in model organisms databases such as FlyBase is based on high-throughput screens, which may reflect nonspecific phenotypes that were caused by genetic background or other technical issues such as experimental artifacts.
When and where the gene of interest is expressed is also an important piece of information that should be taken into consideration when using model organisms for functional studies. Although gene expression data alone do not provide functional information, one can utilize this information to design probing experiments, especially when cell type– or tissue-specific functional information is needed. FlyBase integrates gene and protein expression information for individual genes from various sources (see Supplemental Text 2). Such data could be compared with gene expression data in human (e.g., GTEx [The GTEx Consortium 2013], CZ CELLxGENE [CZI Cell Science Program et al. 2025]), other vertebrate model systems (e.g., mouse, rat, zebrafish), as well as phenotypic data from rare disease patients to generate hypotheses regarding molecular mechanisms of pathogenicity. In some cases, the expression level of the fly gene of interest is below the detection level of single-cell RNA sequencing technology. T2A-GAL4 and Kozak-GAL4 lines are genetic tools that have been developed over the last decade that can simultaneously provide both expression and phenotypic data for a given gene of interest with high specificity and sensitively (see Supplemental Text 3; Lee et al. 2018; Kanca et al. 2022). In addition, these reagents can be used to “humanize” Drosophila genes, which will be discussed in the following sections.
Identifying similarities between patient and fly mutant phenotypes
When the human gene of interest is a GUS, variants that are highly likely to function as LOF alleles (e.g., nonsense, frameshift, variants that disrupt canonical splice sites, deletions) may be flagged as VUS, especially when there is limited to no information about the function of the gene of interest in vertebrate systems. LOF human variants can often be studied by investigating the effect of LOF of the orthologous fly gene (Fig. 3A). If similarities between phenotypes of Drosophila mutants and rare disease patients can be identified, such data can be utilized to provide supporting data on pathogenicity (Fig. 2). In addition, this can lead to deeper understanding of the human gene's function and potential disease mechanisms in an in vivo model. This approach seems to be the most common strategy used in recent publications (72% of papers) (Fig. 1C), likely due to the large amount of publicly accessible fly reagents that are LOF alleles.
Overview of methods used to study GUS and VUS in Drosophila. Multiple complementary techniques can be used to study the function of candidate genes and variants identified as a novel disease candidate in patients. (A) Loss-of-function (LOF) studies look at the consequences of complete loss or reduction of the fly ortholog's function. This can happen through (1) mutant alleles that limit/eliminate protein function, (2) T2A-GAL4 cassettes inserted into the first intron, causing only a short portion of the protein to be made that is likely LOF, or Kozak-GAL4 cassettes that replace the coding sequence of the gene and either knock down or knock out the gene, and (3) transgenic RNAi lines to knock down the endogenous transcripts. (B) Humanization studies replace the expression of the fly ortholog with a human ortholog. This can either be accomplished by (1) combining a fly ortholog LOF strategy with cDNA expression, or (2) using CRISPR to knock the human cDNA with the variant into the endogenous fly locus. (C) Overexpression assays express the human/fly cDNA ubiquitously or in a tissue of interest using a GAL4 line, without manipulating the endogenous fly ortholog of the human gene of interest. (D) When the variant of interest affects a conserved amino acid, the fly analogous variant approach can be utilized. This can be accomplished by (1) combining a fly ortholog LOF strategy with fly cDNA expression, (2) generating a knock-in allele by directly targeting the endogenous fly ortholog gene based on genome editing, or (3) overexpressing the fly cDNA ubiquitously or tissue-specifically using various GAL4 lines.
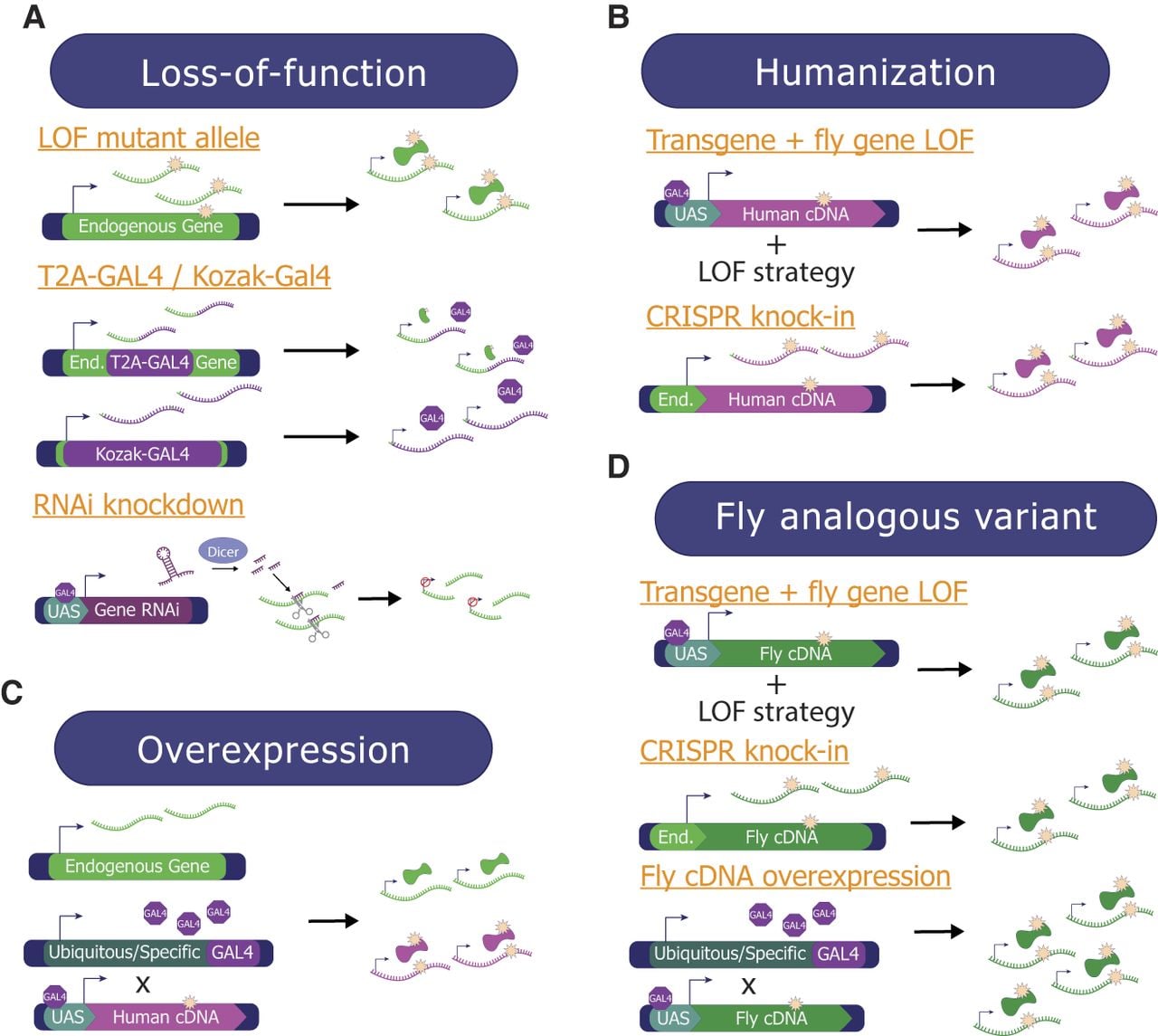
Currently, over 62,000 fly lines covering 12,500+ genes that are designed to function as LOF alleles (e.g., mutant alleles including lines from forward genetic screens, T2A-GAL4 and Kozak-GAL4 lines, deletions, RNAi lines, and gRNA lines) are readily available through public fly stock centers, making LOF studies more easily accessible than strategies that require new reagents (see Supplemental Text 4). For example, one study investigated the role of the microRNA processor encoded by DROSHA using previously generated fly stocks with EMS (ethyl methanesulfonate)-induced LOF mutations (Smibert et al. 2011; Pressman et al. 2012; Luhur et al. 2014) of its fly ortholog drosha (Barish et al. 2022). The small brain phenotype that was documented in the fly drosha mutants provided in vivo information to support the hypothesis that the microcephaly phenotype found in patients with rare genetic variants in DROSHA was likely caused by LOF of this gene. In another study, a homozygous mutant allele of fly Sodh2 (ortholog of human SORD) generated in a large-scale gene disruption project (Bellen et al. 2011) showed loss of synaptic terminals in the visual system and age-dependent worsening of locomotor activities accompanied by increased sorbitol levels (Cortese et al. 2020). This recapitulated some of the key phenotypes seen in patients with LOF variants in SORD, showing that this LOF fly reagent can be used as a model to study the disease mechanism and molecular pathogenesis. Indeed, this study identified aldose reductase inhibitors (e.g., Epalrestat, Ranirestat) as a potential therapeutic approach because fly mutant phenotypes were significantly improved by drug treatment. In more recent years, researchers have been using CRISPR-based approaches to proactively generate strong LOF alleles in genes that have not yet been well characterized in flies. Deletion alleles generated using CRISPR have been used in studies to support the pathogenicity of LOF function variants in multiple GUS (Kim et al. 2020; Wormser et al. 2021; Mattioli et al. 2023). T2A-GAL4 (Lee et al. 2018) and Kozak-GAL4 (Kanca et al. 2022) lines mentioned above have also been used as LOF alleles to provide supporting evidence for novel human disease discovery papers (Fig. 3A; Supplemental Table 1).
It is important to note that there are some drawbacks of studying strong LOF alleles of fly genes in the context of VUS and GUS research. For example, ∼30% of fly genes are estimated to be essential for life, and strong LOF alleles may not allow the study of tissue-specific functions of genes and variants that are clinically relevant. These limitations can be overcome by techniques that allow investigation of tissue- or cell type–specific function of genes such as RNAi (RNA interference) (Fig. 3A; see Supplemental Text 5). Several large collections of transgenic fly lines that allow the conditional expression of long double-stranded RNA or short hairpin sequences based on the UAS/GAL4 system have been made publicly available from Bloomington Drosophila Stock Center, Vienna Drosophila Resource Center, and the National Institute of Genetics in Japan. When crossed to GAL4 lines that are expressed in specific cell populations, one can reveal cell type– or tissue-specific function of essential genes that may have been masked by lethality. For example, neuron-specific knockdown of fly Isha (ortholog of human SCAF4 as well as SCAF8) caused phenotypes affecting locomotor function, learning, and short-term memory, which was accompanied by structural defects in synapse development (Fliedner et al. 2020). This finding helped to establish SCAF4 as a novel neurodevelopmental disease gene in humans.
Studying the functional impact of coding variants based on “humanizing” fly genes
Whereas studying phenotypes of existing or new LOF fly mutants can help us understand the function of GUS and mechanisms underlying diseases linked to human LOF variants, the main limitation of this approach is that it can only provide information at the gene level. The functional impact of missense alleles and late-truncation alleles that escape nonsense-mediated decay are much more difficult to predict compared to early-truncations, canonical splicing variants, and deletion alleles. “Humanization” of an orthologous fly gene is a powerful approach to providing functional information for these types of variants (Fig. 3B), which have been used in 29% of recent publications (Fig. 1C). This can be achieved by rescuing the fly mutant phenotype with reference or variant human protein (often introduced as a transgene that carries a human cDNA) and comparing their effectiveness. When a reference human protein can rescue a fly mutant phenotype, one can say that the molecular function of the human and fly genes is conserved. In addition, VUS that affect amino acids which are not conserved between flies and humans can be studied using this approach.
Humanization of a fly gene can be conducted based on a rescue paradigm that combines T2A-GAL4 alleles with UAS-human cDNA lines. Using this strategy, the human protein of interest can be expressed in the same spatiotemporal expression pattern as the fly protein in a LOF mutant background. If the reference and variant UAS-human cDNA transgenes are integrated into the identical genomic locus using a site-specific transgenesis system based on phiC31 (Venken et al. 2006), one can assess whether the reference human protein can rescue the fly mutant phenotype and further assess how the variant human proteins perform in comparison (Fig. 4A; Pineda et al. 2021; Pan et al. 2023). Although the rescue of the fly mutant phenotype may not always be complete, valuable information can be obtained as long as one can observe a functional difference between the reference and the variant of interest. In one study, reference human TIAM1 was shown to be able to partially rescue the semilethality caused by loss of fly sif (ortholog of human TIAM1 as well as TIAM2). In contrast, a missense variant (p.R23C) in this gene found in monozygotic twins with developmental delay, intellectual disability, and seizures was not able to rescue this defect, leading to the conclusion that this VUS is a strong LOF allele (Lu et al. 2022). Kozak-GAL4 lines can also be combined with UAS-human cDNA lines to achieve a similar goal. For example, reference human TOMM70 cDNA was able to rescue the lethality in homozygous Kozak-GAL4 mutants of fly Tom70 (orthologous to human TOMM70), whereas the two rare variants (p.T607I and p.I554F) found in patients with severe neurological symptoms were classified as partial LOF alleles because they rescue less efficiently. This study also performed rescue of eye morphology and synaptic transmission phenotypes caused by eye-specific knockdown of Tom70 (GMR-GAL4, UAS-Tom70-RNAi) using reference or variant UAS-TOMM7 transgenes. The two variants again showed less potency in their ability to rescue these defects compared to the reference allele, indicating these variants are likely to be hypomorphs (Dutta et al. 2020). Humanization of fly genes can be achieved by combining other genetic methodologies (see Supplemental Text 6), making this a powerful methodology to study GUS and VUS.
Functional classification of VUS based on examining the phenotypic difference between the reference and variant alleles in flies. Both rescue and overexpression-based approaches can utilize various phenotypes such as lethality, fertility, lifespan, morphological defects, or behavioral phenotypes as functional readouts of gene function. (A) Rescue-based approaches to assess patient variants require a scorable phenotype caused by LOF of the fly ortholog that is rescued by expression of the reference allele. If the VUS is a LOF, expression of the variant would be unable to rescue to the same level as the reference. (B) Overexpression strategies can be used to classify the variant of interest based on comparing the phenotypes induced by each transgene. LOF alleles are determined by a phenotype that is less severe than the reference phenotype, indicated by “>”, whereas GOF alleles are determined by a phenotype that is more severe than the reference phenotype, indicated by “<”.
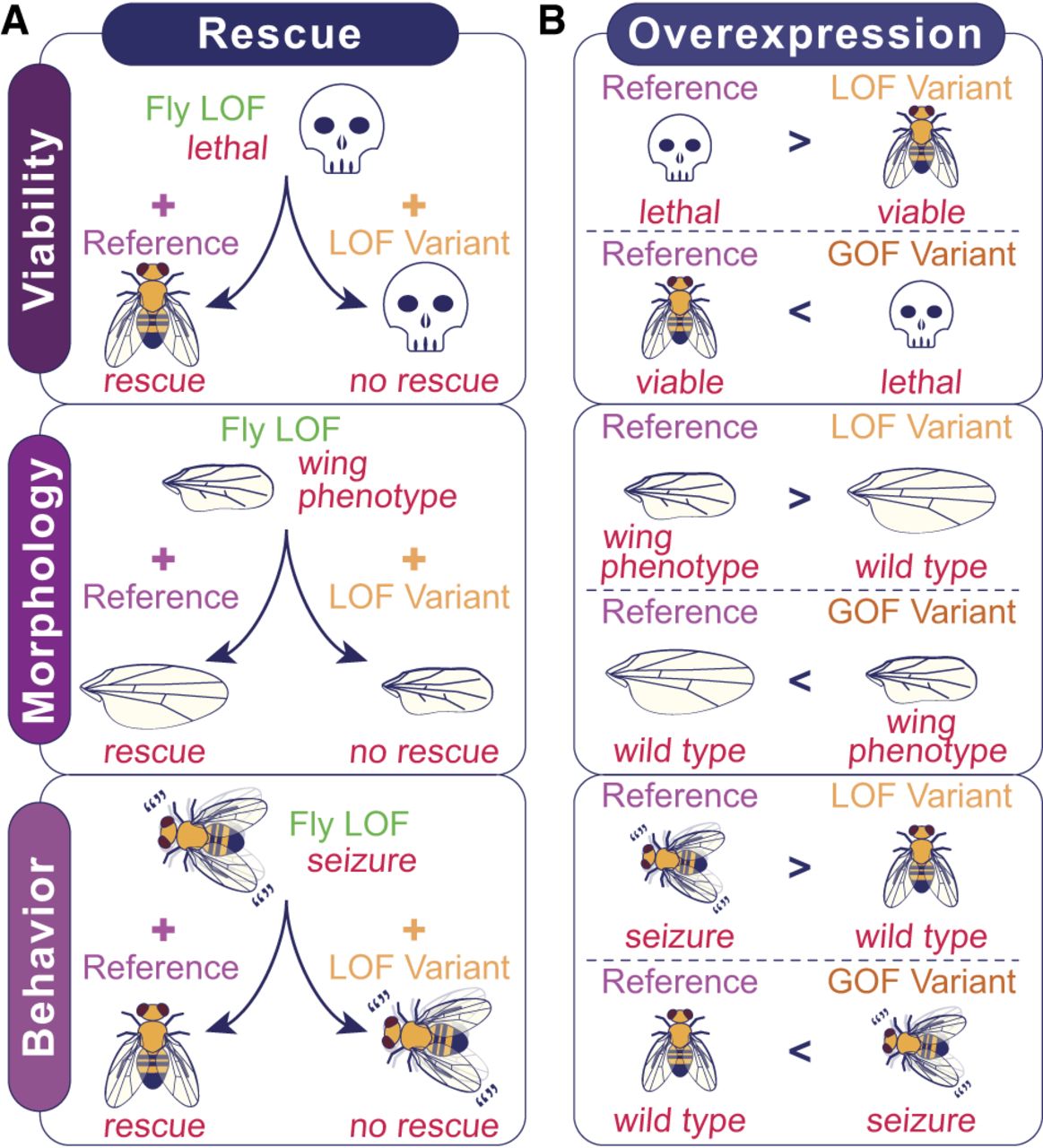
For cases in which the human reference does not rescue loss of fly gene function, there may still be useful functional data that can be retrieved when comparing the phenotypes caused by introducing the reference or variant human transgenes into a fly mutant background. For example, two missense variants in MRTFB (p.R104G and p.A91P) that were found in patients with a novel neurodevelopmental disorder were initially assessed using a rescue-based paradigm (Andrews et al. 2023). Whereas both reference and variant human MRTFB cDNA expression failed to rescue a bristle morphology phenotype caused by hypomorphic alleles of fly Mrtf (ortholog of MRTFB as well as MRTFA and MYOCD), expression of the variants induced a more severe phenotype causing lethality, which was not observed with the reference allele. Based on this and other data, these missense variants were classified as GOF (hypermorphic) alleles.
Studying the functional impact of coding variants based on overexpression of human proteins
Based on our and others’ experiences (Marcogliese et al. 2022; Yamamoto et al. 2024), 40%–60% of human genes are likely to fully or partially rescue a fly ortholog mutant phenotype based on rescue experiments using T2A-GAL4/Kozak-GAL4 lines and UAS-human cDNA transgenes. One reason for failure may be that human proteins cannot function in the context of the fly cell. Another reason could be that heterologous expression of reference human proteins in a fly cell could induce a phenotype through GOF mechanisms (hypermorph, antimorph, neomorph), especially for dosage-sensitive genes, because the level of protein expression achieved using the GAL4/UAS system tends to be higher than the endogenous level. In the latter case, one could take advantage of any scorable phenotypes induced by ectopic human protein expression and use them as readouts of protein function (Fig. 3C; Her et al. 2024). This approach has been used in 35% of publications (Fig. 1C) and is also often used in combination with other strategies (Fig. 1D; Supplemental Table 1). In one scenario, if ubiquitous overexpression of the human reference protein causes lethality, but the overexpression of the variant protein produces viable flies, this variant can be classified as a LOF allele (Fig. 4B). In another scenario, if neuronal overexpression of the variant protein causes a behavioral phenotype that is not observed when the reference protein is expressed, this could be considered as some type of a GOF allele (Fig. 4B). Whereas additional experiments are required to further subclassify LOF variants into amorphic or hypomorphic alleles and the GOF variants into hypermorphic, antimorphic, or neomorphic alleles, overexpression experiments can be integrated into a rapid screening pipeline.
It is important to reemphasize that phenotypes that are screened in fly models do not necessarily have to correlate well to patient symptoms. For example, morphological defects observed when human proteins are overexpressed in the fly eye or wing are often used as a phenotypic readout because development of these tissues is sensitive to alterations in fundamental cellular and developmental pathways (Bier 2005). An overexpression-based approach has been useful in characterizing the functional difference between two missense variants (p.D413G and p.S1522L) in ROBO1 that have been found to cause different neurodevelopmental phenotypes in patients with different inheritance patterns using a T2A-GAL4 allele of fly robo1 (ROBO1 ortholog) as a driver and screening for lethality as well as neuroanatomical defects (Huang et al. 2022a). Through this experiment, the variant linked to a recessive disorder was classified as a hypomorphic LOF allele whereas the variant linked to the dominant disorder was identified to be a neomorphic GOF allele. Hypermorphic and antimorphic variants can also be revealed using overexpression approaches, as in the case of MTSS2 (Huang et al. 2022b).
Once a variant is determined to have a functional consequence, additional experiments can be designed to probe into the disease mechanism (Sandberg et al. 2020; Wilson et al. 2020; Ganguly et al. 2021; Marmion et al. 2021; Gignac et al. 2023; Srivastava et al. 2023). Follow-up biochemical or cell biological studies using tagged-transgenes or specific antibodies can be used to further determine the functional consequence of VUS on protein expression or localization. For example, immunohistological studies of DXH9 identified that two disease-associated variants that were classified as hypomorphic alleles based on overexpression assays show different subcellular localization patterns compared to the reference allele (Yamada et al. 2023). Reporter assays and genetic interaction studies can also be integrated to determine the functional impact of specific variants in the pathway of interest. One example is an experiment that was performed to determine how different variants in PTEN affect the PI3K pathway using wing sizes of flies as a readout of tissue growth (Ganguly et al. 2021). Overexpression studies can also provide a platform to test potential personalized drug treatments that are translatable to rare disease patients (Chung et al. 2020; Bakkar et al. 2021). An example of this is the pharmacological rescue of GABA transport deficiency seen with overexpression of the p.A288V variant in GAT1 (Kasture et al. 2023). This study provided valuable in vivo functional data on drug treatments that could improve the individual's symptoms and set up assays that can be used to assess drug-based rescue of additional GAT1 variants in the future.
It is important to note that not all functions of a protein of interest can be revealed by a single assay. For example, functional assessment of 18 different known pathogenic variants in KDM6B linked to neurodevelopmental diseases was performed using ubiquitous and wing-specific overexpression of the human protein. The authors concluded that the phenotypic assay they selected can be used to determine the function of the protein carried out by the C-domain of the protein but not for variants in the N-domain (Rots et al. 2023). Therefore, whereas positive data can be used as strong supporting evidence that the variant has functional consequences supporting pathogenicity, one should not categorize a variant as “benign” based on negative data.
Overexpression experiments can also provide functional information on variants in human genes that are not conserved in Drosophila (Fig. 2). For example, orthologous genes that correspond to SNCA (Parkinson's disease), PRNP (prion diseases), and MECP2 (Rett syndrome) are not found in the Drosophila genome. However, ectopic overexpression of these human proteins can cause scorable phenotypes in flies (Feany and Bender 2000; Cukier et al. 2008; Myers et al. 2022). In addition, some human genes that are not considered to be conserved in flies based on bioinformatic algorithms can be found to rescue LOF mutant phenotypes in related fly genes, as reported for human APOE (rescuing the fly Glaz mutant) and LEP (rescuing the fly upd2 mutant) (Rajan and Perrimon 2012; Liu et al. 2017). Hence, if a phenotypic difference can be seen upon reference and variant protein overexpression and/or through rescue experiments of functionally related genes, one can study VUS in nonconserved genes. These experiments further provide an entry point to further study the underlying mechanism of these disorders, even when the obvious ortholog of the human gene of interest is not found in the fly genome.
Studies of VUS in human genes through analogous variant modeling in the fly ortholog
For cases in which the variant of interest affects an amino acid that is conserved between humans and flies, studies of the analogous (equivalent/homologous) variant in the context of the fly protein can provide useful information (Figs. 2, 3D). This approach is particularly useful when the human protein fails to function in the context of a fly cell or when human cDNA is not publicly or commercially available. Such an analogous fly variant strategy has been utilized in 35% of the papers we analyzed, suggesting its versatility (Fig. 1C; Supplemental Table 1). Ideally, the amino acid of interest should be identical; however, modeling is still feasible when amino acids differ, provided that their biochemical characteristics are similar. For instance, a recent study employed a fly analogous variant strategy to model a VUS in NKX2.5 (Lovato et al. 2023). In this study, the authors studied the impact of p.K158N by introducing a corresponding p.R321N mutation in the fly ortholog tin (NKX2.5 ortholog), leveraging the fact that both lysine (K) and arginine (R) are basic residues with similar biochemical properties. This approach underscores the potential for cross-species genetic modeling, even when exact amino acid matches are not present.
The most straightforward approach to studying the human variant using this analogous variant strategy is to generate a knock-in allele by directly targeting the fly ortholog gene locus using gene editing technology such as CRISPR-Cas9. This approach is particularly beneficial when: (1) the gene is dosage-sensitive, so maintaining or minimally affecting the endogenous gene expression level is crucial; (2) no available mutant flies or efficient RNAi lines exist; or (3) whole-organism studies are preferred over tissue-specific experiments. In a recent study on missense variants in CYFIP1 found in individuals with a neurodevelopmental condition, the authors generated two different analogous mutant alleles (p.I471V and p.P760L) in fly Cyfip (ortholog of CYFIP1 as well as CYFIP2), corresponding to two missense variants in CYFIP1 (p.I476V and p.P742L) that were found in a trans-heterozygous state using CRISPR-based scarless gene editing (Mariano et al. 2024). The authors found that flies with biallelic mutations mimicking the individual's genotype (Cyfipp.I471V/Cyfipp.P760L) recapitulated the actin polymerization defect identified in patient-derived cells. Biallelic mutant flies also showed defective neuronal morphology and behavioral defects, suggesting that the biallelic variants in CYFIP could contribute to the clinical phenotype in the probands and that the two missense variants are likely pathogenic. Additional strategies to study VUS in the context of the fly gene/protein are discussed in Supplemental Text 7.
Using Drosophila to further classify pathogenic variants based on functional consequences and to discover complex properties of some alleles
As we have seen, different genetic strategies in Drosophila can be used to not only determine that the variant of interest has significant functional consequences but also to provide information regarding its directionality. Understanding whether a variant is a LOF or GOF allele can have direct clinical implications because therapeutic strategies should be tailored to the type of variant an individual carries. For example, if a patient carries a pathogenic LOF variant, strategies to reintroduce or increase the expression of the gene of interest, for example, through gene therapy, could be considered. In contrast, approaches to reduce or abolish the function of the pathogenic allele using antisense oligonucleotides (ASO) or CRISPR-mediated genome editing would be preferred if the pathogenic variant is a GOF allele. Classifying variants into specific Muller's morphs can further help scientists begin to develop and test specific hypotheses regarding the molecular mechanisms of pathogenesis and assist clinicians to further refine therapeutic approaches. As an example, if the patient is heterozygous for an antimorphic allele, therapies to increase the expression of the wild-type copy of this gene could have a beneficial effect. However, such manipulation is likely to be deleterious for individuals who carry hypermorphic alleles and would likely have no beneficial effect for individuals who possess a neomorphic allele. Although some bioinformatic tools are being developed to predict whether a variant may be a LOF or GOF allele (Stein et al. 2023), these tools need further development. Additionally, none of the existing bioinformatics programs have the capacity to assign pathogenic variants into different Muller's morphs; therefore, experimental data are critical for such classification. Of 134 recent papers that have used Drosophila for functional studies of VUS and GUS, 95 papers have annotated the variants of interest as LOF or GOF (Fig. 1E; Supplemental Table 1). Furthermore, 32 of the 43 papers that designated the variants of interest as GOF alleles further provided data to classify these variants into specific Muller's morphs (hypermorph, antimorph, neomorph) (Fig. 1F; Supplemental Table 1), underscoring the value of in vivo functional studies using Drosophila.
Genes and proteins are often pleiotropic and can have more than one molecular function. Also, a single variant can have different functional consequences depending on the context, because proteins can be engaged in different molecular interactions in different cell types, organ systems, or developmental stages. There have been several studies in Drosophila that revealed such unique properties of a handful of variants, and these intriguing biological insights have been obtained based on studying the functional consequences of variants in different contexts. In one study, several variants in DVL1 associated with Robinow syndrome were found to cause both a LOF of canonical Wnt signaling and a GOF of noncanonical (JNK/PCP [c-Jun N-terminal Kinase/Planar Cell Polarity]) Wnt signaling by assessing multiple parameters that can be measured in the fly wing (Gignac et al. 2023). In another study, two de novo missense variants found in patients with autism spectrum disorders found in EPHB1 and MAP4K1 had different functional consequences in different tissues. The EPHB1 (p.V916M) variant behaved as a LOF allele when the overexpression-based functional assay was performed in the fly eye but behaved as a GOF allele in the fly wing. The MAP4K1 (p.T725T) variant had the opposite functional consequence: GOF when tested in the eye and LOF when assessed in the wing (Marcogliese et al. 2022). A similar tissue-specific LOF and GOF effect was found for a missense variant in TNPO2 (p.W727C) that was reported in a new disease gene discovery paper (Goodman et al. 2021) and a missense variant in AXIN2 (p.E66K) linked to phenotypic expansion (Aceves-Ewing et al. 2024). Whereas additional studies are required to clarify which functional effect is most relevant to each clinical phenotype of interest and the molecular mechanisms behind these peculiar behaviors need to be investigated, it is possible that a subset of pathogenic variants may cause different functional effects in different organ systems or cell types. In these cases, therapeutic approaches should be further customized because an approach to correct GOF or LOF aspects of the disorder may have unexpected side effects that may impact a certain organ system.
Collaborations between fly researchers and MAVE developers can facilitate the deciphering of the human variome
Clinical geneticists who analyze exome or genome sequencing data in diagnostic laboratories or research settings often encounter a number of VUS or rare variants in GUS. Whereas these individuals can provide high-quality sequence information and variant interpretation based on bioinformatic analysis (e.g., known pathogenic and benign variant databases, population genomic data, in silico prediction algorithms, experiment-based evidence based on previous publication), they often do not have the capacity to further resolve a VUS in a laboratory setting. Whereas “functional assay” is one of the key criteria that clinicians use while classifying the likely pathogenicity of a variant based on the ACMG/AMP sequence interpretation guideline (PS3 and NS3 codes) (Richards et al. 2015), clinical diagnostic laboratories do not carry out these types of studies. For genes in which the functionality of proteins with relevance to disease mechanism can be quantified, MAVE assays based on cell models or recombinant proteins could be established by basic scientists to assess the functional consequences of all possible variants in vitro (which need to be interpreted with caution [Gelman et al. 2019]). In addition, variants in noncoding (or coding) regions of the gene that may affect transcription and splicing could be studied in large scale based on high-throughput reporter assays using cell lines or transcriptomic assays based on patient-derived materials (Walker et al. 2023). For the clinical genetics field to obtain a well-defined set of MAVE data that could be used in a clinical setting to classify the VUS as pathogenic or benign, model organism researchers can provide valuable in vivo data to fill the knowledge gap that still exists for many clinically relevant genes. For example, ACMG/AMP recommends that more than 10 clearly defined pathogenic and benign variants should be used to determine the sensitivity and specificity of MAVE-type assays (Brnich et al. 2020). Because many ultrarare diseases lack well-defined pathogenic alleles and disease mechanisms, basic researchers with expertise on homologous genes, including Drosophila biologists, could contribute to establishing these foundations. Especially for genes that encode multifunctional proteins or those that function in non-cell-autonomous manners, data obtained from in vivo model systems would be extremely valuable to determine what in vitro (e.g., specific protein-protein interactions that are relevant to pathogenesis) or cell-based (e.g., co-culture system or organoid based) assay could be used to establish MAVEs that are most relevant to the clinical cases/phenotypes of interest.
Basic scientists who can provide valuable insights or are willing to perform functional assays can be identified through literature searches or matchmaking tools such as ModelMatcher (Harnish et al. 2022). Because the name of orthologous genes often differ in different species, and valuable data are scattered across diverse databases on the internet, integrated knowledge bases that combine information from multiple species and sources such as MARRVEL (Wang et al. 2017, 2019), Alliance of Genome Research (Alliance of Genome Research Consortium 2020), Monarch Initiative (Putman et al. 2024), and Gene2Function (Hu et al. 2017) could be used to facilitate this process. Some model organism geneticists have unique technical capability to efficiently generate mutant and transgenic animals for GUS that have never been studied in any organism and to further perform cursory functional assays in moderate scales (e.g., UDN MOSC, CPMM) (Baldridge et al. 2021). These scientists could establish a functional study pipeline for many genes to resolve VUS in a mid-throughput manner, potentially in collaboration with a group of clinical researchers, patient organizations, and/or biotech companies that have strong motivations to facilitate research on specific genes towards their ultimate goal of establishing a treatment and cure.
In summary, Drosophila has been a powerful research tool to reveal the function of GUS and VUS that are relevant to rare genetic disorders. Scientists who have expertise in using this model organism to answer diverse biological questions have the potential to address an unmet need: resolve VUS found in patients who are still undiagnosed by providing functional data that permit the classification of these variants into LOF or GOF alleles and specific Muller's morphs. If a VUS can be proven to be pathogenic, this will end the patient's diagnostic odyssey, helping them financially by reducing the constant medical tests and mentally by providing answers. Importantly, in cases in which there are approved treatments available, a definitive molecular diagnosis backed up by solid functional data can allow them to receive authorization and can help insurance cover the cost of drugs. For cases in which there are potential treatments that are in clinical trials, a solid molecular diagnosis could make them eligible to participate in such studies. For diseases that do not have any drugs that are in development, biological information obtained from model organisms including flies can be leveraged to identify potential therapeutic avenues. Hence, Drosophila research can have a true impact on clinical care, and more stakeholders, especially translational scientists who are developing MAVE-type assays, could benefit by becoming aware of its potential. In addition, considering that studies of rare diseases can often reveal pathogenic mechanisms that underlie more common diseases (Yamamoto et al. 2024) and the Drosophila strategies discussed here can be applied to understand the functional consequences of variants associated with a variety of disorders (Bellen and Yamamoto 2015), collaborative initiatives that include fly geneticists can also facilitate research on common diseases.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Drs. Dustin Baldridge, Hugo J. Bellen, Oguz Kanca, and Michael F. Wangler for valuable discussions. We also thank all individuals with rare diseases and their family members who participated in clinical research cited in this Perspective. J-W.M. has been supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2021R1A6A3A14044510). S.B.G. has been supported by The Cullen Foundation. H.A.D. has been supported by the National Research Service Award (NRSA) training grant T32GM139534 from the National Institutes of Health (NIH, National Institute of General Medical Sciences) and The Cullen Foundation. S.Y. has been supported by research grants from the NIH (Office of Research Infrastructure Programs: R24OD022005, U54OD030165; National Institute on Aging: RF1AG071557, R01AG071557; National Institute of Neurological Disorders and Stroke: U2CNS132415; National Human Genome Research Institute: R01HG011795), the Chan Zuckerberg Initiative (#2023-32824, #2023-332162), and institutional funds from Baylor College of Medicine and Texas Children's Hospital.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278291.123.
References
- ↵Aceves-Ewing NM, Lanza DG, Marcogliese PC, Lu D, Hsu C-W, Gonzalez M, Christiansen AE, Rasmussen TL, Ho AJ, Gaspero A, 2024. Uncovering phenotypic expansion in AXIN2-related disorders through precision animal modeling. medRxiv 10.1101/2024.12.05.24318524
- ↵Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, 2000. The genome sequence of Drosophila melanogaster. Science 287: 2185–2195. 10.1126/science.287.5461.2185
- ↵Alliance of Genome Research Consortium. 2020. Alliance of Genome Resources Portal: unified model organism research platform. Nucleic Acids Res 48: D650–D658. 10.1093/nar/gkz813
- ↵Andrews JC, Mok J-W, Kanca O, Jangam S, Tifft C, Macnamara EF, Russell BE, Wang L-K, Undiagnosed Diseases Network, Nelson SF, 2023. De novo variants in MRTFB have gain-of-function activity in Drosophila and are associated with a novel neurodevelopmental phenotype with dysmorphic features. Genet Med 25: 100833. 10.1016/j.gim.2023.100833
- ↵Bakkar N, Starr A, Rabichow BE, Lorenzini I, McEachin ZT, Kraft R, Chaung M, Macklin-Isquierdo S, Wingfield T, Carhart B, 2021. The M1311V variant of ATP7A is associated with impaired trafficking and copper homeostasis in models of motor neuron disease. Neurobiol Dis 149: 105228. 10.1016/j.nbd.2020.105228
- ↵Baldridge D, Wangler MF, Bowman AN, Yamamoto S, Undiagnosed Diseases Network, Schedl T, Pak SC, Postlethwait JH, Shin J, Solnica-Krezel L, 2021. Model organisms contribute to diagnosis and discovery in the undiagnosed diseases network: current state and a future vision. Orphanet J Rare Dis 16: 206. 10.1186/s13023-021-01839-9
- ↵Barish S, Senturk M, Schoch K, Minogue AL, Lopergolo D, Fallerini C, Harland J, Seemann JH, Stong N, Kranz PG, 2022. The microRNA processor DROSHA is a candidate gene for a severe progressive neurological disorder. Hum Mol Genet 31: 2934–2950. 10.1093/hmg/ddac085
- ↵Bellen HJ, Yamamoto S. 2015. Morgan's legacy: fruit flies and the functional annotation of conserved genes. Cell 163: 12–14. 10.1016/j.cell.2015.09.009
- ↵Bellen HJ, Levis RW, He Y, Carlson JW, Evans-Holm M, Bae E, Kim J, Metaxakis A, Savakis C, Shulze K, 2011. The Drosophila gene disruption project: progress using transposons with distinctive site specificities. Genetics 188: 731–743. 10.1534/genetics.111.126995
- ↵Bellen HJ, Wangler MF, Yamamoto S. 2019. The fruit fly at the interface of diagnosis and pathogenic mechanisms of rare and common human diseases. Hum Mol Genet 28: R207–R214. 10.1093/hmg/ddz135
- ↵Benito-Lozano J, López-Villalba B, Arias-Merino G, Posada de la Paz M, Alonso-Ferreira V. 2022. Diagnostic delay in rare diseases: data from the Spanish rare diseases patient registry. Orphanet J Rare Dis 17: 418. 10.1186/s13023-022-02530-3
- ↵Bier E. 2005. Drosophila, the golden bug, emerges as a tool for human genetics. Nat Rev Genet 6: 9–23. 10.1038/nrg1503
- ↵Boycott KM, Campeau PM, Howley HE, Pavlidis P, Rogic S, Oriel C, Berman JN, Hamilton RM, Hicks GG, Lipshitz HD, 2020. The Canadian rare diseases models and mechanisms (RDMM) network: connecting understudied genes to model organisms. Am J Hum Genet 106: 143–152. 10.1016/j.ajhg.2020.01.009
- ↵Boycott KM, Azzariti DR, Hamosh A, Rehm HL. 2022. Seven years since the launch of the Matchmaker Exchange: the evolution of genomic matchmaking. Hum Mutat 43: 659–667. 10.1002/humu.24373
- ↵Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, Kanavy DM, Luo X, McNulty SM, Starita LM, 2020. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med 12: 3. 10.1186/s13073-019-0690-2
- ↵Chung H-L, Wangler MF, Marcogliese PC, Jo J, Ravenscroft TA, Zuo Z, Duraine L, Sadeghzadeh S, Li-Kroger D, Schmidt RE, 2020. Loss- or gain-of-function mutations in ACOX1 cause axonal loss via different mechanisms. Neuron 106: 589–606.e6. 10.1016/j.neuron.2020.02.021
- ↵Cortese A, Zhu Y, Rebelo AP, Negri S, Courel S, Abreu L, Bacon CJ, Bai Y, Bis-Brewer DM, Bugiardini E, 2020. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat Genet 52: 473–481. 10.1038/s41588-020-0615-4
- ↵Cotton RGH, Auerbach AD, Axton M, Barash CI, Berkovic SF, Brookes AJ, Burn J, Cutting G, den Dunnen JT, Flicek P, 2008. GENETICS. The Human Variome Project. Science 322: 861–862. 10.1126/science.1167363
- ↵Cukier HN, Perez AM, Collins AL, Zhou Z, Zoghbi HY, Botas J. 2008. Genetic modifiers of MeCP2 function in Drosophila. PLoS Genet 4: e1000179. 10.1371/journal.pgen.1000179
- ↵CZI Cell Science Program, Abdulla S, Aevermann B, Assis P, Badajoz S, Bell SM, Bezzi E, Cakir B, Chaffer J, Chambers S, 2025. CZ CELLxGENE Discover: a single-cell data platform for scalable exploration, analysis and modeling of aggregated data. Nucleic Acids Res 53: D886–D900. 10.1093/nar/gkae1142
- ↵Dutta D, Briere LC, Kanca O, Marcogliese PC, Walker MA, High FA, Vanderver A, Krier J, Carmichael N, Callahan C, 2020. De novo mutations in TOMM70, a receptor of the mitochondrial import translocase, cause neurological impairment. Hum Mol Genet 29: 1568–1579. 10.1093/hmg/ddaa081
- ↵Ellwanger K, Brill JA, de Boer E, Efthymiou S, Elgersma Y, Icmat M, Lecoquierre F, Lobato AG, Morleo M, Ori M, 2024. Model matchmaking via the Solve-RD Rare Disease Models & Mechanisms Network (RDMM-Europe). Lab Anim 53: 161–165. 10.1038/s41684-024-01395-2
- ↵Esposito D, Weile J, Shendure J, Starita LM, Papenfuss AT, Roth FP, Fowler DM, Rubin AF. 2019. MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol 20: 223. 10.1186/s13059-019-1845-6
- ↵EveryLife Foundation for Rare Diseases. 2023. The cost of delayed diagnosis in rare disease: a health economic study. EveryLife Foundation for Rare Diseases, Washington, DC. https://everylifefoundation.org/wp-content/uploads/2023/09/EveryLife-Cost-of-Delayed-Diagnosis-in-Rare-Disease_Final-Full-Study-Report_0914223.pdf
- ↵Feany MB, Bender WW. 2000. A Drosophila model of Parkinson's disease. Nature 404: 394–398. 10.1038/35006074
- ↵Fliedner A, Kirchner P, Wiesener A, van de Beek I, Waisfisz Q, van Haelst M, Scott DA, Lalani SR, Rosenfeld JA, Azamian MS, 2020. Variants in SCAF4 cause a neurodevelopmental disorder and are associated with impaired mRNA processing. Am J Hum Genet 107: 544–554. 10.1016/j.ajhg.2020.06.019
- ↵Fortini ME, Skupski MP, Boguski MS, Hariharan IK. 2000. A survey of human disease gene counterparts in the Drosophila genome. J Cell Biol 150: F23–F30. 10.1083/jcb.150.2.F23
- ↵Ganguly P, Madonsela L, Chao JT, Loewen CJR, O'Connor TP, Verheyen EM, Allan DW. 2021. A scalable Drosophila assay for clinical interpretation of human PTEN variants in suppression of PI3K/AKT induced cellular proliferation. PLoS Genet 17: e1009774. 10.1371/journal.pgen.1009774
- ↵Gasperini M, Starita L, Shendure J. 2016. The power of multiplexed functional analysis of genetic variants. Nat Protoc 11: 1782–1787. 10.1038/nprot.2016.135
- ↵Gelman H, Dines JN, Berg J, Berger AH, Brnich S, Hisama FM, James RG, Rubin AF, Shendure J, Shirts B, 2019. Recommendations for the collection and use of multiplexed functional data for clinical variant interpretation. Genome Med 11: 85. 10.1186/s13073-019-0698-7
- ↵Ghosh R, Oak N, Plon SE. 2017. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol 18: 225. 10.1186/s13059-017-1353-5
- ↵Gignac SJ, MacCharles KR, Fu K, Bonaparte K, Akarsu G, Barrett TW, Verheyen EM, Richman JM. 2023. Mechanistic studies in Drosophila and chicken give new insights into functions of DVL1 in dominant Robinow syndrome. Dis Model Mech 16: dmm049844. 10.1242/dmm.049844
- ↵Goodman LD, Cope H, Nil Z, Ravenscroft TA, Charng W-L, Lu S, Tien A-C, Pfundt R, Koolen DA, Haaxma CA, 2021. TNPO2 variants associate with human developmental delays, neurologic deficits, and dysmorphic features and alter TNPO2 activity in Drosophila. Am J Hum Genet 108: 1669–1691. 10.1016/j.ajhg.2021.06.019
- ↵Gorzynski JE, Goenka SD, Shafin K, Jensen TD, Fisk DG, Grove ME, Spiteri E, Pesout T, Monlong J, Baid G, 2022. Ultrarapid nanopore genome sequencing in a critical care setting. N Engl J Med 386: 700–702. 10.1056/NEJMc2112090
- ↵Graungaard AH, Skov L. 2007. Why do we need a diagnosis? A qualitative study of parents’ experiences, coping and needs, when the newborn child is severely disabled. Child Care Health Dev 33: 296–307. 10.1111/j.1365-2214.2006.00666.x
- ↵GTEx Consortium. 2013. The genotype-tissue expression (GTEx) project. Nat Genet 45: 580–585. 10.1038/ng.2653
- ↵Haendel M, Vasilevsky N, Unni D, Bologa C, Harris N, Rehm H, Hamosh A, Baynam G, Groza T, McMurry J, 2020. How many rare diseases are there? Nat Rev Drug Discov 19: 77–78. 10.1038/d41573-019-00180-y
- ↵Hamosh A, Amberger JS, Bocchini C, Scott AF, Rasmussen SA. 2021. Online Mendelian Inheritance in Man (OMIM®): Victor McKusick's magnum opus. Am J Med Genet A 185: 3259–3265. 10.1002/ajmg.a.62407
- ↵Hamosh A, Wohler E, Martin R, Griffith S, Rodrigues ES, Antonescu C, Doheny KF, Valle D, Sobreira N. 2022. The impact of GeneMatcher on international data sharing and collaboration. Hum Mutat 43: 668–673. 10.1002/humu.24350
- ↵Harnish JM, Li L, Rogic S, Poirier-Morency G, Kim S-Y, Network UD, Boycott KM, Wangler MF, Bellen HJ, Hieter P, 2022. Modelmatcher: a scientist-centric online platform to facilitate collaborations between stakeholders of rare and undiagnosed disease research. Hum Mutat 43: 743–759. 10.1002/humu.24364
- ↵Her Y, Pascual DM, Goldstone-Joubert Z, Marcogliese PC. 2024. Variant functional assessment in Drosophila by overexpression: what can we learn? Genome 67: 158–167. 10.1139/gen-2023-0135
- ↵Herskowitz I. 1987. Functional inactivation of genes by dominant negative mutations. Nature 329: 219–222. 10.1038/329219a0
- ↵Holland PWH. 2003. More genes in vertebrates? J Struct Funct Genomics 3: 75–84. 10.1023/A:1022656931587
- ↵Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, Mohr SE. 2011. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12: 357. 10.1186/1471-2105-12-357
- ↵Hu Y, Comjean A, Mohr SE, Consortium FB, Perrimon N. 2017. Gene2Function: an integrated online resource for gene function discovery. G3 (Bethesda) 7: 2855–2858. 10.1534/g3.117.043885
- ↵Huang Y, Lemire G, Briere LC, Liu F, Wessels MW, Wang X, Osmond M, Kanca O, Lu S, High FA, 2022a. The recurrent de novo c.2011C>T missense variant in MTSS2 causes syndromic intellectual disability. Am J Hum Genet 109: 1923–1931. 10.1016/j.ajhg.2022.08.011
- ↵Huang Y, Ma M, Mao X, Pehlivan D, Kanca O, Un-Candan F, Shu L, Akay G, Mitani T, Lu S, 2022b. Novel dominant and recessive variants in human ROBO1 cause distinct neurodevelopmental defects through different mechanisms. Hum Mol Genet 31: 2751–2765. 10.1093/hmg/ddac070
- ↵Kanca O, Zirin J, Hu Y, Tepe B, Dutta D, Lin W-W, Ma L, Ge M, Zuo Z, Liu L-P, 2022. An expanded toolkit for Drosophila gene tagging using synthesized homology donor constructs for CRISPR-mediated homologous recombination. eLife 11: e76077. 10.7554/eLife.76077
- ↵Kasture AS, Fischer FP, Kunert L, Burger ML, Burgstaller AC, El-Kasaby A, Hummel T, Sucic S. 2023. Drosophila melanogaster as a model for unraveling unique molecular features of epilepsy elicited by human GABA transporter 1 variants. Front Neurosci 16: 1074427. 10.3389/fnins.2022.1074427
- ↵Kim JH, Singh M, Pan G, Lopez A, Zito N, Bosse B, Ye B. 2020. Frameshift mutations of YPEL3 alter the sensory circuit function in Drosophila. Dis Model Mech 13: dmm042390. 10.1242/dmm.042390
- ↵Kuang D, Weile J, Kishore N, Nguyen M, Rubin AF, Fields S, Fowler DM, Roth FP. 2021. Maveregistry: a collaboration platform for multiplexed assays of variant effect. Bioinformatics 37: 3382–3383. 10.1093/bioinformatics/btab215
- ↵Kuzniar A, van Ham RCHJ, Pongor S, Leunissen JAM. 2008. The quest for orthologs: finding the corresponding gene across genomes. Trends Genet 24: 539–551. 10.1016/j.tig.2008.08.009
- ↵Lee P-T, Zirin J, Kanca O, Lin W-W, Shulze KL, Li-Kroeger D, Tao R, Devereaux C, Hu Y, Chung V, 2018. A gene-specific T2A-GAL4 library for Drosophila. eLife 7: e35574. 10.7554/eLife.35574
- ↵Linard B, Ebersberger I, McGlynn SE, Glover N, Mochizuki T, Patricio M, LeCompte O, Nevers Y, Thomas PD, Gabaldón T, 2021. Ten years of collaborative progress in the quest for orthologs. Mol Biol Evol 38: 3033–3045. 10.1093/molbev/msab098
- ↵Liu L, MacKenzie KR, Putluri N, Maletić-Savatić M, Bellen HJ. 2017. The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab 26: 719–737.e6. 10.1016/j.cmet.2017.08.024
- ↵Lovato TL, Blotz B, Bileckyj C, Johnston CA, Cripps RM. 2023. Modeling a variant of unknown significance in the Drosophila ortholog of the human cardiogenic gene NKX2.5. Dis Model Mech 16: dmm050059. 10.1242/dmm.050059
- ↵Lu S, Hernan R, Marcogliese PC, Huang Y, Gertler TS, Akcaboy M, Liu S, Chung H-L, Pan X, Sun X, 2022. Loss-of-function variants in TIAM1 are associated with developmental delay, intellectual disability, and seizures. Am J Hum Genet 109: 571–586. 10.1016/j.ajhg.2022.01.020
- ↵Luhur A, Chawla G, Wu Y-C, Li J, Sokol NS. 2014. Drosha-independent DGCR8/Pasha pathway regulates neuronal morphogenesis. Proc Natl Acad Sci 111: 1421–1426. 10.1073/pnas.1318445111
- ↵Marcogliese PC, Deal SL, Andrews J, Harnish JM, Bhavana VH, Graves HK, Jangam S, Luo X, Liu N, Bei D, 2022. Drosophila functional screening of de novo variants in autism uncovers damaging variants and facilitates discovery of rare neurodevelopmental diseases. Cell Rep 38: 110517. 10.1016/j.celrep.2022.110517
- ↵Mariano V, Kanellopoulos AK, Ricci C, Di Marino D, Borrie SC, Dupraz S, Bradke F, Achsel T, Legius E, Odent S, 2024. Intellectual disability and behavioral deficits linked to CYFIP1 missense variants disrupting actin polymerization. Biol Psychiatry 95: 161–174. 10.1016/j.biopsych.2023.08.027
- ↵Marmion RA, Yang L, Goyal Y, Jindal GA, Wetzel JL, Singh M, Schüpbach T, Shvartsman SY. 2021. Molecular mechanisms underlying cellular effects of human MEK1 mutations. Mol Biol Cell 32: 974–983. 10.1091/mbc.E20-10-0625
- ↵Mattioli F, Worpenberg L, Li C-T, Ibrahim N, Naz S, Sharif S, Firouzabadi SG, Vosoogh S, Saraeva-Lamri R, Raymond L, 2023. Biallelic variants in NSUN6 cause an autosomal recessive neurodevelopmental disorder. Genet Med 25: 100900. 10.1016/j.gim.2023.100900
- ↵McGary KL, Park TJ, Woods JO, Cha HJ, Wallingford JB, Marcotte EM. 2010. Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proc Natl Acad Sci 107: 6544–6549. 10.1073/pnas.0910200107
- ↵Morgan TH. 1910. Sex limited inheritance in Drosophila. Science 32: 120–122. 10.1126/science.32.812.120
- ↵Muller HJ. 1932. Further studies on the nature and causes of gene mutations. Int Congr Genet 1932: 213–255.
- ↵Myers RR, Sanchez-Garcia J, Leving DC, Melvin RG, Fernandez-Funez P. 2022. New Drosophila models to uncover the intrinsic and extrinsic factors that mediate the toxicity of the human prion protein. Dis Model Mech 15: dmm049184. 10.1242/dmm.049184
- ↵Osmond M, Hartley T, Johnstone B, Andjic S, Girdea M, Gillespie M, Buske O, Dumitriu S, Koltunova V, Ramani A, 2022. PhenomeCentral: 7 years of rare disease matchmaking. Hum Mutat 43: 674–681. 10.1002/humu.24348
- ↵Pan X, Alvarez AN, Ma M, Lu S, Crawford MW, Briere LC, Kanca O, Yamamoto S, Sweetser DA, Wilson JL, 2023. Allelic strengths of encephalopathy-associated UBA5 variants correlate between in vivo and in vitro assays. eLife 12: RP89891. 10.7554/eLife.89891
- ↵Pandey AK, Lu L, Wang X, Homayouni R, Williams RW. 2014. Functionally enigmatic genes: a case study of the brain ignorome. PLoS One 9: e88889. 10.1371/journal.pone.0088889
- ↵Pineda S, Nikolova-Krstevski V, Leimena C, Atkinson AJ, Altekoester A-K, Cox CD, Jacoby A, Huttner IG, Ju Y-K, Soka M, 2021. Conserved role of the large conductance calcium-activated potassium channel, KCa1.1, in sinus node function and arrhythmia risk. Circ Genom Precis Med 14: e003144. 10.1161/CIRCGEN.120.003144
- ↵Post KL, Belmadani M, Ganguly P, Meili F, Dingwall R, McDiarmid TA, Meyers WM, Herrington C, Young BP, Callaghan DB, 2020. Multi-model functionalization of disease-associated PTEN missense mutations identifies multiple molecular mechanisms underlying protein dysfunction. Nat Commun 11: 2073. 10.1038/s41467-020-15943-0
- ↵Pressman S, Reinke CA, Wang X, Carthew RW. 2012. A systematic genetic screen to dissect the microrna pathway in Drosophila. G3 (Bethesda) 2: 437–448. 10.1534/g3.112.002030
- ↵Putman TE, Schaper K, Matentzoglu N, Rubinetti VP, Alquaddoomi FS, Cox C, Caufield JH, Elsarboukh G, Gehrke S, Hegde H, 2024. The Monarch Initiative in 2024: an analytic platform integrating phenotypes, genes and diseases across species. Nucleic Acids Res 52: D938–D949. 10.1093/nar/gkad1082
- ↵Rajan A, Perrimon N. 2012. Drosophila cytokine unpaired 2 regulates physiological homeostasis by remotely controlling insulin secretion. Cell 151: 123–137. 10.1016/j.cell.2012.08.019
- ↵Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. 2001. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res 11: 1114–1125. 10.1101/gr.169101
- ↵Richards S, Aziz N, Bale S, Bick S, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. 10.1038/gim.2015.30
- ↵Rots D, Jakub TE, Keung C, Jackson A, Banka S, Pfundt R, de Vries BBA, van Jaarsveld RH, Hopman SMJ, van Binsbergen E, 2023. The clinical and molecular spectrum of the KDM6B-related neurodevelopmental disorder. Am J Hum Genet 110: 963–978. 10.1016/j.ajhg.2023.04.008
- ↵Sandberg A, Ling H, Gearing M, Dombroski B, Cantwell L, R'Bibo L, Levey A, Schellenberg GD, Hardy J, Wood N, 2020. Fibrillation and molecular characteristics are coherent with clinical and pathological features of 4-repeat tauopathy caused by MAPT variant G273R. Neurobiol Dis 146: 105079. 10.1016/j.nbd.2020.105079
- ↵Seal RL, Braschi B, Gray K, Jones TEM, Tweedie S, Haim-Vilmovsky L, Bruford EA. 2023. Genenames.org: the HGNC resources in 2023. Nucleic Acids Res 51: D1003–D1009. 10.1093/nar/gkac888
- ↵Smibert P, Bejarano F, Wang D, Garaulet DL, Yang J-S, Martin R, Bortolamiol-Becet D, Robine N, Hiesinger RP, Lai EC. 2011. A Drosophila genetic screen yields allelic series of core microRNA biogenesis factors and reveals post-developmental roles for microRNAs. RNA 17: 1997–2010. 10.1261/rna.2983511
- ↵Srivastava S, Shaked HM, Gable K, Gupta SD, Pan X, Somashekarappa N, Han G, Mohassel P, Gotkine M, Doney E, 2023. SPTSSA variants alter sphingolipid synthesis and cause a complex hereditary spastic paraplegia. Brain 146: 1420–1435. 10.1093/brain/awac460
- ↵Starita LM, Ahituv N, Dunham MJ, Kitzman JO, Roth FP, Seelig G, Shendure J, Fowler DM. 2017. Variant interpretation: functional assays to the rescue. Am J Hum Genet 101: 315–325. 10.1016/j.ajhg.2017.07.014
- ↵Stein D, Kars ME, Wu Y, Bayrak CS, Stenson PD, Cooper DN, Schlessinger A, Itan Y. 2023. Genome-wide prediction of pathogenic gain- and loss-of-function variants from ensemble learning of a diverse feature set. Genome Med 15: 103. 10.1186/s13073-023-01261-9
- ↵Takahashi Y, Date H, Oi H, Adachi T, Imanishi N, Kimura E, Takizawa H, Kosugi S, Matsumoto N, Kosaki K, 2022. Six years’ accomplishment of the Initiative on Rare and Undiagnosed Diseases: nationwide project in Japan to discover causes, mechanisms, and cures. J Hum Genet 67: 505–513. 10.1038/s10038-022-01025-0
- ↵Venken KJT, He Y, Hoskins RA, Bellen HJ. 2006. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 314: 1747–1751. 10.1126/science.1134426
- ↵Wakap SN, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, Murphy D, Cam YL, Rath A. 2020. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Gen 28: 165–173. 10.1038/s41431-019-0508-0
- ↵Walker LC, de la Hoya M, Wiggins GAR, Lindy A, Vincent LM, Parsons MT, Canson DM, Bis-Brewer D, Cass A, Tchourbanov A, 2023. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: recommendations from the ClinGen SVI Splicing Subgroup. Am J Hum Genet 110: 1046–1067. 10.1016/j.ajhg.2023.06.002
- ↵Wang J, Al-Ouran R, Hu Y, Kim S-Y, Wan Y-W, Wangler MF, Yamamoto S, Chao H-T, Comjean A, Mohr SE, 2017. MARRVEL: integration of human and model organism genetic resources to facilitate functional annotation of the human genome. Am J Hum Genet 100: 843–853. 10.1016/j.ajhg.2017.04.010
- ↵Wang J, Liu Z, Bellen HJ, Yamamoto S. 2019. Navigating MARRVEL, a web-based tool that integrates human genomics and model organism genetics information. J Vis Exp (150): e59542. 10.3791/59542
- ↵Wilson C, Kavaler J, Ahmad ST. 2020. Expression of a human variant of CHMP2B linked to neurodegeneration in Drosophila external sensory organs leads to cell fate transformations associated with increased Notch activity. Dev Neurobiol 80: 85–97. 10.1002/dneu.22722
- ↵Wormser O, Levy Y, Bakhrat A, Bonaccorsi S, Graziadio L, Gatti M, AbuMadighem A, McKenney RJ, Okada K, Riati SE, 2021. Absence of SCAPER causes male infertility in humans and Drosophila by modulating microtubule dynamics during meiosis. J Med Genet 58: 254–263. 10.1136/jmedgenet-2020-106946
- ↵Yamada M, Nitta Y, Uehara T, Suzuki H, Miya F, Takenouchi T, Tamura M, Ayabe S, Yoshiki A, Maeno A, 2023. Heterozygous loss-of-function DHX9 variants are associated with neurodevelopmental disorders: human genetic and experimental evidences. Eur J Med Genet 66: 104804. 10.1016/j.ejmg.2023.104804
- ↵Yamamoto S, Kanca O, Wangler MF, Bellen HJ. 2024. Integrating non-mammalian model organisms in the diagnosis of rare genetic diseases in humans. Nat Rev Genet 25: 46–60. 10.1038/s41576-023-00633-6