
The impact of long-read sequencing on human population-scale genomics
- 1European Molecular Biology Laboratory (EMBL), Genome Biology Unit, 69117 Heidelberg, Germany;
- 2Institute for Medical Biometry and Bioinformatics, Medical Faculty and University Hospital Düsseldorf, Heinrich Heine University, 40225 Düsseldorf, Germany;
- 3Center for Digital Medicine, Heinrich Heine University, 40225 Düsseldorf, Germany
Abstract
Long-read sequencing technologies, particularly those from Pacific Biosciences and Oxford Nanopore Technologies, are revolutionizing genome research by providing high-resolution insights into complex and repetitive regions of the human genome that were previously inaccessible. These advances have been particularly enabling for the comprehensive detection of genomic structural variants (SVs), which is critical for linking genotype to phenotype in population-scale and rare disease studies, as well as in cancer. Recent developments in sequencing throughput and computational methods, such as pangenome graphs and haplotype-resolved assemblies, are paving the way for the future inclusion of long-read sequencing in clinical cohort studies and disease diagnostics. DNA methylation signals directly obtained from long reads enhance the utility of single-molecule long-read sequencing technologies by enabling molecular phenotypes to be interpreted, and by allowing the identification of the parent of origin of de novo mutations. Despite this recent progress, challenges remain in scaling long-read technologies to large populations due to cost, computational complexity, and the lack of tools to facilitate the efficient interpretation of SVs in graphs. This perspective provides a succinct review on the current state of long-read sequencing in genomics by highlighting its transformative potential and key hurdles, and emphasizing future opportunities for advancing the understanding of human genetic diversity and diseases through population-scale long-read analysis.
The promise of long genomic reads for human population-scale studies
The advent of long-read sequencing technologies, exemplified by platforms such as Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), is reshaping the genomics field. Unlike short-read sequencing, which struggles to resolve repetitive and complex regions, long-read sequencing offers unparalleled accuracy and resolution in these areas of the genome, providing insights into human genetic diversity. These platforms have recently gained in accuracy, enabling applications from assembling human chromosomes from telomere-to-telomere (Nurk et al. 2022; Rautiainen et al. 2023; Cheng et al. 2024) to the construction of pangenomes with advanced computational methods, allowing to represent the genetic variation of numerous assembled human genomes in a compact and extendable graph (Liao et al. 2023; Hickey et al. 2024). Advances have been particularly prominent with respect to understanding structural variants (SVs) in the genome, including in regions and variant classes previously largely inaccessible to genomic ascertainment.
The data production throughput of long-read platforms has recently further increased, offering the opportunity to go beyond assembling a small to intermediate number of samples to enable studying genetic variation in the context of population-scale and disease studies. This capacity of long-read sequencing to detect genetic variants in regions inaccessible by short-read sequencing is positioning long-read technologies as a pivotal tool for human genetics, revealing connections between previously unexplored genetic variations and human phenotypes. For instance, a recent preprint from the SOLVE-RD consortium reports up to a 13% improvement in diagnostic yield using long-read sequencing, highlighting the technology's potential clinical relevance for genetic diagnoses (Steyaert et al. 2025). In the future, careful interpretation will be required to accurately assess the incremental diagnostic yield of long-read sequencing per genetic variant class, with a subset of the mutations detected by Steyaert et al. comprising single-nucleotide variants (SNVs) which could potentially be detected using short-read platforms in combination with advanced analytical approaches. Nevertheless, advances in computational methods for long-read data analysis and a better understanding of genomic regions poorly resolved by short-read approaches are likely to further enhance the diagnostic yield of long-read sequencing.
Comparing short- and long-read sequencing with respect to genetic variant discovery
Comparative analyses of genomic sequencing technologies have highlighted that although short-read sequencing continues to be effective in the detection and genotyping of large copy-number variants, long reads are highly beneficial for insertions and complex SVs, primarily due to their ability to uncover SVs and their breakpoints at single-nucleotide resolution and additionally by allowing to assemble complex variant sequences (Chaisson et al. 2019; Ebert et al. 2021; Zhao et al. 2021; Liao et al. 2023). These differences between short- and long-read sequencing are further enhanced through the possibility to resolve the haplotype structure of genetic variation through the use of read-based phasing information carried by long reads. In combination with parent–offspring trios or chromosome-wide haplotyping technologies such as Strand-seq, this allows to haplotype resolve almost the entire genome (Koren et al. 2018; Ebert et al. 2021), with only few regions such as the short arms of acrocentric chromosomes left to be addressed by future technological innovations. These haplotype-resolved genomes offer insights into mobile element insertions (MEIs), inversions, and SVs in regions loaded with complex and tandemly oriented repeats, such as variable number tandem repeats (VNTRs), segmental duplications, telomeres, and centromeres (Ebert et al. 2021; Porubsky et al. 2022; Logsdon et al. 2024b). As such, long-read sequencing has begun to transform the investigation of these variant classes in studies of human genetic diversity and in clinical settings (Fig. 1; Sanford Kobayashi et al. 2022; Mastrorosa et al. 2023; Oehler et al. 2023).
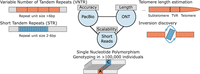
Long-read sequencing facilitates accurate discovery of SVs and their breakpoints at single-nucleotide resolution, with PacBio HiFi sequencing showing very high accuracy in terms of mapping repeat polymorphisms and ONT ultra-long (ONT-UL) being an essential method for determining centromeric or telomeric variant repeat (TVR) structures as well as balanced inversions which are often embedded within large DNA repeats. Short reads remain highly cost-efficient and thus, scalable to tens of thousands of genomes. Notably, some applications such as telomere-to-telomere assembly projects presently require a combination of technologies, with current studies using PacBio Hifi sequencing, ONT-UL, and Strand-seq (short reads) (Logsdon et al. 2024a).
The transformative character of long-read sequencing technologies is particularly apparent when studying heritable classes of genetic variation. Indeed, long-read sequencing typically identifies more than twice the number of germline SVs per individual genome than short reads (Ebert et al. 2021; Zhao et al. 2021). While short reads, due to their low cost and high throughput, remain a valuable tool for applications in large disease cohorts, their limitations are particularly evident in complex regions of the genome—which in studies using short-read sequencing are typically left out through the application of “genome accessibility masks.” Comparing short- and long-read sequencing echoes the historical debate between short-read sequencing and microarrays, where both methods initially complemented one another; yet, short-read whole genome sequencing has ultimately provided much improved sensitivity over microarrays, even with low sequencing coverage (Rubinacci et al. 2023). We anticipate that, with continued advances in computational methods, a similar shift will occur in long-read sequencing. Even when pursued with relatively shallow coverage levels, we expect long reads to significantly surpass short-read sequencing in the systematic detection of SV in clinical cohorts.
When compared to the characterization of heritable genetic variation, advances of long reads are more nuanced in relation to the discovery of somatic mutations in cancer genomes (Choo et al. 2023). This is since positively selected somatic chromosome alterations in cancer genomes typically lead to large segmental copy-number changes, which are sensitively captured with short reads when using read-depth analysis, even when present at a low clonal fraction due to tumor heterogeneity. Generally, the mosaic nature of somatic mutations in human tissues including cancers demands higher sequencing coverages, which currently makes long-read sequencing less cost-effective for clinical laboratories in oncology.
Nonetheless, long-read technologies clearly offer advantages and hold promise for advancing our understanding of cancer genomics, such as in unraveling SV formation mechanisms (Rausch et al. 2023). They enable the discovery of somatic SVs that remain undetectable through read-depth analysis, by providing deeper insights into repetitive regions, such as through revealing somatic alterations affecting telomeric sequences (Kinzig et al. 2024), and by uncovering links between centromeric sequence variants and somatic DNA alteration processes. Additionally, the ability to haplotype resolve and assemble long-read sequences offers advantages in the interpretation of somatic mutations, including linking genetic variants with epigenetic patterns (discussed below) on the same haplotype. This highlights the potential of long reads, not just for germline genetic variation, but also for understanding somatic mutations in cancer genomes.
From study design to current challenges: bringing long-read-based variant discovery to disease cohorts
Advances in long-read sequencing technology and computational tools have raised interest in the study of large human sample cohorts with long-reads to foster population-genetic studies and map complex phenotypes (Beyter et al. 2021; De Coster et al. 2021). Yet, there are current hurdles to scaling haplotype-resolved human genome assembly to large population-scale cohorts. This is because the highest quality genome assemblies currently rely on laborious, and costly, cross-platform data generation efforts, for combining the benefits of the base pair accuracy of PacBio reads with the ultra-long ONT sequencing reads to yield chromosome-scale assemblies.
Designing studies for long-read sequencing, therefore, requires considerations and careful planning to balance cost and coverage—depending on the research objectives, the types of variants under investigation, and the cohort size (Fig. 2). Indeed, the utility of long-read sequencing varies between rare and common disease cohorts. In rare diseases, long-read sequencing is particularly powerful for discovering causal variants that are difficult to capture with short reads, and are often private to the carrier or at very low population allele frequency. Common disease studies, by contrast, typically prioritize the genotyping of a maximally large set of previously known genetic variants across larger populations.
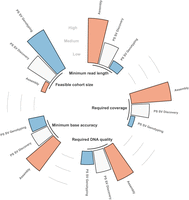
Relationship between sequencing coverage, base accuracy, read length, DNA quality, and feasible cohort size—showing how different study designs affect outcomes such as variant discovery and genotyping accuracy. For rare disease studies, long-read sequencing offers unmatched power in uncovering novel variants, whereas in common diseases, the emphasis will be toward larger sample sizes with lower coverage to increase statistical power in population-scale (PS) genetics and genome-wide association studies (GWAS).
In the light of the significant costs associated with multiplatform sequencing, most recent studies have targeted relatively small sample sizes for this approach. Low-to-intermediate coverage sequencing is more cost-effective and comprehensively yields access to genetic variation present at low allele frequency in the population—thus allowing the inclusion of SVs in studies linking genotype and phenotype, including for performing rare variant association tests (Beyter et al. 2021). This approach is also likely to facilitate the development of new frameworks for harmonizing and prioritizing pathogenic SVs in clinical genetics, in spite of limitations regarding complex genomic regions that will require higher sequencing depth to be fully resolved at the population-scale in the future.
Partially alleviating these well-understood limitations, hybrid approaches to genome inference have been developed—such as PanGenie (Ebler et al. 2022)—which leverage haplotype-resolved whole genome assemblies of a relatively small number of individuals to detect and genotype variants discovered in these samples from short-read sequencing data. This results in a significant increase in SVs that can be captured in large-scale population-based panels. Nonetheless, this currently comes with the trade-off that rare and population-specific SVs are underrepresented until the input human samples sequenced with long reads (i.e., the discovery sample set) reach an adequate size.
The need for open-access reference data sets generated with long DNA reads
Particularly in rare disease studies, population variant reference data sets are paramount for prioritizing disease-causing variants over such present in the normal population. Current population-scale reference catalogs for SVs remain incomplete, primarily because short reads—commonly employed in openly accessible cohorts like the 1000 Genomes Project—fail to fully resolve insertions, inversions, and SVs in repeat-rich regions (Mahmoud et al. 2019). Consequently, there is an urgent need for large-scale sequencing of human genomes using long-read technologies. Such efforts will enable public reference resources like the Genome Aggregation Database (gnomAD) (Chen et al. 2024) to include a much more comprehensive catalog of common and low-frequency SVs across diverse population ancestries.
Given the costs and required efforts for pursuing multiplatform sequencing and assembly, in the foreseeable future, the construction of such reference catalogs is likely to significantly benefit from low-to-intermediate coverage long-read sequencing across larger cohorts. Efforts are currently underway to resequence over a thousand representative samples from the 26 human populations reflected in the 1000 Genomes Project with long reads (Gustafson et al. 2024; Schloissnig et al. 2024). These open data resources are expected to enable the construction of more comprehensive imputation panels including SVs at low allele frequencies, which can benefit association studies that are currently still largely based on microarrays or short-read sequencing technology. Rare disease studies in clinical genetics, performed with relatively high coverage in an effort to maximize the diagnostic yield, are likely to significantly benefit from such expanded rare variant catalogs generated from long-read sequencing of diverse human individuals. These data will enable the filtering of variants unlikely to be responsible for a clinical phenotype, and thus ultimately facilitate genetic diagnoses. Emerging tools that leverage machine learning, such as those using language models for variant effect prediction (Ji et al. 2021; Rozowsky et al. 2023), hold promise in overcoming some of the expected downstream challenges with interpreting variant data from long-read sequencing studies.
Methylation as a parallel readout for assessing genetic variant consequences
Another exciting advance in long-read sequencing is its ability to simultaneously detect genetic variants and DNA methylation patterns, which represent uniquely identifiable base signatures through single-molecule long-read sequencing (Lucas and Novoa 2023). DNA methylation is a key epigenetic modification of the genome that can provide insights into the functional consequences of genetic variants. A recent study on direct haplotype-resolved 5-base HiFi sequencing, which profiled hypermethylation outliers in a rare disease cohort, showcases the potential of long-read sequencing in linking epigenetic modifications to genetic variants (Cheung et al. 2023). DNA methylation patterns captured by long-read sequencing can help reveal the parent of origin of genetic variation extending to chromosome-length haplotypes, as well as detect parent-specific methylation patterns to help resolve genomic imprinting and disease mechanisms (Akbari et al. 2023).
Patterns of DNA methylation also enable biological discovery: For example, the direct detection of episignatures (Barbosa et al. 2018)—defined as aberrant, disease-specific genome-wide DNA methylation changes—through long-read sequencing provides a promising approach to diagnosing a subset of developmental disorders, assessing the pathogenicity of variants of uncertain significance, and understanding disease mechanisms. With long-read sequencing, such episignatures can be identified without the need for separate methylation assays (Geysens et al. 2025), integrating genetic and epigenetic insights into a single streamlined workflow. As another example, in studies of human centromeres (Altemose et al. 2022), it has become apparent that the kinetochore attachment site during cell division is marked by localized changes in methylation, termed the centromere dip region (Gershman et al. 2022; Nurk et al. 2022). Differences in centromere structure and higher-order repeat array composition are occasionally reflected in different attachment sites for the kinetochore (Logsdon et al. 2024a). This suggests that long-read sequencing platforms hold future potential for unraveling the relationships between centromere structure and genomic evolution, as well as the potential impact of centromere architecture on chromosomal instability.
In the future, the integration of methylation data with genetic variation is likely to show value both in population-scale germline as well as somatic genetic variation studies. However, computational approaches for calling and comparing epigenetic alterations of the DNA are under continuous development, and the rebasecalling of existing data sets to include methylation information is labor intensive. Clearly, the development of robust, streamlined methods enhancing epigenomic analyses by long-read sequencing will be crucial as the field moves forward. Ultimately, DNA methylation reference data sets, generated through long-read sequencing across diverse human tissues, will be crucial for the effective interpretation and application of long-read-based epigenetic signals in disease diagnostics.
Current technology and infrastructure challenges
While major recent advances in long-read sequencing technologies bring exciting opportunities, they also reinforce the need to solve computational bottlenecks. The analysis of long-read data requires significant computational resources, particularly for large cohorts and for de novo assembly. This demands continuous development of algorithms in compression, assembly, (graph) alignment, variant calling, phasing, genotyping, and variant interpretation from long-read sequencing.
Presently, there is still a pressing need for more efficient algorithms that can scale with the increasing size of long-read data sets. This particularly holds true for approaches that use pangenome graphs for SV analyses (Liao et al. 2023; Schloissnig et al. 2024), which will need to be developed further to ensure their effective applicability in disease genetics communities. Specific methodological advances that will be required include improving the alignment of long reads to pangenome graphs to facilitate variant calling in a graph-aware manner, including in regions laden with complex repeat sequences (Fig. 3). Being able to work with long reads relative to a pangenome reference promises to be particularly rewarding as long reads have the ability to resolve highly polymorphic regions which may be better represented in pangenomes than in linear references. At the same time, pangenome analyses come with considerable computational challenges, with certain basic operations that are critical for the widespread use of human genomic references, such as liftover in structurally polymorphic loci, currently incompletely solved. Addressing these methodological developments will be critical for the field moving forward.
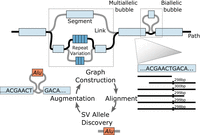
Pangenome graphs require methodological developments for scalable graph construction, accurate long-read alignments, graph-based variant discovery, and flexible graph augmentation. The upper graph shows a small portion of a pangenome graph with segments representing nucleotide sequences and links delineating possible paths through the graph (one possible haplotype path shown in black). Biallelic bubbles have two possible paths, while multiallelic bubbles with more than two paths pose significant graph construction challenges, especially for highly polymorphic and multiallelic variable number of tandem repeats (Li et al. 2020). Population-scale long-read sequencing efforts enable iterative cycles of alignments to a pangenome graph to facilitate genetic variant discovery (exemplified by a new Alu element insertion) with subsequent graph augmentation using new alleles.
The pace of technological advancement in long-read sequencing platforms and algorithmic development benefits genomics research, while at the same time representing a challenge for the translation of these sequencing technologies to clinical applications. As error rates decrease due to the application of more accurate basecalling algorithms using advanced machine learning, we foresee increased interest in rebasecalling older data sets in the future, which will come with additional data science infrastructure challenges. Projects spanning multiple years such as the Human Pangenome Reference Consortium (HPRC) and the Human Genome Structural Variation Consortium (HGSVC) will face the challenge of maintaining consistency in data interpretation as sequencing technologies evolve.
As long-read sequencing scales up, the sheer volume of raw data generated will present significant challenges for storage and archival. Ensuring that these large data sets remain accessible under the FAIR principles of findability, accessibility, interoperability, and reusability is a major hurdle for the field. Provision of the data from these projects, including raw data, on widely accessible, public compute clouds will be paramount to maximize their long-term utility.
Moving forward, the utilization of long-read sequencing technologies in disease studies will benefit from continuous improvements in sample preparation protocols. Beyond fresh-frozen samples, improved protocols for subjecting formalin-fixed paraffin-embedded (FFPE) clinical samples to long-read sequencing could unlock a treasure trove of historical clinically relevant specimens. This could enable joint analyses of genome and epigenetic changes in disease tissues (Afflerbach et al. 2024), and facilitate retrospective studies on disease biology, prolonged treatment outcomes, and temporal patterns of disease progression.
Conclusions and future perspectives
The future of long-read sequencing in population-scale studies is bright. As costs continue to decrease and computational tools improve, long-read sequencing will become a standard tool in large-scale genomics research in the future. The ability to phase entire haplotypes, detect previously hidden SVs, and integrate DNA methylation signals will revolutionize our understanding of the human genome and its role in health and disease.
Trade-offs between coverage, read length, and cohort size are critical considerations in study design. Furthermore, ongoing development in computational methods operating in the space of genome graphs will be crucial to help advance the field, and fully leverage the benefits that pangenome references can provide. In the future, the integration of long-read sequencing into GWAS will offer ample opportunities for enhancing efforts to decipher the genetic basis of common diseases. Performing GWAS-scale long-read sequencing would capture a more complete spectrum of rare and common genetic variation, helping to uncover risk factors for diseases such as cancer, diabetes, cardiovascular disease, and neurodegenerative disorders as well as their interaction with environmental variables.
In conclusion, long-read sequencing has already made significant contributions to the field of genomics, particularly in the areas of SV characterization, de novo assembly, and pangenome construction. As the technology continues to evolve, its potential to uncover novel genetic and epigenetic variation in population-scale and cancer genomic studies will further grow. The challenges of cost, data management, and computational bottlenecks are real but surmountable, and the future of long-read sequencing holds the promise of a more complete understanding of human genetic diversity and phenotypes.
Competing interest statement
The authors declare no competing interests.
Footnotes
-
Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280120.124.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








