
Closing the gaps, and improving somatic structural variant analysis and benchmarking using CHM13-T2T
- Luis F. Paulin1,7,
- Jeremy Fan2,7,
- Kieran O'Neill2,
- Erin Pleasance2,
- Vanessa L. Porter2,3,4,
- Steven J.M. Jones2,3 and
- Fritz J. Sedlazeck1,5,6
- 1Human Genome Sequencing Center Baylor College of Medicine, Houston, Texas 77030, USA;
- 2Canada's Michael Smith Genome Sciences Centre at BC Cancer, Vancouver, British Columbia V5Z 1L3, Canada;
- 3Department of Medical Genetics, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada;
- 4Michael Smith Laboratories, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada;
- 5Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA;
- 6Department of Computer Science, Rice University, Houston, Texas 77005, USA
-
↵7 These authors contributed equally to this work.
Abstract
The complexities of cancer genomes are becoming more easily interpreted due to advancements in sequencing technologies and improved bioinformatic analysis. Structural variants (SVs) represent an important subset of somatic events in tumors. While the detection of SVs has been markedly improved by the development of long-read sequencing, somatic variant identification and annotation remain challenging. We hypothesized that the use of a completed human reference genome (CHM13-T2T) would improve somatic SV calling. Our findings in a tumor–normal matched benchmark sample and three patient samples show that the CHM13-T2T improves SV detection accuracy compared to GRCh38 with a notable reduction in false-positive calls, and thus supports improved prioritization. We also overcame the lack of annotation resources for CHM13-T2T by lifting over CHM13-T2T-aligned reads to the GRCh38 genome, therefore combining both improved alignment and advanced annotations. In this process, we assessed the current SV benchmark set for COLO829/COLO829BL across four replicates sequenced at different centers with different long-read technologies. We discovered instability of this cell line across these replicates; 346 SVs (1.13%) were only discoverable in a single replicate. We identify 54 somatic SVs, which appear to be stable as they are consistently present across the four replicates. As such, we propose this consensus set as an updated benchmark for somatic SV calling and include both GRCh38 and CHM13-T2T coordinates in our benchmark. Our work demonstrates new approaches to optimize somatic SV detection in cancer with potential improvements in other genetic diseases.
Advancements in long-read sequencing (LRS) technologies have delivered unprecedented insights into cancer genomics (Aganezov et al. 2020; Thibodeau et al. 2020; Fujimoto et al. 2021; Akagi et al. 2023; Choo et al. 2023). Structural variants (SVs), defined as insertions, deletions, and rearrangements larger than 50 base pairs (bp), represent an important subset of somatic driver events in tumors (Aganezov et al. 2020; Espejo Valle-Inclan et al. 2022). Examples include the amplification of oncogenes (e.g., ERBB2) (Nattestad et al. 2018), generation of oncogenic fusion genes (e.g., BCR-ABL [Druker et al. 2001]), and silencing of tumor suppressor genes (e.g., TP53 [Hernández Borrero and El-Deiry 2021]). Accurate detection of somatic SVs and characterization of their functional effects is thus critical for appropriate cancer diagnosis and selection of therapy. Short-read whole-genome sequencing is routinely used for this purpose (Pleasance et al. 2020; Tsang et al. 2021), and can detect the majority of SVs (Choo et al. 2023). However, short-read sequencing is often unable to fully characterize the function of oncogenic SVs (Pleasance et al. 2020; Thibodeau et al. 2020). Even worse, short-read sequencing reports a high proportion of falsely identified SVs, which can be misleading (e.g., repeat expansions as translocations) (Sedlazeck et al. 2018; Mahmoud et al. 2019, 2024; Thibodeau et al. 2020). Cell lines (e.g., SKBR3) have been analyzed to assess the complexity behind these oncogenic somatic changes. For example, Akagi et al. (2023) recently identified virus-mediated progression in head and neck cancer samples through the generation of unstable human papillomavirus (HPV) integrated molecular structures. Further research is needed to assess the stability of SVs in cancer genomes, which can help direct priority when analyzing genomics data in heterogeneous cancer samples.
A key challenge in disease research is variant prioritization to identify potential causative variants of a condition. This is even more amplified in cancer patients since, depending on the type of cancer, the number of mutations is increased compared to normal tissue. Thus, researchers leverage normal controls (often blood) from patients to identify somatic variants that could potentially be driving the cancer (Mandelker and Ceyhan-Birsoy 2020). This process is efficient but also often complicated by multiple factors such as tumor purity, availability of noncancerous tissue, gene annotation accuracy, and variant comparison/representation (Yoshihara et al. 2013; Salzberg 2019; English et al. 2022). In particular, variant prioritization is impacted by falsely identified variants in either tumor or normal samples. The cause of these falsely identified variants can be attributed to misinterpretation of mapped reads, coverage fluctuations, low-quality mapping in tandem repeat regions, and unresolved regions in the reference genome. To improve these, multiple advancements have been proposed. The first complete reference genome CHM13-T2T recently became available, which showed improvements in variant calling (Aganezov et al. 2022; Nurk et al. 2022), and even corrected misrepresented medically relevant genes (Behera et al. 2023). The CHM13-T2T genome added ∼200 megabase pairs (Mbp) of resolved sequence to the GRCh38 reference build, thus closing reference gaps and completing the centromere and telomere sequences (Nurk et al. 2022). These repetitive regions can play a key role in cancer progression as they include hundreds of protein-coding genes that so far are not well understood (English et al. 2024). Perhaps more importantly, they can lead to the onset of genome instability and thus the formation of variants or inclusions of viruses (e.g., HPV) (Akagi et al. 2023; Porter et al. 2025). Annotating these newly assembled regions is challenging, but such annotations are necessary to understand the impact of mutations in these regions. While there are significant improvements in variant calling when SVs are detected against the CHM13-T2T reference (Aganezov et al. 2022), the benefits of using the complete genome reference have yet to be determined for somatic variants in a cancer context. Additionally, since CHM13-T2T is still being annotated, the annotated reference genomes GRCh37 and GRCh38 still hold value for ranking and characterizing SVs (Collins et al. 2020; Tanner et al. 2024). Using both a complete and annotated reference genome would allow researchers to identify novel oncogenic candidate variants across different cancer types.
Bioinformatics methodologies are continuously undergoing improvements to better utilize these novel reference genomes and enable improved detection of novel alleles, genes, and the variants and mutations that impact them (Majidian et al. 2023; Chen et al. 2024; Smolka et al. 2024). One such improvement was a novel method (LevioSAM2) (Chen et al. 2024) that lifts over the read alignments between two reference genomes, thus gaining benefits of both and resulting in improved variant detection overall. This contrasts to previous methods that instead lifted over variant calls, which often lead to false positive (FP) variant calls, especially with larger rearrangements or duplications (Aganezov et al. 2020). We recently developed the SV caller Sniffles2 to identify SVs using LRS data (Smolka et al. 2024). While Sniffles2 has been applied in germline contexts such as neurological and Mendelian disorders, its utility for cancer is still unproven, and somatic mutation detection often requires additional steps to reduce the false discovery rates. Furthermore, widely available benchmark samples are important instruments for benchmarking approaches (Zook et al. 2020; Wagner et al. 2022; Majidian et al. 2023; Olson et al. 2023). Over the past years, several groups have proposed benchmarks for normal and cancer samples, such as SKBR3 and COLO829 (Craig et al. 2016; Espejo Valle-Inclan et al. 2022). Nevertheless, these require constant vetting as reference genome changes and novel technologies change the downstream results that constitute the established benchmark.
In this work, we investigated novel advancements in SV calling and reference genome mapping to improve somatic variant detection, which leads to lower FP, thus improving prioritization. To accomplish this, we analyzed four replicates of the tumor–normal benchmark samples COLO829/COLO829BL at different laboratories to investigate the genome stability of these samples and also investigate how mapping to a complete reference genome (CHM13-T2T) influences the established benchmark. Nevertheless, the lack of annotations on CHM13-T2T complicates variant prioritization. To overcome this, we propose a liftover approach of aligned reads that combines the benefits from both reference genome versions to deliver less falsely identified variants on GRCh38, further benefiting from the vastly available annotation resources. In addition to this work, we further investigated the genome instability of COLO829/COLO829BL and thus the risk of utilizing previous postulated benchmarks. Furthermore, we propose a corrected, stable benchmark based on the four replicates of COLO829/COLO829BL sequenced across different laboratories. Finally, we utilized this strategy to analyze three patient samples from the long-POG cohort.
Results
An updated COLO829 structural variation benchmark
We investigated the previously established COLO829/COLO829BL benchmark (Espejo Valle-Inclan et al. 2022), as recent reports highlighted discrepancies between the current benchmark and other sequencing data (Shiraishi et al. 2023; Smolka et al. 2024). We utilized four independent COLO829/COLO829BL samples from four sequencing centers profiled using the Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) long-read platforms, including the reads from the most recently established benchmark (Supplemental Table 1; Espejo Valle-Inclan et al. 2022). Figure 1A shows the steps followed to obtain somatic SV calls that were used to investigate the aforementioned benchmark data set (see Methods). Briefly, we aligned the four tumor–normal pairs to the GRCh38 reference genome, then identified SVs using Sniffles2's population call/merge strategy (Smolka et al. 2024). Next, we included SVs from cuteSV (Jiang et al. 2022), Serverus (Keskus et al. 2024), and nanomonSV (Shiraishi et al. 2023). After calling SVs with each tool (cuteSV per sample, Sniffles2, Serverus, and nanomonSV in somatic mode), the SVs were merged into a multisample VCF, then all SVs were genotyped using Sniffles2 genotyper and remerged (see Methods). We selected SVs that were present in the cancer samples with a variant allele frequency (VAF) ≥ 10% and not present in any of the normal samples. These filters were applied as we aim to detect these SVs in 30× sequencing experiments and tools such as Sniffles and Serverus will detect a low-frequency SV with a minimum of three reads by default. We identified 30,526 SVs with the Sniffles2 strategy with 44 being unique to COLO829, and 24,642 SVs with the multi-tool strategy, from which 26 are unique to the COLO829 cancer cell line. We combined these unique SVs and found that 19 SVs were present in both data sets, 25 only in the Sniffles2 strategy and seven only in the multi-tool strategy. We manually reviewed in the Integrative Genomics Viewer (IGV) these SVs and continued with 49 SVs that looked like promising candidates. We denoted this data set as COLO829-GRCh38 (Supplemental Table 2).
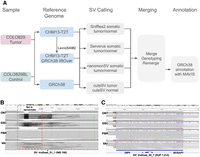
Schematic overview of the benchmark analysis and SVs examples. (A) Schematic overview of our variant calling and comparison methods using CHM13-T2T versus GRCh38. (B) IGV screenshot of an SV located in Chr 14 cataloged as missing (truthset_51_1, purple triangle) according to the benchmark by Valle-Inclan et al. We did not detect any reads supporting it. Moreover, we detected a DEL in all four samples, which is missing from the benchmark. (C) IGV screenshot of a DUP located in Chr 15 that was assigned two distinct SV types (DUP and INS), and thus cataloged as missing according to the benchmark by Valle-Inclan et al. Manual inspection in IGV showed the SV. Notice that these alignments contain mapping artifacts in the ONT samples.
We compared these 49 SVs to a previously established SV benchmark for COLO829 (62 SVs, see Methods) by Espejo Valle-Inclan et al. (2022). Table 1 shows the benchmark summary. When compared to established benchmark SVs, 42/49 SVs were categorized as true positives (TP), seven as FP, and 20 as false negatives (FN) (Supplemental Table 3).
Benchmark results for the three COLO829 data sets based on the reference genome
We proceeded with an in-depth analysis of the FP and FN calls to understand how the previous benchmark compared to the full set of data from four centers. A manual inspection of FP events revealed that 6/7 were falsely classified as FPs as they showed clear evidence in samples from multiple centers (Supplemental Table 4; Supplemental Fig. 1). For example, a heterozygous 69 bases insertion SOMATIC_SV_12, which involved PMS2, was clearly detected in all four cancer samples (Supplemental Fig. 1A) but was not reported in the previous benchmark (Espejo Valle-Inclan et al. 2022). Another interesting case is a deletion that in the benchmark is labeled as an insertion (Fig. 1B). For the 20 SVs classified as FN, we performed a genotyping experiment (Chander et al. 2019) (also known as force-calling, see Methods; Supplemental Table 5; Supplemental Fig. 2) in order to assess if there was any signal in the data to support such SVs. Here, five SVs that were initially missed by our analysis had read the evidence in all four COLO829 replicates (Fig. 1C; Supplemental Fig. 2A–G), one FN SV only was detected in a control sample with one read thus was discarded by our filter that removed SVs with any evidence in the control samples (Supplemental Fig. 2H). In addition, nine FN SVs had variant allele frequencies (VAF) below our threshold of 10% and therefore were not included (VAF 1.4%–6.2%, Supplemental Fig. 2I–N), and finally, four SVs had no read support for the SVs across all four cancer replicates (Supplemental Fig. 2O,P). In total, 51 somatic SVs detected in SV calling in COLO829-GRCh38, excluding one FP, and five somatic SVs called from force-called genotyping and manual review had read the evidence in data sets from all four centers.
Leveraging a complete genome reference for cancer structural variation benchmarking
The recent introduction of CHM13-T2T showed improved variant calling (Aganezov et al. 2022; Nurk et al. 2022) and corrections of a few medically important genes (Behera et al. 2023), for example, the CHM13-T2T reference has corrected such sequences that impact more than 12 medical relevant genes and 33 protein-coding genes. Thus, we examined the impact of using CHM13-T2T for SV calling in cancer research. Using the CHM13-T2T, we observed a high number of SVs in the telomeric and centromeric regions of the chromosomes, the detection of which was aided by a complete reference (Supplemental Fig. 3). Supplemental Table 6 shows the comparison of SVs detected in centromeric and telomeric regions between using the CHM13-T2T reference (labeled as COLO829-T2T) and COLO829-GRCh38. From COLO829-T2T, 18 chromosomes have higher number of SVs in centromeric and telomeric regions (average 16.55% increase), while six chromosomes have higher number of SVs in centromeric and telomeric regions in COLO829-GRCh38 (average 1.76% increase). Next, we identified somatic SVs in the COLO829-T2T data set (Supplemental Table 7), using the same methodology as described for the benchmark SV set. Here, we did not observe an enrichment in centromeric and telomeric regions (red dots in Supplemental Fig. 3), suggesting these events are mostly germline variation. We observed a deviation in the ratio of insertions (INS) and deletions (DEL) with an INS:DEL ratio of 1:1.4 in CHM13-T2T and 1:0.87 in GRCh38 (1:1.59 and 1:1.3 in the multi-tools approach, respectively). The switched INS:DEL ratio has been described before and is thought to be caused by too small tandem repeats reported in GRCh38 (Aganezov et al. 2022). Furthermore, we observed a substantial decrease in the number of interchromosomal events (BND) from 225 in GRCh38 to 83 in CHM13-T2T for COLO829, with no substantial difference in the multi-tool strategy (two and one, respectively). As interchromosomal events are not expected to be found in normal samples, this reduction largely reflects a decrease in noise in variant calling. To enable a comparison between the GRCh38 and CHM13-T2T-based SV calls, we linked SV calls of the same SV type and chromosome using the read names supporting each SV. Since there is no COLO829 SV benchmark with CHM13-T2T coordinates, we used the read names to determine which SVs correspond to the benchmark based on the previous genotyping. We manually reviewed the 49 SVs detected only in tumor samples by merging the SVs from both strategies (Sniffles2 and multi-tool). From there, 41 were initially classified as TP and 8 as FP relative to the comparison of the COLO829-GRCh38 somatic SV benchmark by Valle-Inclan et al. (see Methods). We identified four interesting cases where the somatic SV is present in both GRCh38 and CHM13, but is called as being a different size due to differences in the references (Supplemental Table 8). One such example is the SOMATIC_SV_17 (corresponding to truthset_28_1 in the Valle-Inclan benchmark) differed in size by ∼6 kb (Fig. 2A), which overlaps with the glutamate metabotropic receptor GRM8. Another one is the SOMATIC_SV_19 (corresponding to truthset_30_1 in the Valle-Inclan benchmark) differed in size by 126 kb, which in the case of CHM13-T2T SV is not hitting any genetic element but the GRCh38 does overlap with an olfactory receptor OR2A1 and its antisense RNA OR2A1-AS1. This difference is a consequence of the size difference between both SVs and not given by differences in the annotation. Next, we identified one SV that was deemed an inversion in the CHM13-T2T reference but a translocation in GRCh38 (SOMATIC_SV_33/truthset_52_1). Finally, manual inspection of the remaining FP showed that they actually represent TPs, as we detected supporting reads in all cases (Supplemental Table 8; Fig. 2B; Supplemental Fig. 4). For example, Figure 2B shows a 1357 bp deletion (SOMATIC_SV_29) that can be clearly observed in all four cancer samples. Next, we used the UCSC Genome Browser (Kent et al. 2002) to annotate the somatic SVs for COLO829-T2T. Here, we observed similar results to the annotation of somatic SVs in GRCh38 coordinates (20/31, 64.5% shared genes), with the addition of nine characterized genetic elements and the centromere (Supplemental Table 7B).

Differences between the benchmarks based on the used reference. (A) IGV screenshot of an SV located in Chr7 that was reported with different sizes in CHM13-T2T and GRCh38. (B) IGV screenshot of an SV located in Chr17 cataloged as FP according to the benchmark by Valle-Inclan et al., which we are able to detect in all four cancer samples.
Although SV detection is improved (i.e., no FP SVs detected), using CHM13-T2T limits downstream analysis such as prioritization and interpretation as population frequencies are not available on this reference, and GRCh38 has more informative annotation databases (Collins et al. 2020; Chowdhury et al. 2022; Nicholas et al. 2022). Thus, we used recent advances in liftover of alignments to take advantage of both the improved mapping using a CHM13-T2T reference (Chen et al. 2024) and the years of annotation and curation of the GRCh38 reference genome. Briefly, we took the CHM13-T2T read alignments and used LevioSAM2 (Chen et al. 2024) (v0.4.1) to liftover the alignments from CHM13-T2T to GRCh38 coordinates. The liftover analysis dismisses reads that are not present in GRCh38, thus cleaning alignment from reads that the mapper will attempt to place, incorrectly. This improves the signal-to-noise ratio in difficult genomic regions that may create FP SVs. We denoted this data set as “COLO829-lifted” (Supplemental Table 9). We compared the COLO829-lifted calls to the benchmark by Valle-Inclan et al., alongside the data set COLO829-GRCh38 that was produced earlier. Table 1 shows how leveraging the CHM13-T2T genome improves SV calling and decreases both the number of SV classified as FP and FN when compared to the native GRCh38 alignment, after genotyping and manual review (Supplemental Table 10). For the case of the SVs labeled as FP in comparison to the benchmark by Valle-Inclan et al., we were able to identify all the SVs as TP with high confidence in the cancer samples (Supplemental Table 11; Supplemental Fig. 5).
Next, when analyzing the FN, we identified one SVs that was also missed in the COLO829-GRCh38 (benchmark SVs: truthset_44_1, Supplemental Table 12; Supplemental Fig. 6A,B). In both cases, we applied filters during SV calling that removed them from the final call set (COV_CHANGE for the case of SV:truthset_44_1). Then, one SVs was detected as INS in our callset and as DUP in the benchmark (SOMATIC_SV_10, truthset_15_1). These two types are often hard to distinguish as a duplication is an insertion of the same sequence next to itself (Mahmoud et al. 2019). The rest of the FN are very similar to the COLO829-GRCh38, moreover, we did observe differences in the reported VAF between the GRCh38 alignment, the CHM13-T2T alignment and the liftover. Figure 3 shows an example in which there is no evidence of the SV in all the tumor samples and thus it was removed from the benchmark, although present in the one from Valle-Inclan.

Example of a low-frequency SV. IGV screenshot of an SV located in Chr 10, that was labeled as FN according to the benchmark by Valle-Inclan et al. Our analysis calls removed three SVs due to a lack of evidence in all samples (Supplemental Table 12).
Overall, the approach of incorporating alignment to CHM13-T2T followed by liftover analysis eliminated all FP somatic SV calls for COLO829. Such a reduction in FP has important impacts particularly when analyzing patient samples, where a reduction in FP can have implications for interpretation and necessary follow-up analysis. The SV liftover approach allows for combining the improved accuracy gained by incorporating CHM13-T2T alignments (precision in COLO829-GRCh38 98% vs. COLO829-T2T/COLO829-lifted 100%) with the biological and clinical annotations available on GRCh38. Additionally, we observed a drastic reduction in BND (225 and 130, respectively, for COLO829-GRCh38 and COLO829-lifted), both in tumor and normal samples, indicating improvements in germline SV calling, which has implications for variant prioritization in disease research. We used MAVIS (Reisle et al. 2019) to annotate our proposed somatic SVs for the COLO829 cell line in GRCh38 space. From the 54 somatic SVs detected in COLO829-lifted (53 TP and 1 FN), we observed 31 genes being affected or disrupted by SVs, many of which have been linked to cancer. Some examples are FHIT (near a fragile site), tumor suppressors ITIH5, MAGI2, PTEN, WWOX, and cell-proliferation control gene TMX3 (Zhang et al. 2019; Cao et al. 2020; Bellon et al. 2021; Husanie et al. 2022; Yehia et al. 2023).
We suggest using this call set (54 somatic SVs) as a refined benchmark for this important tumor–normal control cell line (see Data access). The use of data from multiple centers and independent cell line passages is particularly important as we demonstrate differences between samples that are likely to in part represent instability and evolution of this cell line at the SV level in addition to the previously described instability at the SNV level (Craig et al. 2016). Thus, accurate benchmarks that incorporate multiple replicates are necessary to reduce possible sources of error introduced by a single sample.
Utilizing a complete genome reference for cancer SV detection
Our results show the benefit of using the CHM13-T2T reference for SV analysis in cancer. Furthermore, we introduced and assessed a liftover approach that leverages the benefits of both reference genomes to improve the detection and annotation of somatic SVs in COLO829/COLO829BL. SV analysis and prioritization in cancer is complex, as most SVs are not cancer drivers, but instead, passenger mutations picked up in the process of cancer evolution. In addition, cancer samples have diversity in tumor content (fraction of sequenced DNA derived from tumor cells compared to normal cells), which can result in somatic SVs with low VAF. Because of the overall complexity of SV analysis in cancer, a reduction in FP calls is greatly beneficial. Given these results, we next investigated the use of the CHM13-T2T reference genome for the analysis of three different cancer patient samples. In this pilot experiment, we used samples with whole genome coverage over 30× (POG044 63×, POG1022 47×, and POG846 31×) all with normal controls (Supplemental Table 1; O'Neill et al. 2024). When using CHM13-T2T alignment followed by GRCh38-liftover analysis, we observed fewer somatic SVs when compared to GRCh38 (Supplemental Table 13A–F).
Overall, the liftover strategy reduced the number of candidate somatic SVs in the POG044 and POG1022 samples and increased in POG846. Supplemental Figure 7 shows the candidate and curated somatic SVs for the three samples across the 22 autosomes and Chromosome X.
For POG044, we observed 56 somatic SVs in the liftover analysis (Supplemental Table 13A), for the POG1022 62 (Supplemental Table 13C) and for the POG846 72 (Supplemental Table 13E), with over 70% of the candidate DUP, INV, and BND being true somatic SVs. For INS and DEL, the number of candidates being true somatic SVs was close to 10%–30% with many SVs being low frequency in the control samples or in complex regions that prevented us from correctly assessing them. The POG846 has a greater number of candidates (over 2500), and thus additional filtering was applied (SVs needed to overlap with genes). This alone made the correspondence of candidates to curated somatic SVs over 90%.
The liftover analysis not only aided in reducing the number of FP, but for the case of the POG846 also impacted the FN, with six DEL being rescued, four of which were megabase long. Finally, for the POG846, we detected several large SVs (likely genomic rearrangements) with 15 somatic SVs being larger than 100 kb, 9 larger than 1 Mb, and 3 larger than 10 Mb.
While performing the manual curation of the candidate somatic SVs we saw for many INS and DEL, we could detect reads supporting the SV in the control samples. From those, 30 were caused by SV of the same type that were collapsed in the tumor sample but not in the control causing a difference in size and thus two different SV (Supplemental Fig. 8A), 23 were in low-complexity regions where we could observe multiple SVs of the same type (Supplemental Fig. 8B) and 18 have read support in the control with mapping quality lower than out threshold (MQ ≥ 20) and thus were not used during SV calling (Supplemental Fig. 8C). For sample POG1022, MAVIS annotated 184 somatic SVs (100 were filtered or collapsed) from which 129 (70.1%) had reads supporting the SV in the control sample. From those, 64 occurred in low-complexity regions (Supplemental Fig. 9A) and 46 were caused by collapsed SV of the same type (difference in size Supplemental Fig. 9B).
We annotated SVs from the liftover analysis (in GRCh38 coordinates) using MAVIS, but overall did not observe any SV that would be predicted to be causative in tumor formation. MAVIS predicted a nonsynonymous SV on Chr 1 in the sample POG044 (Supplemental Table 14). This SV affects the neuroblastoma breakpoint family gene NBPF20, which has been associated with several types of cancer. This region was affected by several deletion events, moreover, most of them show low mapping quality and thus were not used during SV calling, specially in the control sample (Supplemental Fig. 10).
For sample POG1022, three nonsynonymous coding events were categorized by MAVIS (Supplemental Table 15). The first one, a 24.2 kb inversion in Chromosome 2, which affects the immunoglobulin kappa variable gene IGKV3-11 (Supplemental Fig. 11) which expression has been observed in myelomas (https://www.proteinatlas.org/ENSG00000241351-IGKV3-11/pathology). The second, a 8.9 kb deletion also in Chromosome 2, which affects the ankyrin repeat domain gene ANKRD36 (Supplemental Fig. 12), which has been used as a biomarker of disease progression in Leukemia (Iqbal et al. 2021). Upon further inspection, we detected a larger deletion in the same region (12 kb) with some reads supporting the SV in the control. The third nonsynonymous coding event was a 609 kb deletion in Chromosome 20, which was only detectable in the liftover analysis and not in the GRCh38 alignment. Visual inspection of the region in both the liftover analysis and GRCh38 alignment shows that the latter has a weaker signal, although it is present. This SV affects the prostaglandin synthase PTGIS (Fig. 4A,B), which has been linked with various cancers like prostate cancer (Qiao et al. 2023), colorectal cancer (Ding et al. 2023), and cancer-free trichothiodystrophy (Lombardi et al. 2021).

Example of a large somatic deletion detected in the POG1022 sample. (A) Coverage pattern for a 609 kb somatic deletion detected in POG1022 in Chr 20 that affects the PTGIS gene among other genes. (Supplemental Table 15). (B) IGV screenshot that zooms into the breakpoints of the somatic deletion.
For the POG846 sample, we detected several large events, including deletion and duplication (CNV), some of them spanning complete chromosomes like a 93 Mb duplication of Chr 6, a 53 Mb deletion in Chr 4, a 45 Mb deletion in Chr 12, and a 41.5 Mb deletion in Chr 5 which affected the gene dosage of countless genes (Supplemental Table 15). One gene of interest is TCEA3, which is affected by a deletion that disrupts the two exons (Fig. 5). TCEA3 has been linked to gastric, lung, ovarian, and colorectal cancer, among others (Cha et al. 2013; Li et al. 2015; Hou et al. 2021; Wu et al. 2024). Other genes linked to cancer affected only in the tumor sample are NF1, IFNLR1, RAB11A, CBFA2T2, BOD1L1 (Supplemental Fig. 13; Supplemental Table 16).

Example of a somatic SV in the POG849 sample. A 540 bp somatic deletion in cancer sample POG846 (compared to the control POG846Co) located in Chr 1 that affects two exons of the TCEA3 gene, which has been linked to gastric, lung, ovarian and colorectal cancer, among others.
Discussion
In this work, we assessed the benefits of using a complete reference genome for SV improved calling in tumors. For this, we examined four replicates of COLO829/COLO829BL together with two tumor samples aligned to both CHM13-T2T and GRCh38. We saw lower false-positive somatic SV calls when using CHM13-T2T as the reference. To enable taking advantage of CHM13-T2T mapping, while also benefiting from the richer clinical annotations available for GRCh38, we demonstrated the efficacy of lifting over alignments using LevioSAM2 (Chen et al. 2024), which roughly increases the compute time by 50%, compared to only using GRCh38. Furthermore, we developed a new approach to trace variants across reference genome versions using read names instead of coordinates, which is less sensitive to different allelic representations. Lastly, we identified inconsistencies among replicates of the existing COLO829/COLO829BL benchmark set, likely due to the instability of the COLO829 cell line (Supplemental Fig. 14), compared to, for example HG002, which seems to be more stable. Thus, we propose new consensus somatic SV benchmarks for both GRCh38 and CHM13-T2T that addresses these inconsistencies. Our suggested approach should lead to more accurate and less laborious cancer SV analysis, while the improved benchmark provides a more accurate testbed for the assessment of cancer SV callers.
While the advent of long reads holds promise to improve the detection of complex alleles together with resolution in tandem repeats, it also highlights the problem around variant annotation and prioritizations. In contrast to short reads, we often see many novel variants (e.g., SV) that are not part of public databases such as gnomadSV (Collins et al. 2021), making annotation and prioritization more difficult. To overcome this, analysis often relies on tumor–normal comparison, which streamlines the detection of causative variants (Mandelker and Ceyhan-Birsoy 2020). Short reads tumor–normal analysis often indicates large (>10 kbp) causative SV (Choo et al. 2023) but also has higher FP and FN rates (Mahmoud et al. 2019; Aganezov et al. 2022), which can hinder rapid SV prioritization during clinical workup of tumors. An example is the reporting of repeat expansions as translocation/BND events (Sedlazeck et al. 2018). We find that using long reads on different reference genomes appears to reduce the FP calls and can thus improve variant prioritization. In this paper, we observed an important decrease in the number of BND when utilizing CHM13-T2T compared to the GRCh38 genome. Sniffles2 falsely called 130 SVs for GRCh38, 84 for CHM13-T2T, and 83 for CHM13-T2T lifted over to GRCh38. FP SV calls are often caused by misassembled and incomplete reference regions (Mahmoud et al. 2019)]; the completeness of CHM13-T2T reduces these FP. However, clinical annotations of SVs remain incomplete for CHM13-T2T. We therefore suggest a liftover mapping approach that combines the increased accuracy of CHM13-T2T together with the utility of annotations across GRCh38 itself. Furthermore, this liftover approach does not double the analytical time nor the costs for analysis as it utilizes a chain file to rapidly liftover the read alignments without additional pairwise alignments needed (Chen et al. 2024). This also permits downstream utilization of annotation databases and approaches such as gnomadSV (Collins et al. 2021) and STIX (Chowdhury et al. 2022) to further filter and improve variant prioritization.
COLO829 and its matched control COLO829BL are well-established cancer cell lines that provide advantages for testing and benchmarking of sequencing and analysis approaches (Pleasance et al. 2010; Craig et al. 2016; Espejo Valle-Inclan et al. 2022). However, our study clearly demonstrates that caution should be used when interpreting and comparing results from a single analysis, sequencing run, or even biological sample. When we compared our benchmark to the most up-to-date benchmark from (Espejo Valle-Inclan et al. 2022), we identified multiple SVs that did not appear in any other replicates of COLO829 sequenced at other centers, and some of which were not identified in the reanalysis of the original Nanopore data. For example, four SV present in the benchmark from Valle-Inclan could not be identified in any of the cancer replicates, which includes a replicate from the aforementioned benchmark. Moreover, fourteen SVs could not be identified in all cancer replicates or had low VAF (<10%), which complicates the interpretation of the results. Moreover, we aim to produce a benchmark that can be tested with ∼30× coverage and thus should be accessible for different callers and studies. This suggests caution should be used when interpreting results in comparison to the previous COLO829/COLO829BL benchmark (Espejo Valle-Inclan et al. 2022). Furthermore, across the different replicates, we could demonstrate and manually validate differences between cancer replicate samples (i.e., SVs uniquely identified per replicate), similar to changes documented for somatic SNVs observed in different COLO829 passages (Craig et al. 2016). One interesting case was an inversion in the previous benchmark, that in the data looked like a duplication, (likely an INV/DUP) moreover the signal was not present in all samples for neither SV (Supplemental Fig. 14). These changes clearly highlight the instability of COLO829/COLO829BL and need to be taken into consideration across benchmarks. Otherwise, there is potential for incorrect interpretation of the accuracy of sequencing and analysis techniques evaluated against the benchmark. To aid this, we identified a core set of SVs that appear to be maintained stably across independent sequencing replicates, and we propose this set to be used as the updated version of the COLO829/COLO829BL benchmark for the detection of somatic cancer SVs. It is interesting to note that some differences in the replicates of COLO829/COLO829BL can be attributed to technical artifacts, as the data set includes Nanopore data generated with prior versions of the approach that might suffer from base calling biases (i.e., deletions) that have since been improved (Kolmogorov et al. 2023). Nevertheless, biological artifacts are clearly present given the evolution of the cell line.
Our work demonstrates new approaches to optimize somatic SV detection, which leads to improved prioritization in cancer with potential improvements in other genetic diseases. We demonstrate this over patient samples but further over COLO829, where we introduce a new benchmark due to its variability. Given all these artifacts, it's still important to note the importance of the widely available COLO829/COLO829BL cell line for technology and analytical development.
Methods
Samples
We used the COLO829 cancer cell line (melanoma) and its germline control COLO829BL (Blood, B lymphoblast) to assess somatic SV calling with whole genome long reads sequencing. We sequenced the tumor–normal samples with different technologies (ONT PromethION, ONT MinION, and PacBio Revio) at different sites: Canada's Michael Smith Genome Sciences Centre, BC, Canada (labeled GSC) sequenced with ONT PromethION; Oxford Nanopore Technologies (labeled ONT) sequenced with ONT PromethION; Pacific Biosciences of California, Inc. (labeled REV) sequenced with PacBio Revio; Center for Molecular Medicine and Oncode Institute, UMC Utrecht, Utrecht, the Netherlands (labeled VAI) sequenced with ONT MinION.
Cancer samples were derived from the Personal Oncogenomics (POG) program (Pleasance et al. 2020), clinical trial NCT02155621, approved by and conducted under the University of British Columbia—BC Cancer Research Ethics Board (H12-00137, H14-00681). POG1022 is a tumor sample (diploid) from a metastatic diffuse large B cell lymphoma, with the control sample taken from a skin biopsy (POG1022SK). POG044 is a tumor sample (diploid) taken from a recurrent anaplastic oligodendroglioma, with a blood control sample (POG044BL). POG846 is a tumor (triploid) from an ovarian cancer, with a matched tissue control (ovary, diploid). Supplemental Table 1 summarizes the samples used in this study and includes links for accessing the data. Two human reference genomes were utilized: GRCh38.p13 and CHM13-T2T v2.
Alignment
minimap2 (Li 2021) (version 2.24-r1122) was used to align the long reads to the human genome. We utilized two different versions of the human genome: GRCh38.p13 and CHM13-T2T v2. We used default parameters, with the output to be in the SAM format (-a) and preset for both Oxford Nanopore reads (-x map-ont) and PacBio reads (-x map-hifi). Additionally, we converted the alignment to BAM format, sorted and indexed using SAMtools (Danecek et al. 2021) (version 1.16.1).
Liftover
We used LevioSAM2 (Chen et al. 2024) (version 0.4.1) to liftover alignments from CHM13-T2T into GRCh38. We ran LevioSAM2 using the provided chain files for CHM13-T2Tv2. We differentiated between sequencing technologies (ONT and PacBio) in the configuration file and mapping presets for minimap2 (map-ont and ont_all.yaml for ONT and map-hifi and pacbio_all.yaml for PacBio). Additionally, we used specific allowed gaps (-g) and edit distance (-H) values for each technology: -g 1500 -H 6000 for ONT and -g 1000 -H 100 for PacBio.
SV calling
Sniffles2 (Smolka et al. 2024) (version 2.2) was used to call SVs. Each sample had three reference backgrounds: GRCh38, CHM13-T2T, and the liftover alignment (CHM13-T2T to GRCh38). For both GRCh38 alignments (native and liftover), we added a tandem repeat annotation file during the run (‐‐tandem-repeats file.bed). This file is provided alongside Sniffles (https://zenodo.org/records/8121996). In all cases, the reference genome was provided (‐‐reference) and the SNF file was produced (‐‐snf file). The rest of the parameters were left as default. Next, we performed population SV calling with Sniffles2 using the SNF files produced in the previous step. Each population call was done by reference genome, such that we produced three population files, one for GRCh38, one for CHM13, and one for the liftover. In this step, Sniffles2 was used with default parameters.
We also ran cuteSV to all the samples using the recommended parameters for ONT and PacBio, respectively. Next, we ran both nanomonSV and Severus to call somatic SV unique in the tumor samples. In both cases using default parameters and following each author's usage guide. During the analysis, we ran into an issue with cuteSV (latest released version v2.1.1, April 2024) in which it failed to complete the run on four different occasions with two files (ONT sample on GRCh38 coordinates), so they were removed from the analysis.
Somatic SV detection in COLO289
We used the fully genotyped population VCF file generated with Sniffles to assess the SVs that were unique to the cancer samples. We used the support vector provided in the VCF file (SUPP_VEC) to assess the presence/absence of each variant. We used BCFtools (version 1.16) (Li 2011) to extract SVs that were tumor-only (using the ‐‐include option, example: bcftools view ‐‐include “SUPP_VEC = ‘11110000’”). We denoted a somatic-cancer variant if the variant was only present in the cancer samples, had a VAF ≥ 10%, and had a minimum of 10 supporting reads. Initially, we excluded any SV that had any read support in any of the control replicates. Manual inspection overrode cases where a single read was detected in a control sample.
Next, we included SV from cuteSV, Serverus, and nanomonSV. After calling SVs with each tool (cuteSV per-sample, Sniffles2, Serverus, and nanomonSV in somatic mode), the SVs were merged into a multisample VCF with BCFtools (1.16) and truvari 4.2.2 (English et al. 2022). Next, we genotype INS, DEL, INV, and DUP for each sample independently using Sniffles2 genotyper to finally remerge with BCFtools and truvari. This method gives us a set of SV that all have an assigned genotype so they can be compared, moreover, it also gives us some variants of the same type that differ in a couple bases in size with similar sequence. Next, we selected candidates SVs those with no reads in the control samples and a minimum of 3 reads and 10% VAF in the tumor samples. These filters were applied as we aim that these SVs can be detected from 30× coverage data from the current tools, which for the case of Sniffles2 and Serverus have a minimum of three reads to call an SV. We selected SV that were detected by at least 50% of the tool + data set to be added to the benchmark. Finally, we combined the SVs of the same type if they were in the same chromosome, at a distance smaller equal to 1 kb. That left us with 30 SVs, from which 22 were in the Sniffles2 initial benchmark and 8 were new for the liftover sample (37 total in GRCh38 and 16 new in CHM13-T2T). Instead of reducing the number of SVs, we only added the SVs not detected by our initial attempt using only Sniffles to increase the benchmark data set. Furthermore, as the BND cannot be genotyped, we only used the four from the somatic calling with Sniffles.
SV annotation
Postprocessing of SVs of the GRCh38 and CHM13-T2T to GRCh38 call sets was conducted with MAVIS (Reisle et al. 2019) (version 3.1.0). Briefly, we collapsed duplicate SVs, and merged SVs by breakpoint proximity (100 bp) and type. Next, we used the RefSeq curated gene annotation track in the UCSC Genome Browser (Kent et al. 2002) to annotate the impacted genes from the somatic SVs for COLO829-T2T. SV subtype-specific analyses also incorporated RepeatMasker (version 4.1) (Tarailo-Graovac and Chen 2009) to annotate the events. Events flagged as nonsynonymous coding variants by MAVIS were manually reviewed.
SV benchmark
We compared the two COLO829 somatic SV data sets that are in GRCh38 coordinates (COLO829-GRCh38 and COLO829-lifted) to a published COLO829 SV benchmark by Espejo Valle-Inclan et al. (2022), COLO829-VAI hereafter. The COLO829-VAI benchmark consists of 68 SVs, with representation from all five SV types (INS, DEL, DUP, INV, and BND). We removed six SVs whose length was smaller than 50 bp (default reported by Sniffles2), leaving 62 SVs in the COLO829-VAI benchmark. We used BEDTools (version 2.31) (Quinlan and Hall 2010) to compare the SV coordinates of the COLO829-VAI benchmark to our COLO829-GRCh38 and COLO829-lifted somatic SV data sets.
For the case of the COLO829-T2T data set, we included the ‐‐output-rnames parameter in Sniffles to output the read names supporting each SV. We then used these read names along with the chromosome and the SV type as a proxy for matching the SV from CHM13-T2T to GRCh38 coordinates to perform a partial benchmark (FP only). Only for one case, we used the same procedure to investigate a FN call in the COLO829-T2T data set (DEL in Chr 16), because Sniffles made the call and reported the read names. However, the SV was removed from the final call set by the COV_MIN filter, thus it was still considered a FN.
SV genotyping (force-calling)
We used the Sniffles2 ‐‐genotype-vcf option to look for all the FN calls in the COLO829-GRCh38 and COLO829-lifted data sets. This option takes a VCF as input (including the FN calls for our case) and uniquely searches for the SV present in the VCF input and updates the genotype according to what Sniffles detects in the alignment file. For the case of BNDs, we additionally took all the reads overlapping with the coordinates provided by the COLO829-VAI benchmark and searched for reads that had supplementary alignments (SA; flag 2048) and compared the coordinates from the SA tag in the alignment to the “CHR2” value from the INFO field of the COLO829-VAI benchmark. We matched chromosomes and allowed for a 10 kb distance between the positions.
Somatic SV detection in POG samples
We used the fully genotyped population VCF file generated by merging tumor and normal samples using Sniffles2 SNF files (sniffles ‐‐input tumor.snf normal.snf ‐‐vcf merge.vcf.gz). Once merged, we used the support vector provided in the VCF file (SUPP_VEC) to assess the presence/absence of each SV. We used BCFtools (version 1.16) to extract SVs that were tumor-only (using the ‐‐include option, example: bcftools view ‐‐include “SUPP_VEC = ‘10’”). We denoted a somatic-cancer variant if the SV was only present in the cancer samples, a VAF ≥ 10% and a minimum of 10 read support.
Data access
The COLO829/COLO829BL proposed benchmark and nutty, a Sniffles2 companion app for parsing the VCF, can be found at Zenodo (https://doi.org/10.5281/zenodo.14367730) and as Supplemental Material.
Competing interest statement
K.O., V.L.P., L.F.P., and S.J.M.J. received travel funding from Oxford Nanopore Technologies to present at conferences in 2022 and/or 2023. F.J.S. receives research support from ONT, PacBio, Illumina, and Genentech. L.F.P. received research support from Genentech from 2021 to 2023.
Acknowledgments
This study was in part supported by funding from the Canada Research Chairs Program, Terry Fox Research Institute Marathon of Hope, and the British Columbia Cancer Foundation. F.J.S. and L.F.P. were supported by NIH (UM1DA058229, 1UG3NS132105-01, and 1U01HG011758-01). This study was conducted with the financial support of The Terry Fox Research Institute and the Terry Fox Foundation. The views expressed in the publication are the views of the authors and do not necessarily reflect those of the Terry Fox Research Institute or the Terry Fox Foundation.
Author contributions: S.J.M.J. and F.J.S. conceptualized the project. L.F.P. and J.F. performed the analysis and wrote the manuscript. K.O., E.P., and V.L.P. wrote the manuscript and provided insights to the patient samples.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279352.124.
- Received March 15, 2024.
- Accepted January 6, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








