
Transcriptomic and proteomic effects of gene deletion are not evolutionarily conserved
- Yang Li and
- Jianzhi Zhang
Abstract
Although the textbook definition of gene function is the effect for which the gene was selected and/or by which it is maintained, gene function is commonly inferred from the phenotypic effects of deleting the gene. Because some of the deletion effects are byproducts of other effects, they may not reflect the gene's selected-effect function. To evaluate the degree to which the phenotypic effects of gene deletion inform gene function, we compare the transcriptomic and proteomic effects of systematic gene deletions in budding yeast (Saccharomyces cerevisiae) with those effects in fission yeast (Schizosaccharomyces pombe). Despite evidence for functional conservation of orthologous genes, their deletions result in no more sharing of transcriptomic or proteomic effects than that from deleting nonorthologous genes. Because the wild-type mRNA and protein levels of orthologous genes are significantly correlated between the two yeasts and because transcriptomic effects of deleting the same gene strongly overlap between studies in the same S. cerevisiae strain by different laboratories, our observation cannot be explained by rapid evolution or large measurement error of gene expression. Analysis of transcriptomic and proteomic effects of gene deletions in multiple S. cerevisiae strains by the same laboratory reveals a high sensitivity of these effects to the genetic background, explaining why these effects are not evolutionarily conserved. Together, our results suggest that most transcriptomic and proteomic effects of gene deletion do not inform selected-effect function. This finding has important implications for assessing and/or understanding gene function, pleiotropy, and biological complexity.
The current gold standard of inferring the function of a gene requires measuring the phenotypic effect of deleting the gene or mutating the gene in such a way that its function is completely abolished (Alberts et al. 2002). For an essential gene, one may lower its expression or activity to a nonzero level instead of deleting it. The function of the gene is then inferred to be preventing the occurrence of the mutant phenotype from the wild-type phenotype. For example, if deleting a mouse gene causes deafness, the gene is said to prevent deafness or to function in hearing. Although phenotypes at the organismal level are traditionally the focus in inferring gene function (Liao et al. 2010), recent years have seen a surge in the analysis of cellular and molecular phenotypes (Camp et al. 2019) such as the mRNA and protein concentrations of all genes in a genome, thanks to the rapid progress in high-throughput molecular phenotyping of mutants such as Perturb-seq and variants (Adamson et al. 2016; Dixit et al. 2016; Jaitin et al. 2016; Datlinger et al. 2017). It is commonly thought that studying the molecular phenotypic effects of gene deletion helps understand the molecular basis of gene function (Camp et al. 2019).
Does the phenotypic effect of deleting a gene faithfully inform the function of the gene? According to the definition of the “proper biological function” or “selected-effect function,” the function of a gene is the effect for which the gene was selected and/or by which it is maintained (Doolittle et al. 2014; Graur et al. 2016). Hence, it is possible that some phenotypic effects of gene deletion are byproducts of other effects (He 2016) and therefore do not reflect the selected-effect function of the gene. Specifically, cell physiology is often grossly disturbed upon gene deletion; as a result, many phenotypic responses may occur that have nothing to do with what the deleted gene normally does in the wild type. Consider, for example (He 2016), gene A whose product, protein A, binds to protein B, which induces downstream phenotypic effects that are referred to as set I. Let us assume that because of the tight binding between A and B, B is unavailable for binding to protein C. Upon the deletion of A, B becomes available for binding to C, which induces a different set of downstream phenotypic effects, referred to as set II. It is possible that the function of A is simply to bind to B to induce I and that the prevention of the B–C binding that induces II is a byproduct of the A–B binding rather than another function of A. Under the above scenario, although deleting A causes the loss of I and gain of II, only the induction of I, but not the prevention of II, should be considered A’s function, because A is likely subject to natural selection for inducing I but not for preventing II (regardless of whether II has a fitness effect). Therefore, under the assumption that A’s function is evolutionarily conserved and when the effects of deleting A are examined in multiple species, the loss of I but not the gain of II is expected to be conserved (He 2016). Indeed, deleting the transcription factor gene HAP4 in the budding yeast Saccharomyces cerevisiae (Sce) results in mRNA expression changes of 65 genes concentrated in related Gene Ontology (GO) terms as well as those of 130 genes distributed rather randomly across diverse biological processes; the former expression changes upon HAP4 deletion tend to be evolutionarily conserved in related species, whereas the latter tend not to be conserved (Liu et al. 2020). These and other observations support the hypothesis that many phenotypic effects of deleting HAP4 do not inform the function of HAP4 (Liu et al. 2020). It is, however, unknown whether HAP4 is representative of yeast genes.
To address the above question, we compare the transcriptomic and proteomic effects of systematic gene deletions in Sce with those effects in the fission yeast Schizosaccharomyces pombe (Spo). Although Sce (Duina et al. 2014) and Spo (Vyas et al. 2021) were separated from each other ∼500 mya (Kumar et al. 2017), they are both unicellular, free-living fungi belonging to the phylum Ascomycota. Although gene essentiality can vary across strains and species (Liao and Zhang 2008; Dowell et al. 2010; Koo et al. 2017; Rousset et al. 2021; Chen et al. 2022), systematic gene deletions showed that 83% of orthologous genes between the two fungi share gene essentiality (Kim et al. 2010). In fact, in 47% of cases examined, Sce cells lacking an essential gene could be rescued by the expression of its human ortholog (Kachroo et al. 2015), indicating that about one-half of orthologous genes more or less share their functions even between human and Sce, which diverged from each other ∼1300 mya (Kumar et al. 2017) and belong to different kingdoms. It is thus safe to assume that the functions of orthologous genes are largely conserved between Sce and Spo (Gabaldón and Koonin 2013). Hence, the transcriptomic and proteomic effects of deleting a Sce gene and those of deleting its Spo ortholog are expected to be similar if most of such effects reflect gene functions.
Results
Transcriptomic and proteomic effects of gene deletion are not conserved
Using publicly available transcriptomic and proteomic data of wild-type and (nonessential) gene deletion strains (Supplemental Table S1), we systematically compared the effects of gene deletion between Sce and Spo. The transcriptomic data were, respectively, generated by microarray and RNA-seq in Sce (Kemmeren et al. 2014) and Spo (Öztürk et al. 2022) and were available from deletions of 41 one-to-one orthologous genes (Fig. 1, top left). These data included estimated mRNA concentrations of 4142 orthologous genes of the two species (Fig. 1, top right) The proteomic data were generated by mass spectrometry in Sce (Messner et al. 2023) and Spo (Öztürk et al. 2022) and were available from deletions of 2242 one-to-one orthologous genes in the two yeasts (Fig. 1, bottom left). These data contained estimated protein concentrations of 1478 orthologous genes of the two species (Fig. 1, bottom right). In the following, only the set of 4142 orthologs and the set of 1478 orthologs were considered in the analyses of transcriptomic and proteomic effects, respectively.
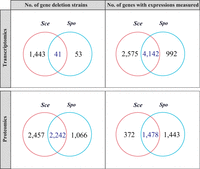
Comparing transcriptomic and proteomic effects of gene deletions in the budding yeast Saccharomyces cerevisiae (Sce) with those in the fission yeast Schizosaccharomyces pombe (Spo). On the left side, each circle represents genes deleted in each species, whereas the intersection represents orthologous genes deleted in both species. On the right side, each circle represents genes with expressions measured in gene deletion strains of each species, whereas the intersection represents orthologous genes with expressions measured in the deletion strains of both species. Gene numbers are indicated for overlapping and nonoverlapping areas of various circles.
The original studies that generated these data provided the adjusted P-value that the mRNA (or protein) concentration of each gene has changed in a gene deletion strain compared with that in the
wild type. However, because of methodological differences and sample size differences between the Sce and Spo studies, the number of genes with significant expression changes differed widely between the two yeasts, making the inter-specific
comparison potentially inappropriate (Supplemental Fig. S1). We thus performed the following adjustment. Let NSce and NSpo be the numbers of genes with significant expression changes upon a gene deletion in Sce and Spo, respectively, in the original studies. We considered 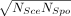 genes with the smallest adjusted P-values in each species to be significant. After this normalization, the median fraction of genes showing significant mRNA
(or protein) concentration changes upon a gene deletion is 10.4% (or 0.8%) in each species (Supplemental Fig. S1). The drastically smaller fraction of genes with protein level changes than that with mRNA level changes is at least in part
owing to the lower sensitivity of proteomic than transcriptomic measurements (Ponomarenko et al. 2023).
genes with the smallest adjusted P-values in each species to be significant. After this normalization, the median fraction of genes showing significant mRNA
(or protein) concentration changes upon a gene deletion is 10.4% (or 0.8%) in each species (Supplemental Fig. S1). The drastically smaller fraction of genes with protein level changes than that with mRNA level changes is at least in part
owing to the lower sensitivity of proteomic than transcriptomic measurements (Ponomarenko et al. 2023).
Upon deleting a gene in Sce and its ortholog in Spo, we computed the fraction of shared expressional effects between the two species (Fortho) by dividing the number of genes whose expression levels are significantly affected in both species by the number of genes whose expression levels are significantly affected in Sce. In theory, Fortho varies between 0% and 100%, with 0% indicating no sharing and 100% indicating a complete overlap of the genes whose expression levels are significantly influenced by the gene deletion between the two yeasts.
From the transcriptomic data, we found Fortho to vary from 0% to 39.5% (median = 11.1%) for the 41 orthologous gene deletions in the two yeasts (Fig. 2A; Supplemental Table S2). That the median Fortho is similar to the median fraction of genes (10.4%) showing significant mRNA concentration changes upon a gene deletion suggests that most of the sharing of transcriptomic effects between deleting orthologous genes is by chance. To rigorously assess the expectation under no evolutionary conservation of the transcriptomic effects of gene deletion, we randomly paired the 41 Sce gene deletions with the 41 Spo gene deletions and recomputed the 41 shared fractions of genes with significant mRNA effects between Sce and Spo; these fractions are referred to as Fnon-ortho. Indeed, Fortho and Fnon-ortho (median = 10.8%) are similar (P = 0.45, Mann–Whitney U test) (Fig. 2A), suggesting no evolutionary conservation of the transcriptomic effects of gene deletion between the two yeasts.
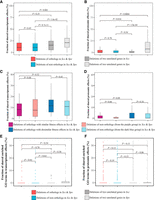
Transcriptomic and proteomic effects of gene deletions are not conserved between the two yeasts. (A,B) Fractions of shared transcriptomic (A) or proteomic (B) effects between deletions of two genes of four types: orthologous genes in the two yeasts (n = 41 pairs for transcriptomics and 2242 pairs for proteomics), corresponding nonorthologous genes in the two yeasts, randomly paired genes in the orthologous group in Sce, and randomly paired genes in the orthologous group in Spo. (C,D) Fractions of shared transcriptomic (C) or proteomic (D) effects between deletions of two genes of two types in two subsets of data: orthologous genes with similar fitness effects of deletion in the two yeasts (n = 17 pairs for transcriptomics and 1150 pairs for proteomics) and corresponding nonorthologous genes; orthologous genes with dissimilar fitness effects of deletion in the two yeasts (n = 12 pairs for transcriptomics and 426 pairs for proteomics) and corresponding nonorthologous genes. Orthologous genes included in C and D must have information of the fitness effect of deletion in both yeasts. (E,F) Fractions of shared enriched GO terms of transcriptomic (E) or proteomic (F) effects between deletions of orthologous genes, nonorthologous genes, randomly paired unrelated Sce genes, and randomly paired unrelated Spo genes. Most shared enriched GO terms are related to stress response. P-values are based on Mann–Whitney U tests. In all panels, the lower and upper edges of a box represent the first (qu1) and third (qu3) quartiles, respectively; the horizontal line inside the box indicates the median (md); and the whiskers extend to the most extreme values inside inner fences; md ± 1.5(qu3−qu1). Dots in E and F show outliers.
We also examined the shared fraction of genes exhibiting significant mRNA level changes between deletions of two unrelated Sce genes (FSce) and that between deletions of two unrelated Spo genes (FSpo). FSce (median = 15.8%) and FSpo (median = 21.2%) are both significantly greater than Fnon-ortho (Fig. 2A), suggesting species-specific transcriptomic effects of gene deletion such that deleting two unrelated genes in the same species yielded more similar effects than deleting two unrelated genes in different species.
Furthermore, FSce and FSpo both exceed Fortho, although the difference between FSce and Fortho is not statistically significant (Fig. 2A), suggesting that deleting unrelated genes in the same species yielded more similar effects than deleting orthologous genes in different species. Given the lack of evolutionary conservation and the presence of species specificity in transcriptomic effects of gene deletion, the above observation is unsurprising.
The analysis of proteomic data yielded similar results (for examples, see Supplemental Fig. S2) as those of transcriptomic data, although various F-values are substantially lower for proteomic than transcriptomic effects of gene deletion. For instance, Fortho (median = 0) for the 2242 orthologous gene deletions in the two yeasts is not significantly different from Fnon-ortho (median = 0) (Fig. 2B; Supplemental Table S2). FSce (median = 0) and FSpo (median = 9.1%) are greater than both Fnon-ortho and Fortho, with three of the four comparisons exhibiting significant differences (Fig. 2B).
To evaluate the robustness of the above results, we conducted three analyses. First, we assumed that the number of genes with significant transcriptomic (or proteomic) effects in Spo equals that reported in Sce. Second, we assumed that the number of genes with significant transcriptomic (or proteomic) effects in Sce equals that reported in Spo. Third, we simply considered the significant expression effects reported in the original studies without equalizing the numbers of significant cases between the two yeasts. In all analyses, Fortho is not significantly different from Fnon-ortho in both transcriptomic and proteomic analyses (Supplemental Fig. S3). Of course, without the equalization, FSce and FSpo are not comparable with Fortho or Fnon-ortho. Furthermore, although our main analysis (Fig. 2A,B) regarded an expression effect in Sce and that in Spo as shared without considering the directions of these effects (i.e., upregulation or downregulation), qualitatively similar results were obtained even when expression effects in the two species must be of the same direction to be considered shared (Supplemental Fig. S4).
A minority of orthologous genes are essential in Sce but nonessential in Spo or vice versa (Kim et al. 2010). This group of genes was excluded from the above analyses because the transcriptomic and proteomic data were collected from strains lacking only nonessential genes. However, even for nonessential genes, their fitness effects upon deletion could differ substantially between the two yeasts (Qian and Zhang 2014). To evaluate if our observation of a lack of evolutionary conservation of transcriptomic and proteomic effects of gene deletion is caused by such genes, we focused on a subset of nonessential genes whose fitness effects of deletion in the two yeasts differ by no more than 0.1 (referred to as orthologs with similar fitness effects). The subset of nonessential genes whose fitness effects of deletion in the two yeasts differ by more than 0.1 are referred to as orthologs with dissimilar fitness effects. We found no significant difference in Fortho between the two subsets of orthologous genes in transcriptomic (Fig. 2C) and proteomic effects (Fig. 2D). Furthermore, in both the transcriptomic (Fig. 2C) and proteomic (Fig. 2D) analyses, we found no significant difference between Fortho and Fnon-ortho for each subset.
As mentioned, although most of the transcriptomic effects of deleting HAP4 are not evolutionarily conserved, a subset of the effects in enriched GO terms appears to be conserved (Liu et al. 2020). To evaluate the generality of this finding, for each orthologous gene pair, we, respectively, identified the enriched GO terms of the transcriptomic effects in Sce and Spo using an adjusted P = 0.20 as the cutoff. We then computed the fraction of enriched GO terms shared between the two yeasts. For comparison, we computed the corresponding fraction for nonorthologs between the two yeasts. We found no significant difference between the orthologs and nonorthologs in the fraction of shared enriched GO terms (Fig. 2E). Similar results were obtained when the GO analysis was based on proteomic effects (Fig. 2F). For comparison, we also computed the fraction of enriched GO terms shared between deletions of unrelated genes in Sce or in Spo (Fig. 2E,F). We varied the cutoff in the GO enrichment analysis to an adjusted P-value of 0.05 or 0.50, but the results were not qualitatively different (Supplemental Fig. S5).
To probe the types of genes showing relatively high Fortho, we focused on the proteomic data because they include a 55-fold larger number of orthologous gene deletions than that in the transcriptomic data. Specifically, we conducted a GO analysis of the top 10% of genes in Fortho against the bottom 90% of genes. The top 10% of genes tend to be associated with catalytic activity, hydrolase activity, and UDP-forming activity (Supplemental Fig. S6; Supplemental Table S2).
The scarcity of shared transcriptomic effects of orthologous gene deletions cannot be fully explained by experimental noise or stereotypical response
A potential cause of the apparent lack of conservation of transcriptomic and proteomic effects of gene deletion between the two yeasts is transcriptome and proteome measurement errors, because these errors could cause under- or misidentification of genes with significant mRNA or protein effects upon gene deletion (i.e., false-negative or false-positive errors), which would lower Fortho. In particular, for both transcriptomics and proteomics, the Sce and Spo data were generated in different laboratories and thereby might contain laboratory-specific biases. Therefore, it would be ideal to perform a control analysis by comparing two laboratories’ data of the transcriptomic (or proteomic) effects of deleting the same gene of the same species. Below, we describe such a comparison of the transcriptomic effects of 119 gene deletions in the same Sce genetic background reported by different laboratories (Hu et al. 2007; Kemmeren et al. 2014). We found that the fraction of transcriptomic effects of deleting the same Sce gene shared between the two studies (median = 53.5%) is significantly greater than that of deleting different genes between the two studies (median = 5.9%), as well as that between deleting two different genes in each study (Fig. 3A). Although deleting the same gene in the two studies did not yield exactly the same detected transcriptomic effects, indicative of the presence of technical noise, the fraction of shared effects (Fig. 3A) is substantially greater than that between orthologous gene deletions in the two yeasts (Fig. 2A). Our analysis of enriched GO terms further supports this conclusion (Fig. 3B; Supplemental Fig. S7). Note that Reimand et al. (2010) reanalyzed Hu et al.’s data and detected substantially more transcriptomic effects than reported by the original authors. Our use of Reimand et al.’s results instead of Hu et al.’s results did not qualitatively alter our conclusion.
Comparison of the transcriptomic effects of budding yeast gene deletions detected in different studies. (A) Fraction of shared transcriptomic effects of Sce gene deletions between two microarray-based studies: Kemmeren et al. (2014) and Hu et al. (2007). (B) Fraction of shared enriched GO terms of transcriptomic effects between the above two studies. (C) Fraction of shared transcriptomic effects of four gene deletions (HAP2, HAP3, HAP4, and HAP5) between two microarray-based studies (Hu et al. 2007; Kemmeren et al. 2014). (D) Fraction of shared transcriptomic effects of the same four gene deletions between a microarray study (Kemmeren et al. 2014) and an RNA-seq study (Liu et al. 2020). (C,D) Each dot represents one comparison. In each panel, the lower and upper edges of a box represent the first (qu1) and third (qu3) quartiles, respectively; the horizontal line inside the box indicates the median (md); and the whiskers extend to the most extreme values inside inner fences; md ± 1.5(qu3−qu1). P-values are based on Mann–Whitney U tests.
However, when juxtaposed with our main analysis (Fig. 2A), the above control (Fig. 3A) has a difference. Specifically, although the main analysis compared two studies that, respectively, used microarray and RNA-seq to detect transcriptomic effects of gene deletion, the control compared two studies that each employed the microarray technology. Although RNA-seq is superior to the microarray for comparing expression levels of different genes (Xiong et al. 2010), the two technologies are both reliable and commonly used for comparing the expression levels of the same gene between samples, which is what is needed for detecting transcriptomic effects of gene deletion. Notwithstanding, we further verified our control by examining four Sce gene deletion strains in Figure 3A that had also been transcriptomically profiled by RNA-seq (Liu et al. 2020). We first compared the transcriptomic effects of these four Sce gene deletions respectively assessed by two laboratories both using microarray (Fig. 3C; Hu et al. 2007; Kemmeren et al. 2014), and then compared these effects assessed by two laboratories respectively using microarray (Kemmeren et al. 2014) and RNA-seq (Fig. 3D; Liu et al. 2020). These two comparisons yielded highly similar results: The fraction of shared transcriptomic effects between two studies is much greater for the same gene than for different genes, regardless of whether the two studies used the same transcriptomic method. The observation in Figure 3D that the transcriptomic effects of the same gene deletion overlap by ∼50% even when the data were generated by different technologies in different laboratories suggests that the low Fortho of ∼10% in Figure 2A is not owing to the use of different technologies but the use of different species. In other words, the lack of evolutionary conservation in transcriptomic effects of gene deletion cannot be explained by transcriptome measurement errors/biases.
A prior analysis of Sce gene deletions identified stereotypical transcriptomic effects that do not reflect the specific function of the gene deleted but the fitness effect of the deletion (O'Duibhir et al. 2014), meaning that Fortho in Figure 2, A and B, might have been underestimated for orthologous gene deletions with dissimilar fitness effects in the two yeasts. Nevertheless, our analysis of the subset of orthologous gene deletions with similar fitness effects in the two yeasts still found Fortho to be similar to Fnon-ortho (Fig. 2C,D). Furthermore, even after the removal of the stereotypical effects previously identified (i.e., genes with stereotypical transcriptomic changes and their Spo orthologs) (Kovács et al. 2021), Fortho is not significantly different from Fnon-ortho (Supplemental Fig. S8). Together, these results indicate that the observation of a lack of conservation of transcriptomic/proteomic effects of gene deletion is not explainable by stereotypical effects of gene deletions.
Transcriptomic/proteomic effects of gene deletion are sensitive to the genetic background
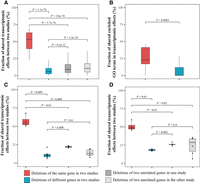
Another potential cause of the apparent lack of evolutionary conservation of transcriptomic and proteomic effects of gene deletion is that most transcriptomic and proteomic effects of deleting a gene do not inform the function of the gene, a hypothesis mentioned in the Introduction. Consistent with this hypothesis is the report that transcriptomic and proteomic effects of metabolic gene deletions in Sce are highly sensitive to the genetic background (Alam et al. 2016). Specifically, Alam et al. (2016) measured the transcriptomic and proteomic effects of deleting HIS3 in the eight genetic backgrounds in which zero to all of LEU2, URA3, and MET17 (also known as MET15) are absent. Note that whenever a metabolic gene (e.g., LEU2) is absent in the genome, the corresponding nutrient (e.g., leucine) is supplied to the medium such that the absence of the gene is not lethal (Alam et al. 2016). We found that the shared fraction of transcriptomic effects of HIS3 deletion between two genetic backgrounds varies from 12% to 46.8% (median = 25.5%) (Fig. 4A; Supplemental Table S3). Similarly, the shared fraction of proteomic effects of HIS3 deletion between two genetic backgrounds varies from 0% to 34.8%, with a median of 15.0% (Fig. 4B; Supplemental Table S3). That is, deleting the same gene in different genetic backgrounds causes largely nonoverlapping transcriptomic/proteomic effects, even though these backgrounds differ by no more than the presence/absence of three genes whose metabolic functions are compensated by the additives in the media. Furthermore, the fraction of transcriptomic/proteomic effects shared does not decrease significantly with the dissimilarity in genetic background (P > 0.30 and P > 0.078 for each comparison between all dots of one color with those of another color in each of the first four bars; Mann–Whitney U test) (Fig. 4A,B, respectively), suggesting the possibility that even a slight difference in genetic background drastically lowers the fraction of transcriptomic/proteomic effects shared. For comparison, we computed the fraction of transcriptomic/proteomic effects shared between deleting two different genes such as HIS3 and LEU2 in the same wild-type background. Regarding transcriptomic effects, this fraction varies from 12.9% to 35.6% for different gene pairs, with a median of 29.3% (Fig. 4A). Regarding proteomic effects, this fraction varies from 0% to 28.3%, with a median of 8.7% (Fig. 4B). Thus, the difference in transcriptomic/proteomic effects of gene deletion between different genetic backgrounds is as large as that between deleting different genes from the same genetic background. Because the genetic backgrounds of Sce and Spo differ by orders of magnitude more than three genes, it is unsurprising that the transcriptomic and proteomic effects of gene deletion are not conserved between the two yeasts.
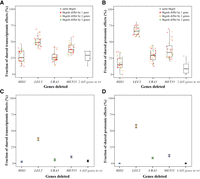
Transcriptomic and proteomic effects of gene deletions in budding yeast are sensitive to the genetic background. (A,B) Fractions of shared transcriptomic (A) or proteomic (B) effects of deleting a gene in two genetic backgrounds or deleting two different genes in the same background. Specifically, each of four focal genes (HIS3, LEU2, URA3, and MET15) is deleted in eight genetic backgrounds that vary in the presence/absence of the other three genes. For example, the focal gene HIS3 is deleted in the eight strains lacking zero to three of LEU2, URA3, and MET15. Shown for comparison are fractions of shared transcriptomic/proteomic effects of deleting two different genes in the wild-type background. In the first four bars, each dot represents a comparison between two genetic backgrounds, in which different colors indicate that the two genetic backgrounds differ by the presence/absence of one (red), two (yellow), or three (green) genes. In the last bar, each dot represents a comparison between two gene deletions in the wild type. (C,D) Fractions of shared transcriptomic (C) or proteomic (D) effects of deleting a gene between four genetic backgrounds (the wild type plus three genetic backgrounds, each lacking one of the other three genes). Shown for comparison are fractions of shared transcriptomic/proteomic effects of deleting the four different genes in the wild type. Error bars show binomial sampling-based 95% confidence intervals. In all panels, the lower and upper edges of a box represent the first (qu1) and third (qu3) quartiles, respectively; the horizontal line inside the box indicates the median (md); and the whiskers extend to the most extreme values inside inner fences; md ± 1.5(qu3−qu1).
It is plausible that the transcriptomic effects of gene deletion shared across multiple genetic backgrounds reflect the true biological function of the gene. We thus compared the shared fraction of transcriptomic effects of deleting the same metabolic gene (e.g., HIS3) in four different genetic backgrounds (e.g., wild type, ΔLEU2, ΔURA3, and ΔMET15) with a negative control, which is the fraction of effects shared among the deletions of the four different metabolic genes (HIS3, LEU2, URA3, and MET15) in the same wild-type background (Fig. 4C). For HIS3 (2.5%), URA3 (5.5%), and MET15 (9.9%), the shared fraction of transcriptomic effects of gene deletion is similar to the negative control (3.7%), suggesting the possibility that few if any of the shared transcriptomic effects reflect the functions of these genes. In contrast, the shared fraction of transcriptomic effects of deleting LEU2 (37.2%) is greater than the negative control by an order of magnitude, suggesting that the sizable fraction of the shared transcriptomic effects reflects the true function of LEU2. Similar inferences can be made based on the analysis of proteomic effects (Fig. 4D).
Discussion
Although multiple lines of evidence support that functions of orthologous genes are generally conserved between budding yeast and fission yeast, we observed no evolutionary conservation in the transcriptomic and proteomic effects of gene deletion between these species. A similar conclusion can be drawn even for the enriched GO terms of the transcriptomic/proteomic effects. Further analysis excluded poor transcriptomic measurements and stereotypical effects of gene deletion as main causes of this apparent lack of conservation. Rather, our results are consistent with the hypothesis that most transcriptomic/proteomic effects of gene deletion do not reflect selected-effect function and thereby are not evolutionarily conserved. At least in theory, gene regulation can evolve rapidly even when gene function is conserved. Because changes in transcriptomic/proteomic effects should largely reflect changes in gene regulation, rapid changes in transcriptomic/proteomic effects in evolution are not incompatible with the conservation of gene function. In other words, our finding is not surprising mechanistically. The nonfunctional transcriptomic/proteomic effects are presumably idiosyncratic, depending on the presence/absence of countless other compounds and interactions in the cell. In theory, even a small change in the genetic background could alter these compounds and interactions, triggering a different set of transcriptomic/proteomic effects of gene deletion. Thus, the finding that transcriptomic/proteomic effects of gene deletion are highly sensitive to the genetic background supports our conclusion that most transcriptomic/proteomic effects of gene deletion are nonfunctional. Our finding and interpretation are further supported by the recent report that the transcriptomic effects of deleting transcription factor genes are largely nonoverlapping between two Sce strains (Liu et al. 2024).
There are several caveats in our analyses that warrant discussion. First, our comparison of the transcriptomic effects of gene deletion between Sce and Spo (Fig. 2A) was based on only 41 orthologous gene deletions. Nevertheless, this small sample is compensated by the large number of orthologous gene deletions (2242) studied in proteomic comparisons (Fig. 2B). Furthermore, the similar trends revealed in Figure 2, A and B, suggest that the transcriptomic results are reliable despite the relatively small number of gene deletions examined.
Second, we were able to perform a control analysis to exclude the possibility that transcriptomic measurement error causes our main finding. Such a control was unavailable for our proteomic analysis owing to the lack of suitable data. However, the similarity between our transcriptome- and proteome-based results suggests that our proteomic results are unlikely to be primarily caused by poor data quality, but this inference should be verified in the future.
Third, the phenotypic effects of deleting a gene can vary with the environment in which the phenotype is measured. Although the media used for collecting the transcriptomic and proteomic data in Sce and Spo were not identical, they were similar (see Methods). Evidence from mRNA and protein expression levels, fitness effects of gene deletions, and a control experiment performed in different media all suggests that the apparent lack of conservation of transcriptomic/proteomic effects of gene deletion cannot be explained by the slight difference in the media used for phenotyping Sce and Spo (see Methods).
Fourth, the two yeast species compared here are separated by hundreds of millions of years of independent evolution, and their orthologous genes cannot all be functionally identical. Consequently, some phenotypic effects of gene deletion are expected to differ between the two yeasts even if all phenotypic effects genuinely reflect gene function. This said, the virtually equal degrees of sharing of transcriptomic/proteomic effects between deleting orthologous genes and deleting nonorthologous genes in the two yeasts are unexplainable if most of these effects reflect gene function.
Our conclusion has multiple implications. First, it suggests that surveying the transcriptomic/proteomic effects of gene deletion yields many nonfunctional effects. Because high-throughput approaches such as Perturb-seq and its variants almost exclusively measure the transcriptomic effects of gene deletion or mutation (Adamson et al. 2016; Dixit et al. 2016; Jaitin et al. 2016; Datlinger et al. 2017), care should be taken in interpreting the results. That is, a transcriptomic/proteomic effect should not be automatically regarded as a function. Our results further imply that transcriptomic/proteomic effects of gene deletion in model organisms are frequently different from those in humans, which should be verified in the future. Of course, we are not saying that transcriptomic/proteomic effects inform nothing about gene function. For example, some genes encoding members of the same protein complex or participating in the same biological pathway show similar transcriptomic effects when deleted, which may genuinely reflect these genes’ functions (Kemmeren et al. 2014).
Second, because of the unavailability of systematically measured nonmolecular phenotypic effects of gene deletion in multiple species, we do not know whether our finding applies to nonmolecular phenotypes such as morphological and physiological traits observable at the organismal level. Previous empirical results (Ho et al. 2017) and theoretical considerations (Zhang 2018) suggested the possibility that, when phenotypic traits are stratified according to a hierarchy of biological organization, the proportion of phenotypic evolutionary changes that are adaptive rises with the phenotypic level considered (i.e., from molecular to organismal). In other words, it is plausible that nonfunctional effects of gene deletion are more common at the level of molecular traits than at the level of organismal traits. This said, empirical studies are necessary to verify this prediction.
Third, some important quantities in genetics are measured by phenotypic effects of gene deletion or mutation. For example, the pleiotropic level of a gene or mutation is commonly estimated by the number or fraction of phenotypic traits influenced by gene deletion or mutation (Zhang 2023). One wonders whether nonfunctional phenotypic effects should be counted toward pleiotropy. If the answer is yes, one consequence is that the pleiotropy of a gene or mutation would be highly sensitive to the genetic background and would not be evolutionarily conserved. Related to the concept of pleiotropy is the notion that most traits are each influenced by many genes/mutations (Ho and Zhang 2014). In the example provided in the Introduction, if the selective maintenance of gene A is solely for its protein product to bind to B to induce phenotype set I, should A be regarded as a gene underlying phenotype set II (simply because II is affected when A is deleted)? If the relationship between A and II is not functional, is not selected, is not conserved, and is highly dependent on the genetic background, would knowing this relationship help or hinder our understanding of biology? Reasonable people may have different answers to these questions, but they are questions worth pondering.
Fourth, one could extend the thread of thought from one gene to all genes in a genome and ask whether our understanding of biological complexity, which depends on the concepts of pleiotropy and complex traits, has been misled by the numerous nonfunctional effects of gene deletion that have been discovered (He 2016). Should we consider only functional effects when studying biological complexity, and would biology be simpler if only functional effects are considered?
Fifth, deleting the same gene in budding and fission yeasts yields a smaller fraction of shared transcriptomic/proteomic effects than deleting different genes in the same yeast (Fig. 2A,B). This unexpected result suggests that budding and fission yeasts have evolved general, species-specific responses to gene deletion as a result of their high evolutionary divergence. These general responses are likely relevant to the species of interest but not to the gene of focus (O'Duibhir et al. 2014; Kovács et al. 2021). In other words, they probably do not inform gene function.
Because functional effects are selected, they can presumably be detected by examining the shared effects of orthologous gene deletions in multiple species, under the assumption that the gene function is conserved. The evolutionarily conserved effects likely reflect selected-effect functions. One caveat of our study is that we compared two distantly related species: budding and fission yeasts. If comparable data from additional, more closely related species are available, we could quantify the evolutionary rate of transcriptomic/proteomic effects of gene deletion and estimate the fraction of transcriptomic/proteomic effects that are functional. The results in Figure 4, C and D, although based on only four genes, suggest the possibility that this rate is high and that the functional fraction is small. These predictions, strongly supported by the low overlap between two Sce strains in transcriptomic effects of transcription factor gene deletions (Liu et al. 2024), would be important to verify in other taxa when relevant data become available in the future.
Methods
Orthologous genes between Sce and Spo
Orthologous genes between Sce and Spo were downloaded on October 19, 2023, from PomBase (Lock et al. 2018; https://www.pombase.org/data/orthologs/pombe-cerevisiae-orthologs-one-line-per-gene.tsv). Only one-to-one orthologs were subsequently considered.
Transcriptomic and proteomic effects of gene deletion
Transcriptomic effects of gene deletion in Sce were determined from four replicates per mutant based on microarray expression measures of cells cultured in synthetic complete (SC) medium (Kemmeren et al. 2014). Proteomic effects of gene deletion in Sce were estimated by mass spectrometry in four replicates of cells grown in synthetic minimal (SM) medium (Messner et al. 2023). Transcriptomic effects of gene deletion in Spo were determined by RNA-seq using 388 wild-type replicates and one replicate per mutant grown in yeast extract with supplement (YES) medium (Öztürk et al. 2022). Proteomic effects of gene deletion in Spo were determined by mass spectrometry using 461 wild-type replicates and one replicate per mutant in YES medium (Öztürk et al. 2022). Although the media used in Sce and Spo experiments are not identical, they are similar. Furthermore, mRNA (Supplemental Fig. S9A) and protein (Supplemental Fig. S9B) expression levels are significantly correlated between orthologous genes of the two yeasts even when they are cultured in different media. Specifically, in Supplemental Figure S9A, the RNA-seq data of Sce were generated in the rich medium YPD (Chou et al. 2017), similar to SC, whereas those of Spo were from YES (Öztürk et al. 2022). In Supplemental Figure S9B, the proteomic data of Sce (Messner et al. 2023) and Spo (Öztürk et al. 2022) were from the studies mentioned above. Additionally, growth assays of Sce gene deletion strains in three media (SC, SM, and YPD) (Messner et al. 2023) revealed a strong correlation between the relative growth rate of a strain in one medium with that in another (pairwise r = 0.70 between SC and YPD, 0.74 between SM and YPD, and 0.88 between SC and SM, respectively; P < 2.2 × 10−16 in all cases). Additional control experiments are described in the section Comparing the Effects of Gene Deletion on Microarray-Based mRNA Levels between Two Sce Studies.
Significant transcriptomic and proteomic effects
The original studies provided the P-value (upon corrections for multiple testing) that the mRNA (or protein) concentration of a gene differs between a gene deletion
strain and the wild-type strain. However, because of methodological differences and sample size differences between studies,
the number of genes exhibiting significant expression effects from gene deletion differed between the two yeasts, making the
inter-specific comparison potentially inappropriate. We thus made the following adjustment. Let NSce and NSpo be the numbers of genes with significant effects in Sce and Spo in the original studies, respectively. We, respectively, considered 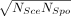 genes with the smallest P-values in each species to be significant in subsequent analyses. For instance, if NSce = 40 and NSpo = 10, we consider
genes with the smallest P-values in each species to be significant in subsequent analyses. For instance, if NSce = 40 and NSpo = 10, we consider 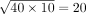 genes with the smallest P-values in Sce to be significant in Sce; we similarly consider the 20 genes with the smallest P-values in Spo to be significant in Spo. When one, but not both, of NSce and NSpo equals zero, we treated zero as one in applying the above formula. When both NSce and NSpo equal zero, the number of significant effects is zero in both species, and the fraction of effects shared between species
is also considered to be zero (only one orthologous gene pair belongs to this category, and excluding this case does not alter
our conclusion). This adjustment was applied in all comparisons unless otherwise noted. To assess the robustness of our results,
we also, respectively, considered NSce genes with the smallest P-values in each species to be significant; respectively, considered NSpo genes with the smallest P-values in each species to be significant; or, respectively, considered NSce genes in Sce and NSpo genes in Spo with the smallest P-values to be significant.
genes with the smallest P-values in Sce to be significant in Sce; we similarly consider the 20 genes with the smallest P-values in Spo to be significant in Spo. When one, but not both, of NSce and NSpo equals zero, we treated zero as one in applying the above formula. When both NSce and NSpo equal zero, the number of significant effects is zero in both species, and the fraction of effects shared between species
is also considered to be zero (only one orthologous gene pair belongs to this category, and excluding this case does not alter
our conclusion). This adjustment was applied in all comparisons unless otherwise noted. To assess the robustness of our results,
we also, respectively, considered NSce genes with the smallest P-values in each species to be significant; respectively, considered NSpo genes with the smallest P-values in each species to be significant; or, respectively, considered NSce genes in Sce and NSpo genes in Spo with the smallest P-values to be significant.
Fitness effects of gene deletions
For each orthologous gene pair between Sce and Spo, we computed Δf = ∣fSce − fSpo∣, where fSce and fSpo are the fitness of the gene deletion strain relative to that of the wild type in Sce (Costanzo et al. 2010) and Spo (Han et al. 2010), respectively. These fitness values were acquired from a previous study (Qian and Zhang 2014), which showed that these fitness values of Sce and Spo gene deletion strains are comparable. An orthologous gene pair is considered to have similar fitness effects of gene deletion in the two yeasts when Δf < 0.1; otherwise, it is considered to have dissimilar fitness effects.
GO analysis
For Supplemental Figure S6, we used GO Term Finder (Boyle et al. 2004), a tool available in the Saccharomyces Genome Database (Cherry et al. 1998; https://www.yeastgenome.org/goTermFinder) to identify enriched GO terms in the set of genes against other genes. This tool calculates the adjusted P-value upon corrections for multiple comparisons to assess the statistical significance of enrichment of a term.
GO term enrichment analysis
Biological process GO term enrichment was assessed using the clusterProfiler R package (Yu et al. 2012). We applied three different cutoffs (adjusted P-value = 0.05, 0.20, and 0.50) to identify enriched GO terms for a set of transcriptomic or proteomic effects and then compared enriched GO terms between orthologs or between nonorthologs.
Transcriptomic and proteomic effects of metabolic gene deletions in different Sce genetic backgrounds
According to the original study (Alam et al. 2016), 16 yeast strains were constructed with combinatorial auxotrophies in histidine (his3Δ), leucine (leu2Δ), uracil (ura3Δ),
and methionine (met15Δ) biosynthesis pathways. Strains were cultured in SC medium supplemented with nutrients corresponding
to their auxotrophies. In analyzing RNA-seq data, we excluded genes with very low read counts (fewer than 50 total counts
across all replicates, both raw and normalized). A total of 5923 expressed genes were considered. Normalization and P-value calculation for differential gene expression between strains were performed using DESeq2 (Love et al. 2014) in R. P-values were adjusted using the Benjamini–Hochberg method (Benjamini and Hochberg 1995), and an adjusted P < 0.05 was used as the cutoff to call differentially expressed mRNAs. Protein concentrations were provided in the original
proteomic study (Alam et al. 2016). Differential protein expression analysis was then conducted using DESeq2 (Love et al. 2014), and an adjusted P < 0.05 was used as the threshold for identifying significant protein concentration changes. Again, when comparing two sets
of transcriptomic/proteomic effects, if N1 and N2 are the numbers of genes with significant effects in the two sets, we, respectively, considered 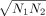 genes with the smallest adjusted P-values in each set to be significant in subsequent analyses. When comparing four sets of transcriptomic/proteomic effects,
if N1, N2, N3, and N4 are the numbers of genes with significant effects in the four sets, we, respectively, considered (N1N2N3N4)0.25 genes with the smallest adjusted P-values in each set to be significant in subsequent analyses.
genes with the smallest adjusted P-values in each set to be significant in subsequent analyses. When comparing four sets of transcriptomic/proteomic effects,
if N1, N2, N3, and N4 are the numbers of genes with significant effects in the four sets, we, respectively, considered (N1N2N3N4)0.25 genes with the smallest adjusted P-values in each set to be significant in subsequent analyses.
Comparing the effects of gene deletion on microarray-based mRNA levels between two Sce studies
To assess the reliability of using microarrays to detect the effects of gene deletion on mRNA levels, we compared the results of two independent studies that examined the transcriptomic effects of an overlapping set of 119 Sce gene deletions in the same genetic background (Hu et al. 2007; Kemmeren et al. 2014). The strains were, respectively, cultivated in SC (Kemmeren et al. 2014) and YPD (Hu et al. 2007); YPD is equivalent to YES used for collecting the Spo transcriptomic data (Öztürk et al. 2022). The original studies, respectively, identified genes with significant changes of mRNA levels. We then employed the method of normalization as shown in Supplemental Figure S1 to identify genes with significant mRNA level changes before the comparison between the two studies. The comparison between Figure 3, A and B, and Figure 2, A, C, and E, suggests that the nonoverlap in the transcriptomic effects of orthologous gene deletions between Sce and Spo cannot be explained by the slight difference in the media for culturing Sce and Spo.
Comparing expressions of orthologous genes between Sce and Spo
We compared the mRNA concentrations of orthologous genes between Sce and Spo using RNA-seq data of wild-type Sce cells grown in YPD (Chou et al. 2017) and those of wild-type Spo cells grown in YES (Öztürk et al. 2022). We similarly compared the proteomic data of wild-type Sce cells grown in SM (Messner et al. 2023) and those of wild-type Spo cells grown in YES (Öztürk et al. 2022). Gene expression levels were first loge-transformed and then standardized by subtracting the minimum observed value followed by dividing by the maximum observed value. The standardized expression levels are thus between zero and one.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Chongwei Shi for technical assistance; the Ralser, Holstege, Butter, and Du laboratories for kindly providing data and answering our inquiries about their methods and data; and D. Jiang, S. Song, M. Sun, and H. Xu for valuable discussions and comments. We thank the three anonymous reviewers for their constructive comments. This work was supported by the research grant R35GM139484 from the U.S. National Institutes of Health to J.Z.
Author contributions: J.Z. conceived the study. Y.L. and J.Z. designed the study. Y.L. analyzed the data. Y.L. and J.Z. wrote the paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280008.124.
-
Freely available online through the Genome Research Open Access option.
- Received September 7, 2024.
- Accepted February 7, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








