
Proxy panels enable privacy-aware outsourcing of genotype imputation
- 1Department of Bioinformatics and Systems Medicine, D. Bradley McWilliams School of Biomedical Informatics, University of Texas Health Science Center, Houston, Texas 77030, USA;
- 2Department of Health Data Science and Artificial Intelligence, D. Bradley McWilliams School of Biomedical Informatics, University of Texas Health Science Center, Houston, Texas 77030, USA
Abstract
One of the major challenges in genomic data sharing is protecting participants’ privacy in collaborative studies and in cases when genomic data are outsourced to perform analysis tasks, for example, genotype imputation services and federated collaborations genomic analysis. Although numerous cryptographic methods have been developed, these methods may not yet be practical for population-scale tasks in terms of computational requirements, rely on high-level expertise in security, and require each algorithm to be implemented from scratch. In this study, we focus on outsourcing of genotype imputation, a fundamental task that utilizes population-level reference panels, and develop protocols that rely on using “proxy panels” to protect genotype panels, whereas the imputation task is being outsourced at servers. The proxy panels are generated through a series of protection mechanisms such as haplotype sampling, allele hashing, and coordinate anonymization to protect the underlying sensitive panel's genetic variant coordinates, genetic maps, and chromosome-wide haplotypes. Although the resulting proxy panels are almost distinct from the sensitive panels, they are valid panels that can be used as input to imputation methods such as Beagle. We demonstrate that proxy-based imputation protects against well-known attacks with a minor decrease in imputation accuracy for variants in a wide range of allele frequencies.
Decreasing costs of DNA sequencing and genotyping brought about a massive increase in the number of personal genomes (Muir et al. 2016). Starting with the small-scale population-wide sequencing (Chen et al. 2022) efforts such as The HapMap Consortium (International HapMap Consortium 2005; Locke et al. 2006), The 1000 Genomes Project (The 1000 Genomes Project Consortium 2015), and the population-scale projects such as UK Biobank, Genomics England, Trans-omics for Precision Medicine (TOPMed) (Kowalski et al. 2019), and All of Us research program (All of Us Research Program Investigators et al. 2019), millions of personal genomes are available for analysis including underrepresented populations that are vital for increasing diversity in research (Popejoy and Fullerton 2016; Bentley et al. 2017; Matalon et al. 2023) and for the inclusion of underrepresented populations (Stark et al. 2019; Choudhury et al. 2020).
Some of the most critical uses of the large data sets are their secondary usage as reference data sets (Martyn et al. 2024), for example, to evaluate allele counts via beacon servers (Fiume et al. 2019) and variant databases (e.g., gnomAD [Karczewski et al. 2020] and dbSNP [Sherry et al. 2001]), and for building genotype imputation outsourcing services (Das et al. 2016; Sun et al. 2022). For example, NIH's TOPMed (Kowalski et al. 2019; Taliun et al. 2021) and the Haplotype Reference Consortium (HRC) (McCarthy et al. 2016) serve as reference panels for genotype imputation, which is a fundamental step in genetic analysis and a computationally resource intensive process that requires access to large protected reference panels. It is therefore performed using an outsourcing approach via “imputation servers,” for example, the Michigan Imputation Server (Loh et al. 2016). A client (query site) has a sparsely genotyped panel (e.g., genotyping arrays). The client wants to impute the genotypes for the remaining set of “untyped” variants in the reference panel, for example, TOPMed panel. Most popular imputation algorithms (Van Leeuwen et al. 2015; Browning et al. 2018) use hidden Markov models (HMMs; Li–Stephens model) for imputation. Imputation servers offer convenient services to perform imputation in which the client submits the typed variant genotypes to the server, and the server imputes the variants exclusive to the reference panel and sends the results back to the client (Sun et al. 2022).
Of specific concern to our study are the numerous privacy-related risks in this basic outsourcing protocol (Bonomi et al. 2020): The servers share the alleles and variant coordinates with the client without a tunable protection mechanism, which may pose unexplored risks. For example, the reference panels contain a very large number of rare untyped variants. The knowledge of the coordinates for these rare variants can be directly used in a beacon-type attack (Shringarpure and Bustamante 2015) to reidentify individuals, even without the knowledge of the alleles. This risk is currently not considered in many data analysis methods and data reporting policies (e.g., gnomAD, 1000 Genomes, All of US), and substantial changes may be required to how variant coordinates are reported and shared. Additionally, the client submits the typed variant genotypes in cleartext form to the imputation server, which may pose a risk to the confidentiality of these subjects, which complicates the compliance with regulations and may require further agreements (Rayner et al. 2024). These agreements only set a point of accountability, rather than protecting the data meaningfully. Because the privacy risks are multidimensional and complex (Erlich and Narayanan 2014; Erlich et al. 2014; Hubaux et al. 2017; Wan et al. 2022), for example, usage for forensic purposes and concerns about discrimination (Niemiec and Howard 2016; Pulivarti 2023), it is important to build the technological means to improve data protection and protect against unforeseen attack surfaces. As public awareness of genetic privacy is becoming more evident (Jamal et al. 2014; Sherburn et al. 2023), it is important to develop these techniques to be as transparent and easy to use as possible while considering the factors around other ethical concerns about discrimination (2010; Garrison 2013). Although GDPR and HIPAA consider genetic data as identifying information, their interpretation is not clear about protection because genetic data are very hard to irreversibly deidentify (Cohen and Mello 2018), and consent may not be sufficient for the protection of downstream and secondary tasks (Greenbaum et al. 2011).
The privacy risks related to genetic information stem from its high dimensionality and complex correlative structure (Lin et al. 2004), reidentification (Greenbaum et al. 2011; Gymrek et al. 2013; Shabani and Marelli 2019), and linking (Supplemental Methods; Harmanci and Gerstein 2016, 2018). Even at summary statistics can lead to membership inference attacks, including Homer's t-statistics (Homer et al. 2008) and Sankararaman's LRT (Sankararaman et al. 2009) and their extensions (Visscher and Hill 2009; Im et al. 2012), beacon-type data release mechanisms (Shringarpure and Bustamante 2015; Fiume et al. 2019), and knowledge of haplotype information (Jacobs et al. 2009; Bu et al. 2021). Privacy risks can also impact relatives (Telenti et al. 2014; Branum and Wolf 2015; Ayday and Humbert 2017; Thenen et al. 2018; Ayoz et al. 2021). Although privacy risks have been excessively studied, retrospective studies argued that some of these risks may be overestimated because these attacks must be applied in well-controlled scenarios and may lack formal treatment of the false-positive rates (Egeland et al. 2012) when the assumptions are not satisfied (Sampson and Zhao 2009).
In the context of imputation outsourcing, the imputation server (which are sometimes referred to as the “processors”) and the data owners/controllers (query and reference data owners) must make sure to take precautions to minimize the risks and evaluate them effectively. This process is very challenging when individual-level data sets (from both the client and the reference panel owners) are shared with the imputation servers. For instance, even the knowledge of rare variant positions can be used to identify an individual's participation in the reference panels (e.g., TOPMed Imputation Server).
The most popular genetic data-sharing model is the restricted access model, which relies on users signing agreements for each new data set. Differential privacy (DP) (Dwork 2014; Dwork and Roth 2014) and homomorphic encryption–based approaches (Gentry 2009; Kim and Lauter 2015) are the most rigorous route to share genetic data securely. Cryptographic approaches were applied to the field of genotype imputation (Kim et al. 2021; Yang et al. 2022), association studies (Orlandi 2011; Cho et al. 2018; Zhao et al. 2019; Blatt et al. 2020; Froelicher et al. 2021; Li et al. 2023b), database sequence queries (Shimizu et al. 2016), and read mapping (Popic and Batzoglou 2017; Nakagawa et al. 2022). These methods require careful reformulation of algorithms under cryptographic primitives (Dowlin et al. 2017) and require large storage, network, and maintenance costs.
Synthetic data sets can be useful for protecting privacy (Gonzales et al. 2023), in which the entities generate a representative synthetic data set using their locally sensitive data sets. This approach was used in genotype imputation (Yelmen et al. 2021; Cavinato et al. 2024) and for analysis of ancestral simulations (Wohns et al. 2022; Anderson-Trocmé et al. 2023). Although promising, privacy is not explicitly introduced into the synthetic data generation models. For example, RESHAPE (Cavinato et al. 2024) uses a sampling procedure to build a “mosaicized” panel starting from the reference data sets under the assumption that the mosaic panel protects all reference panel participants, which can be shared publicly and used as an imputation panel. However, the allele frequencies and coordinates for the variants at the rare and ultrarare categories (including singletons that leak immediate membership information) are preserved in the synthetic data. This may make the synthetic data sets vulnerable to well-known reidentification attacks.
Here, we present ProxyTyper, a framework for generating and using “proxy panels” to develop privacy-preserving genotype imputation protocols that can be used when imputation is being outsourced on an imputation server. Proxy panels are generated through a series of randomized protection mechanisms that anonymize the original sensitive panel's variants, coordinates, genetic maps, and genotypes. Compared with each other, the proxy panels and original panels are distinct in terms of statistical properties, such as allele frequencies and local haplotype frequencies. The proxy-generating mechanisms involve basic operations used in cryptographic schemes such as noise addition, random permutations and augmentations, haplotype resampling, and randomized partitioning.
Our main goal in this study is to demonstrate that proxy panels can be used for outsourcing genotype imputation without any modifications to the existing imputation methods (e.g., Beagle) (Browning et al. 2018, 2021) with only a minor decrease in imputation accuracy. We also aim to highlight the flexibility of the protocols that can be built using proxy panels, which indicates that new mechanisms can improve imputation accuracy.
Results
We first present the mechanisms used by ProxyTyper framework to build proxy panels. Next, we study the characteristics of proxy panels and reidentification attacks, and finally, we present the modified imputation outsourcing protocol and its accuracy. We focus on genotype imputation as the focus task and describe the different ways of protecting the typed and untyped variants and alleles.
Outsourcing of genotype imputation
Three entities are involved in a genotype imputation outsourcing task (Fig. 1). First is the query site (or the client), which initiates the imputation process by sending the typed variant loci to the reference site (Fig. 1). The query site is the data owner and controller for the query panel, which is composed of the typed variants (e.g., from an array platform). The reference site is the data owner/controller for the reference panel with typed and untyped variants. The imputation server provides the computational infrastructure for performing the imputation process using the proxy panels generated from query and reference panels. The imputation server implicitly separates the query and reference sites. After the reference site receives the typed variant loci, it generates and sends the randomized proxy mechanism parameters for generating the typed variant proxy panel to the query site. These parameters serve as the protection “model” parameters (akin to symmetric keys) and generate the proxy panels for the typed variants. Untyped variants are only protected by the reference site.
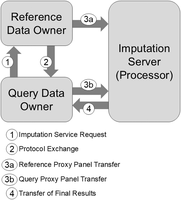
Illustration of the three entities involved in genotype imputation using proxy panel–based outsourcing. Query and reference panels are owners (or controllers) of the respective panels. Imputation server is designated as the processor of the panels. Each step of the protocol is denoted with the corresponding index: (1) The query site initiates the protocol by sending the typed variant loci to the reference site; (2) the reference site generates and sends proxy mechanism parameters; (3a,b) the reference and query sites generate proxy panels and upload to the imputation server; and (4) the imputation server sends the imputed results to the query site.
Proxy panel building
We describe the protection mechanisms used by ProxyTyper for building mosaic panels at the reference and client sites, protecting the variant coordinates and genetic maps and the shared alleles.
Mosaic haplotype generation by resampling original sensitive panels
Resampling a panel disrupts the one-to-one correspondence of subjects in the query and reference panels and reduces the risks
around linking subjects to external sources. Given a panel of haplotypes, a mosaic haplotype panel is generated by the panel's
HMM sampling. In this model, each haplotype represents a state, and transitions are performed probabilistically using modifications
of the Li–Stephens model (Li and Stephens 2003). At each variant, a new haplotype is sampled based on the recombination rates. The allele on the selected haplotype is emitted
as the sampled haplotype's allele (Fig. 2A). Mosaic panel generation uses two tunable parameters for privacy–utility tradeoff: (1) effective population size (Ne), which tunes the average number of recombinations in sampling, and (2) the maximum segment length (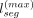 ), which limits the length of the longest continuous haplotype segment.
), which limits the length of the longest continuous haplotype segment.
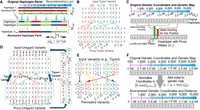
Illustration of proxy panel generation mechanisms. (A) Illustration of typed variant resampling. The original haplotypes are traced from left to right, and recombinations (indicated by switches between haplotypes) are randomly generated using the genetic map (depicted on
top). Recombinations are checked at the recombination loci shown with vertical dashed positions. The consecutive recombination loci have at least 0.001 cM genetic distance between
each other. ProxyTyper constrains the maximal segment length (lseg) to minimize chances of copying of long haplotype segments. (B) Typed variant augmentation. Given three consecutive typed variants (shown in dashed rectangles), each typed variant is copied
to a random position in the vicinity of itself. Each augmented proxy-typed variant is assigned the same genotypes of the original
variant (by default). After augmentation, the number of typed variants increased from three to six, adding three new augmented
variants. (C) Illustration of allele encoding (hashing). Given two windows (depicted with red and green windows), the allele on the proxy
haplotype is calculated as a combination of the alleles on the original haplotype. The two windows have independent encoding
functions. The function takes the alleles and the genetic distance as parameters to calculate the hashed (encoded) alleles
on the proxy haplotype (shown at the bottom). The proxy haplotype is calculated by encoding all windows. (D) Protection of untyped variants by haplotype partitioning. Two variants at positions 3 and 8 are partitioned to four new
proxy variants denoted by 3(1), 3(2), 8(1), and 8(2). The probabilities of imputed alleles for these variants are recovered from proxy variants. Denote the inversion of allele
on 8(2) (Methods) (E) Protection of untyped variants using (rolling) permutation between typed variant blocks. Untyped variant positions are randomly
shuffled between two consecutive typed variants. (F) Protection of the variant coordinates and the genetic distance. The coordinates are normalized to a preselected value lproxy, which obfuscates the typed and untyped variant positions. Genetic map is obfuscated using addition of Gaussian noise with
predefined variants 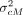 .
.
The query and reference sites perform resampling independently of each other without exchanging any input. This is advantageous because sites can perform resampling offline and store the data for future use, which is especially useful for the reference site.
Typed variant augmentation
ProxyTyper augments typed variants at random positions in the vicinity of the original variant to randomize the number of typed variants used in the imputation (Fig. 2B), which helps to flexibly conceal the exact number of typed variants and their positions. Each round of augmentation probabilistically increases the number of typed variants while placing the new variants at a nearby random position. By default, we use three recursive augmentations with a 0.99 probability of augmentation at each locus.
Protection of typed and untyped alleles
ProxyTyper uses hashing, partitioning, and permutation-based mechanisms to protect the variant alleles.
Hashing typed variant alleles
When phased panels are available on reference and query sites, ProxyTyper can use a locality-sensitive hashing of the alleles to encode the typed variant alleles (not untyped variants) (Fig. 2C). Given variant i on haplotype j, ProxyTyper replaces the original (sensitive) allele of the variant using a randomly weighted combination of the alleles of the typed variants in its vicinity, that is, locality-based hashing (Methods). The hash is calculated in modulo-2 arithmetic, which results in binary “proxy” alleles, and they look like valid alleles, albeit with virtually no correlation to the original (sensitive) alleles. The proxy allele protects the original typed variant allele because it is not easy to invert the proxy allele back to the original allele without the knowledge of the hashing function.
In essence, hashing maps the local haplotypes and their local frequency spectrum to a distinct domain defined by the hashing parameters while protecting the identity of the original allele. For the imputation outsourcing task, the query and reference sites must use the same hashing mechanism to generate hashed panels so that they map their haplotypes concordantly. This property motivates our expectation that the hashed haplotypes should provide utility for imputation.
Partitioning and permutation of untyped variants
ProxyTyper uses partitioning and permutations to protect the untyped variants (Fig. 2D,E). Given an untyped variant i that resides between typed variants a and b, the haplotypes that harbor the alternate allele of the untyped variant are randomly split into two sets. Next, ProxyTyper generates two proxy-untyped variants at random positions i1 and i2 between a and b (Fig. 2D); the alleles in the first haplotype set are assigned to i1, and those in the second set of haplotypes are assigned to i2. This procedure effectively partitions the alleles of the untyped variants into two new proxy-untyped variants such that the allele frequency of the original untyped variant is the sum of the two proxy-variant allele frequencies (Methods). Finally, the partitioned untyped variants between the typed variants are randomly permuted and inverted (Fig. 2E). Although this procedure obfuscates the variant positions and alleles extensively, the imputed genotype probabilities can be mapped back to the original variants perfectly when the partitioning/permuting parameters are known (Methods). These parameters are stored in a file that reference site and are only used for reconstructing the untyped variants after imputation at the imputation server.
Obfuscation of variant positions and genetic maps
Homer (Homer et al. 2008) and LRT-type reidentification attacks (Sankararaman et al. 2009) require matching the variant positions between the reference panel and the mixture panel so that attack statistics can be calculated to identify a target individual with known genotypes (Fig. 2F). To ensure that the proxy-variant positions are not directly linkable to external panels, ProxyTyper obfuscates the variant positions by replacing uniform distribution on an anonymous chromosome with a length of, by default, 108 nucleotides (Methods) (Fig. 2F; Supplemental Methods). Even though the variant positions are obfuscated, genetic maps can be aligned to public sources to match them and reveal exact locations. To protect the genetic maps that are shared among the sites, ProxyTyper perturbs the maps by noise addition with a user-specified deviation, σmap, measured in centiMorgans (cM). ProxyTyper generates anonymized genetic maps only for the typed variants because untyped variant genetic maps are interpolated by imputation software from typed variants. The coordinate anonymization for typed and untyped variants is basically a one-to-one mapping between the original and anonymized coordinates. ProxyTyper stores these in extended BED files. The anonymized genetic maps for typed variants (in anonymized coordinates) are stored in PLINK (Purcell et al. 2007) formatted genetic distance map files.
Genotype imputation outsourcing protocols
We present two imputation outsourcing protocols (Fig. 3) that can be used depending on whether the query site has access to a well-phased panel. When the query panel is phased, both the query and reference sites can use the resampling and hashing mechanisms to protect the typed variant alleles. Otherwise, the unphased-query protocols rely on permutations and augmentations to protect the typed variants (Fig. 3A; Supplemental Methods). The query site initiates the imputation protocols by sending the typed variant loci to the reference site. The reference site initiates all proxy panel generation model parameters (e.g., hashing, permutations, augmentation coordinates, obfuscation of typed variant coordinates) and sends them to the query site. In the phased protocol, sites can protect panels by resampling. Sites protect the typed variants using hashing (Fig. 3B, phased protocol), permutation, and augmentation mechanisms (Fig. 3B,C, for both protocols). Next, the reference site generates the partitioning parameters for the untyped variants and partitions the untyped variants on the reference site. The coordinates are finally anonymized using the obfuscated coordinates on both sites. The proxy panels are sent to the imputation server, which runs Beagle (Browning et al. 2018, 2021) and sends the results back to the query site. When resampling is used in the phased protocol, a local reimputation step is necessary at the query site because the resampled panels do not correspond to the original query panel (Fig. 3B). After imputation is completed, the reference site sends the untyped variant partitioning parameters to the query site. The query site maps the partitioned untyped variants to their original (sensitive) positions and decodes the final imputed genotype values. Finally, the coordinates are deanonymized, and the query site obtains the imputed genotypes.
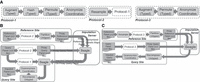
Illustration of phased- and unphased-query protocols using the mechanisms as building blocks that map and mutate panels. (A) Protocol-1 is a subblock to process typed variants in phased panel protocols. It starts by augmenting the typed variants, hashing the alleles, permuting the variants, and finally anonymizing coordinates. Protocol-2 prepends resampling mechanism to Protocol-1. Protocol-3 is similar to Protocol-1 but does not include a hashing step. Protocol-3 is a subblock used in unphased-query protocol. (B) Phased-query protocol. The reference site processes its panel with Protocol-2. Then it protects the untyped variants by the “partition untyped” mechanism. The query also processes its panel with Protocol-2. Both sites send the proxy panels to the imputation server, which runs Beagle and sends the results to query site. Note that imputation server also receives the anonymized genetic maps from the reference site. After receiving the imputed panel from the server, the query site first processes its panel with Protocol-1 (no resampling) and performs local reimputation using this panel as the input to Beagle. After reimputation, the query recomposes untyped variants and deanonymizes coordinates. The final result is a VCF file with imputed variant genotypes. (C) Unphased-query protocol. The reference panel first resamples and processes its panel with Protocol-3. It finally partitions untyped variants. The query site processes its panel with Protocol-3. Both sites send the proxy panels to the imputation server. After running Beagle, the server returns results to the query site, which recomposes the untyped variants and deanonymizes coordinates.
Genotype imputation accuracy
We tested ProxyTyper's protocols using the panels from The 1000 Genomes Project (The 1000 Genomes Project Consortium 2015), GTEx Consortium (Ardlie et al. 2015), and the HRC (McCarthy et al. 2016). In total, we tested six protocols that can be used for outsourcing of genotype imputation tasks: two phased-query protocols with and without augmentation mechanism, and two unphased-query protocols with and without augmentation mechanism. We used imputation with the plaintext query and reference panels with Beagle (Browning et al. 2018, 2021) as the baseline imputation with no protections. We also included RESHAPE as an external method that uses a single sampling step to be applied only to the reference panel. RESHAPE uses only sampling as a way to protect the reference panel and make it publicly available. Therefore, RESHAPE does not provide protection for the query panel and the variant positions and genotypes. ProxyTyper's protocols are more flexible and task specific for genotype imputation, and they aim to protect many more attack surfaces. We nevertheless included RESHAPE as a baseline method that provides a basic level of protection for the reference panel.
First, we tested the accuracy of the protocols with respect to different allele frequencies. Next, we assessed the population-specific variant imputation accuracies described below.
Impact of parameter selections
We used the African genomes from The 1000 Genomes Project and estimated the impact of different parameters (Supplemental Methods; Supplemental Fig. S1) on imputation accuracy with the phased imputation protocol. We found that the weight selection parameters (the first-order
hashing weight, 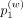 , and the locality constraint parameter [
, and the locality constraint parameter [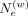 ] used to select the variants used in hashing) have the largest impact on accuracy (Methods) (Supplemental Fig. S1A–E). We found that increasing the vicinity, nvic, size used for hashing increases accuracy before decreasing (Supplemental Fig. S1F). This indicates that integrating vicinity information provides not only protection but also slight accuracy improvement.
The standard deviation for the genetic map anonymizing noise, σg, had observable impact on accuracy below 0.01 cM (Supplemental Fig. S1G). Finally, the permutation window length for protecting untyped variants did not immediately affect imputation accuracy (Supplemental Fig. S1H).
] used to select the variants used in hashing) have the largest impact on accuracy (Methods) (Supplemental Fig. S1A–E). We found that increasing the vicinity, nvic, size used for hashing increases accuracy before decreasing (Supplemental Fig. S1F). This indicates that integrating vicinity information provides not only protection but also slight accuracy improvement.
The standard deviation for the genetic map anonymizing noise, σg, had observable impact on accuracy below 0.01 cM (Supplemental Fig. S1G). Finally, the permutation window length for protecting untyped variants did not immediately affect imputation accuracy (Supplemental Fig. S1H).
Imputation accuracy by minor allele frequency
To assess the imputation accuracy of the protocols by minor allele frequency spectrum of variants, we used the HRC panel, which consists of 27,165 subjects. We randomly divided the subjects across query and reference panels (13,582 and 13,583 subjects, respectively). To decrease the computational requirements, we focused on variants in Chromosome 20:10,000,000–25,000,000, which comprises 6659 typed (matching to Illumina Duo V3 platform) and 210,090 untyped variants. We ran the six imputation protocols and estimated the genotype-level R2 statistic for all untyped variants. We divided the alternate allele frequency spectrum into eight bins (Fig. 4; Supplemental Fig. S2; Supplemental Table S1) to assess the accuracy of the methods.
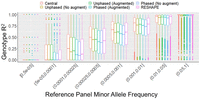
Imputation accuracy (genotype R2) of imputation protocols using random splitting of Haplotype Reference Consortium (HRC) panel as query and reference. The x-axis shows the minor allele frequency bins of the untyped variants. Each box plot shows the genotype-level R2 distribution of imputed variant genotypes. Phased and unphased protocols were run with and without augmentation (augment/no augment), as indicated in the legend. The central protocol corresponds to running Beagle with no protection mechanisms.
Plaintext protocol performed the most accurate imputations across all frequency bins (0.580 vs. 0.548). Among proxy panel–based protocols, the protocols that use augmentation imputed variants more accurately than the protocols that do not use augmentation (phased query with augmentation 0.518 vs. without augmentation 0.506). This suggests that augmentation provides protection and improves imputation accuracy by spreading the variant signal. Among the protocols that use typed variant augmentation, the unphased-query protocol is slightly more accurate than the phased-query protocol across all MAF bins (0.548 vs. 0.518). We also observed that the unphased protocol with augmentation imputed slightly more accurately than RESHAPE (0.535 vs. 0.548).
The methods performed similarly for the common variants (MAF > 5%). RESHAPE and the augmented phased and unphased-query protocols differ by <1% in genotype R2 (plaintext R2 = 0.982, RESHAPE R2 = 0.980 vs. unphased/phased augmented 0.972/0.972) for the common variants. For the uncommon variants (0.05 > MAF > 0.01), the general trend is also similar (plaintext R2 = 0.892, RESHAPE R2 = 0.874, unphased augmented = 0.870, phased augmented = 0.860). For the rare (1% > MAF > 0.1%) and ultrarare (0.1% > MAF) variants, we observed that unphased-query protocol with typed variant augmentation provides slightly higher accuracy than other protocols (ultrarare variants plaintext R2 = 0.395, RESHAPE R2 = 0.333, unphased augmented = 0.358). This may indicate that augmentation improves the rare variant imputation accuracy in proxy panel–based protocols. These results also indicate that the unphased protocol with augmentation step can be a good choice as a low complexity (single step protocol with no local imputation, does not explicitly require query panel to be phased) protocol.
Impact of typed variant sets
To test the impact of the typed variant set that is dependent on the array platform (Infinium Duo V3), we used the typed variants on two other Infinium array platforms: (1) Illumina Infinium Global Diversity Array-8 v1.0 with 1.8 million markers and (2) Infinium Global Screening Array-24 v3.0 with approximately 650 thousand markers. We generated the query and reference panels by equal random splitting of the HRC panel as in the previous experiment. We observed that the comparative accuracy of the protocols is similar for the Duo V3 platform (Supplemental Fig. S3A,B). We observed higher imputation accuracy with Diversity Array, which can be explained by a denser coverage of typed variants. In both cases, the unphased-query protocol imputed rare and ultrarare variants more accurately than did other protocols.
Population-specific variant genotype imputation accuracy
Next, we focused on assessing the accuracy of the population-specific imputation of the protocols. For this case, we first used the HRC panel as the reference panel and used all 1000 Genomes subjects as the query panel: We used 2495 subjects from The 1000 Genomes Project from within the HRC panel as the query panel. The remaining 24,670 subjects were used as the reference site's panel. We used 26,543 variants on Chromosome 20 that overlap with the Illumina Duo array, which were designated as the typed variants. We stratified the remaining 856,199 untyped variants by assigning each variant to one of the five superpopulations (African [AFR], European [EUR], AMR, EAS, SAS) based on their alternate allele frequency in each population (Fig. 5A).
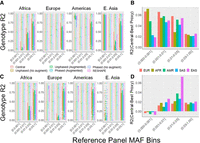
Population specific variant imputation accuracy. (A) Imputation accuracy for the variants on four superpopulations using HRC as the reference and 1000 Genomes as the query panel. Each box plot shows the genotype R2 distribution for the variants most frequently observed on the respective population. (B) The mean R2 difference between plaintext imputation and the unphased protocol at distinct allele frequency bins with HRC reference and 1000 Genomes as the query panel. Each bar corresponds to a population-specific variant set. (C) Imputation accuracy for population-specific variants using 1000 Genomes as the reference panel and GTEx as the query panel. (D) Mean genotype R2 difference between plaintext protocol and unphased protocol with 1000 Genomes as reference and GTEx panel as query.
The plaintext protocol provides the highest imputation accuracy among all methods (plaintext R2 = 0.399, RESHAPE R2 = 0.362, and unphased augmented protocol R2 = 0.358). For all populations, we observed that the protocols that include a typed variant augmentation imputed more accurately than did the protocols without the augmentation step, which holds over all frequency bins.
Among the five populations, variants most observed in African subjects were imputed most accurately. This can be attributed to the high diversity of the African genomes and the more information contained in the African haplotypes, which makes it more accurate to impute variants. We next evaluated the accuracy difference between the plaintext protocol and the best-performing proxy panel–based protocol (unphased query with typed variant augmentation) (Fig. 5B; Supplemental Tables S2–S6). For ultrarare variants (MAF <0.1%), EUR and AFR variants are imputed with 6% lower accuracy than the plaintext protocol. This difference decreases for both populations quickly as the MAF of the variants increases. For America and Asia (AMR, EAS, SAS) populations, the rare variants are imputed with 2% lower accuracy using proxy panels. This difference increases for rare and uncommon categories for these populations (e.g., EAS variants are imputed with 5.3% higher accuracy by plaintext protocol). The imputation accuracy difference for common variants in all populations is fairly uniform at ∼2%–2.5% (Fig. 5B). These results indicate a complex interplay between proxy mechanisms and imputation accuracy in a population-specific manner.
We next used 2504 subjects in The 1000 Genomes Project as the reference panel and used the 635 subjects in the GTEx project as the query panel and ran the six imputation protocols (Fig. 5C). Over most of the MAF ranges, we observed that the African variants were again imputed with highest accuracy, and East Asian–specific variants were imputed with least accuracy, regardless of protocol selection (unphased protocols R2 highest: 0.381 in AFR variants and 0.102 in EAS variants). When comparing the plaintext imputation with the best ProxyType protocol, we observed population-specific differences (Fig. 5D; Supplemental Tables S7–S11). We observed that the East Asian variants were imputed with 7.2% lower accuracy for the uncommon variant category (5% > MAF > 1%) compared with the plaintext imputation protocol. The accuracy difference for the rare variant category is more concordant than the uncommon variants, ranging between a 1% and a 3% difference among all populations.
The population-dependent accuracy of ProxyType protocols is expected to a certain extent because proxy-generating mechanisms mutate and map the haplotypes to a new frequency spectrum. Thus, it is likely that some of the less represented but more informative haplotypes (e.g., Asian- and American-specific haplotypes) will be over- or underrepresented in the proxy panels. Depending on the frequency of variants and the haplotypes, this translates to different imputation accuracies in a population-specific manner.
Time/memory requirements
We finally evaluated the time and memory requirements of the ProxyTyper-based protocols. For this, we used a randomly partitioned HRC panel as the reference site panel (13,582 subjects) and the query site panel (13,583 subjects). We focused on Chromosome 20:10,000,000–15,000,000, which contains 2021 typed variants and 68,443 untyped variants. We ran the phased and unphased query with typed variant augmentation protocols with default configurations using 40 threads.
The longest running portion of both protocols runs Beagle (1216 sec for unphased and 761 sec for phased protocol) (Fig. 6). Most proxy mechanisms take <10 sec to run, except for untyped variant recomposition (or unpartitioning), which takes 58 sec. In terms of memory usage, the usage by the mechanisms was <10 GB (Supplemental Fig. S4). In comparison, RESHAPE's sampling finished in 116 sec. We foresee that these steps can be further tuned to run faster by streamlining multiple steps into one command to decrease the I/O overhead of reading/writing large panels. However, we believe that the current partitioning of the codebase enables more readable protocol scripts and provides a good tradeoff between ease of use and time/memory usage.
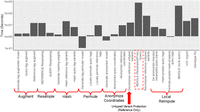
Time requirement for the phased proxy protocol. The bars denote the number of seconds spent at each step (including reference and query sites) of the phased protocol. The steps are grouped below to simplify protocol execution. The dashed rectangle shows the Beagle running step at the imputation server, which takes the longest time. The local reimputation steps consisting of deanonymizing, hashing, permutation, and local Beagle run are shown in one step. The last two steps are for VCF importing and accuracy estimation.
Privacy protection comparisons among protocols
We first compared the original panels and the proxy panels with respect to the allele, linkage, and local haplotype frequencies when hashing is used. To ensure that the analysis was not affected by population-specific factors, we used subjects from the African population in The 1000 Genomes Project (Supplemental Methods).
We found that the allele and haplotype frequencies exhibit very little resemblance to the original and local haplotype frequencies (Supplemental Fig. S5A–C). Next, we calculated the genotype correlations (Pearson R2 between alleles of variants) to quantify the linkage information between typed variants. Compared with the original panel, the linkage patterns did not show immediate relationships (Supplemental Fig. S5D,E). Next, we compared the local haplotype frequencies by calculating the frequency of unique k-mers at each position and comparing these between the original and proxy panels. We observed that hashing expands the local haplotypes in the proxy panels and increases the number of unique local haplotypes (Supplemental Fig. S5F,G). We also observed that when the proxy panels are compared, there is a concordance in the number of unique local haplotypes (Supplemental Fig. S5H,I).
We finally asked if the proxy panels could be used to predict ancestral information from subjects. We found that, without resampling, an adversary can see a separation between major populations using principal component analysis (PCA) (Supplemental Fig. S5J). However, after resampling, PCA-based dimensionality reduction does not provide much information for assessing the populations. Resampling should be used to protect the ancestral information from the proxy panels.
Frequency analysis on local haplotypes
We foresee that an adversarial approach to process proxy panels would involve aiming to decode the alleles of the hashed panels. For this, an adversary can utilize frequency analysis to decode the typed variant alleles of a proxy panel (Supplemental Methods) by striving to match the local haplotype frequencies (i.e., k-mer frequencies) of a public panel (e.g., The 1000 Genomes Panel) and the proxy panel, under the assumption that proxy haplotype frequencies can leak information about the underlying haplotypes. When combined with the constraint that consecutive k-mers match each other with small number of errors, an adversary can try to decode a proxy panel. To test the possibility of reidentifying individuals from proxy panels using a frequency analysis, we implemented a Viterbi-decoder that uses a HMM for identifying the most likely haplotype given a proxy panel by comparing it to a public panel (Supplemental Methods). We observed that this complex decoding attack provides minimal improvement over the assignment of maximum frequency alleles (Supplemental Fig. S6A). Further, the test statistic was uncalibrated when the decoded alleles are used for reidentification with a likelihood-ratio test (Sankararaman et al. 2009). We also did not observe a strong separation between the positive and the hold-out subjects (Supplemental Fig. S6B–D). For this attack to work in practice, the adversary must also match the coordinates between the public reference panel and the proxy panel. Although we assumed coordinate information is roughly available to the attacker, it is not clear how mapping can be performed in practice.
Comparison with sampling-only protection of RESHAPE
When comparing RESHAPE and ProxyTyper's approaches with privacy protection, it is important to consider that these tools assume different threat models: RESHAPE aims to build a semisynthetic reference panel that could be shared publicly under the assumption that sampling haplotypes up to a certain number of generations provides sufficient privacy. RESHAPE may occasionally generate long segments of haplotypes that can be searched for and linked to other databases. Unlike ProxyTyper, RESHAPE does not protect variant coordinates of typed/untyped variants and the alleles of ultrarare untyped variants in the reference panels, which leaves a large attack surface open. Furthermore, RESHAPE does not provide a way to protect the query panel in outsourcing scenarios. We tested RESHAPE-protected panels with respect to the privacy tuning parameter and show that the protected panels may inadvertently leak information through complex attacks and simple attacks that can be performed with a Beagle run followed by processing results with an AWK script (Supplemental Methods; Supplemental Fig. S7). Importantly, we observed that the privacy parameter we used (number of generations equals 128) in the above accuracy comparisons may not be sufficient to guarantee the expected level of privacy protection for the reference panels (Supplemental Fig. S7).
Discussion
We presented the mechanisms for generating proxy panels as a framework to protect large-scale haplotype-level data sets and demonstrated their utility for outsourcing genotype imputation. Proxy panels combine mosaic haplotype generation, noise addition, locality-sensitive random hashing, and random permutations to protect against well-known linking and reidentification of attacks. Of note, releasing rare untyped variant positions alone (even without genotype information) can lead to reidentification using Bustamante's beacon attack, which is currently not considered in the literature. For high-throughput tasks such as genotype imputation, it becomes a major challenge to protect data sets against these risks. The frequencies and alternate alleles of variants, variant coordinates, and genetic maps are protected with different mechanisms through proxy panel generation. These mechanisms add preemptive barriers for deterring the honest-but-curious entities (e.g., white hat hackers, cryptanalysts, and curious researchers) and help defer accidental or intentional reidentifications through linking databases (searching an individual in a forensic database). We therefore believe proxy panels represent an alternative route to anonymized data generation and can be classified as such in the context of personal data sharing regulations, for example, GDPR and HIPAA. Another advantage of proxy panels is that they are optimized for a specific task (e.g., imputation) and are unsuitable for direct secondary analyses. We are optimistic that new proxy panel generation mechanisms can be developed to optimize the panels for task-specific scenarios that provide higher utility with low privacy concerns. One mechanism we did not explore here is partitioning the typed variant alleles, similar to partitioning untyped variants. This mechanism can further protect against haplotype decoding by increasing the local haplotype distribution entropy beyond the mechanisms we used in this study. We foresee that new mechanisms can be designed for other tasks, for example, kinship estimation (Wang et al. 2022), and collaborative GWAS (Li et al. 2023a), as well. These mechanisms can be combined with encryption-based techniques to decrease privacy risks while increasing efficiency.
Our results demonstrate that proxy panels can be directly used in existing imputation methods without modifying the underlying algorithms. This is advantageous compared with, for example, homomorphic encryption–based methods, which require reformulation and careful implementation of the underlying imputation methods. In comparison, proxy panel–based methods require new protocol designs without changes in underlying algorithms.
Our benchmarks also indicate that the accuracy of proxy panel–based protocols is very similar to that of the cleartext protocols. However, there is still a need to assess ultrarare variant imputation accuracy because these variant categories are challenging to impute using the conventional imputation outsourcing scenario we address here. New and highly promising imputation approaches were developed utilizing customized imputation pipelines (Barton et al. 2021). These methods exhibit high imputation accuracy for ultra-ultrarare variants (MAF < 10−5). Future research endeavors can develop new proxy panel–based protocols to implement these customized protocols using ProxyTyper's mechanisms. Overall, we believe new imputation protocol designs and postprocessing tools can help increase the accuracy and efficiency of proxy protocols.
Proxy panels should not be made publicly available because their protection relies on exchanging secret proxy generation parameters between the collaborating entities (akin to symmetric encryption keys). When an adversary gains access to these parameters (e.g., hashing weights) and a proxy panel, they can use the proposed decryption strategy via haplotype-frequency analysis to decode the alleles in the proxy panel. In particular, recent studies showed that similar locality-sensitive hashing approaches (e.g., Google FLoC and Topics) may leak information when the adversary has access to the parameters of the model (Turati et al. 2023). We foresee that proxy panels can enable more collaborative research (rather than public data sharing) because they can be classified as anonymized data sets and exchanged for analysis via noncolluding third parties such as AnVIL and the Michigan Imputation Server (Loh et al. 2016). Implementing the protocols that use proxy panels can be seamlessly accomplished because underlying algorithms are not modified; that is, the analysis server does not have to change its infrastructure to process proxy panel data. It is, however, necessary to establish the infrastructure at each data owner/controller to perform automatic proxy panel generation; that is, every time imputation is initiated, a reference panel must be processed to generate the proxy panels. The successful adoption of proxy panel usage in genotype imputation tasks is implementing an easy-to-use infrastructure. Although we do not anticipate that established servers may be reluctant to adopt proxy panel–based imputation protocols, we believe that localized projects and regional local collaborations can use the proxy-based protocols. To manage the computational load of regenerating proxy panels from large reference data sets, it may be useful to generate multiple proxy panels periodically (weekly or monthly), re-cycle these in this period, and then regenerate the proxy panels. This relieves the burden of resampling and variant partitioning from reference sites. This will be an important factor for developing efficient proxy-based analysis infrastructure.
The adoption also relies heavily on continuous analysis of the attack scenarios, the threat actors, and the practicality of the attacks. Currently, the reidentification attacks of Homer and Sankararaman, which are arguably the most well-known attacks in the literature, are popularized because they can be easily executed by honest-but-curious entities. However, these attacks require precise matching between the variants in the panels and between the ancestral compositions of the reference panel, the target individual, and the “pool” of individuals being tested for the participation of the target individual. The ancestral matching is necessary to ensure that the reference panel appropriately controls the test statistic against population-specific biases. Thus, proxy panels can be designed to exploit this shortcoming of the reidentification attacks: Proxy panels can be generated from randomized fractions of multiancestral samples that the attacker cannot immediately decompose. These multiancestral data sets can be generated once by simulations, for example, SLiM (Haller and Messer 2019), or publicly available panels can be used (e.g., The 1000 Genomes Project). The tunability of proxy panel generation can be used to design new proxy panel generation techniques to thwart future reidentification risks with decoding attacks. Furthermore, ProxyTyper currently protects untyped variants using permutation-based anonymization. When combined with augmentation, permutation, and hashing, the randomization of the ancestral background of the panel would provide strong resilience against potential future reidentification attacks. The accuracy of the attacks is contingent upon the adversary correctly deanonymizing the variant coordinates and unpermuting the variants; that is, in addition to the ancestral composition matching, it is necessary to match or “align” the anonymized coordinates to the reference panel. Because the genetic noise addition (i.e., noise with standard deviation σg) is performed randomly, the coordinates will be randomized at the loci with low recombination rates. However, this mechanism cannot protect the chromosomes as a whole because the genetic length of the anonymized chromosome is sufficient to match it to the original chromosome. It is, therefore, important to make sure that multiple mechanisms are combined to utilize a multilayered deterrence approach and that the proxy panels are not publicly shared.
Regarding the untyped variants, the partitioning provides strong protection using a “hiding-in-the-crowd” approach. Partitioning may, however, introduce a large computational burden because each partitioning step doubles the number of untyped variants. We foresee that it will be necessary to develop new mechanisms that more efficiently protect untyped variants. It may be useful to exploit the redundancy of the untyped variants that share the same haplotype, design a hashing approach, and decrease the redundancies. This way, the mechanisms can also manage the number of untyped variants used in proxy panels. Numerous research directions can use proxy panels with minimal modifications to the underlying imputation methods.
Methods
We describe proxy panel generation, imputation protocol, parameter selections, and decoding attacks.
Adversarial entities
We focus on the “honest-but-curious” adversarial model (most prevalent in genetic research) (Paverd et al. 2014), in which users do not deviate from general protocols of analysis. In collaborative genetic research, the honest-but-curious adversarial model is more prevalent and relevant than malicious entities actively trying to decode proxy panels to execute attacks. Researchers are generally assumed to be well intentioned and to follow prescribed protocols, but they might still be curious about the data they have access to. The data security protocols against malicious entities are computationally challenging to operate and maintain.
Proxy panel generation
Mosaic variant panel generation by resampling
ProxyTyper uses a resampling approach to generate a mosaic panel that removes the one-to-one correspondence of the original
haplotypes to the proxy haplotype panel to provide protection against linking attacks. Given the original haplotype panel
that contains Norig haplotypes over nvar variants (denoted by H(orig) matrix with Norig rows and nvar columns), as well as a genetic map defined on the variant coordinates (i.e., Δg vector of length nvar) as input, resampling generates Nres haplotypes (H(res) matrix with Nres rows and nvar columns) using a Li–Stephens HMM. The sampling starts from the leftmost variant (sorted by coordinates) by selecting a random
haplotype (i.e., state). At variant i, a new state is selected such that the recombination probability determines the transition to a new state: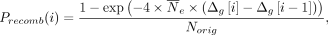 where
where 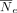 is the normalized effective population size parameter that tunes the number of recombinations in the resampled panel. The
probability of remaining on the same haplotype is calculated as
is the normalized effective population size parameter that tunes the number of recombinations in the resampled panel. The
probability of remaining on the same haplotype is calculated as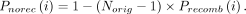
ProxyTyper samples the probabilities over the haplotypes and selects one haplotype as the newly sampled haplotype. The allele
on the sampled haplotype is stored as the allele for the resampled haplotype at variant i; that is, 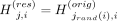 , where jrand(i) < Norig is the sampled haplotype index. After the sampling, the state is updated at variant i, and sampling moves to the next variant. Each haplotype is sampled independently of all other haplotypes. ProxyTyper does
not introduce errors by default in the resampling step but has a parameter to introduce random errors in the resampled panels.
, where jrand(i) < Norig is the sampled haplotype index. After the sampling, the state is updated at variant i, and sampling moves to the next variant. Each haplotype is sampled independently of all other haplotypes. ProxyTyper does
not introduce errors by default in the resampling step but has a parameter to introduce random errors in the resampled panels.
Constraints on maximum segment length
For most variant positions, the nonrecombination probability is larger than the recombination probabilities. This may result
in long consecutive allelic segments getting copied to the resampled panels. To get around this, ProxyTyper uses a parameter
to constrain the length of haplotype segments copied from each haplotype. To ensure that long segments are not copied, ProxyTyper
keeps track of the consecutive segment length sampled from the current haplotype. If the length reaches a user-tunable parameter
(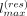 ), the probability of all haplotypes is set uniformly; that is,
), the probability of all haplotypes is set uniformly; that is,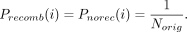
After the probabilities are reset, a new haplotype is sampled uniformly (note that 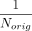 is uniformly set to sampling each of the Norig haplotypes). Sampling is performed until a different haplotype is chosen similar to a rejection sampling.
is uniformly set to sampling each of the Norig haplotypes). Sampling is performed until a different haplotype is chosen similar to a rejection sampling.
Selection of the recombination loci
Note that the resampling is performed independently at the reference and query sites. However, when resampling is performed at the reference sites, the number of variants may be prohibitively large, which may impose a large computational burden. We observed that the positions at which ProxyTyper evaluates a haplotype switch (i.e., recombination) can be constrained using a minimum genetic distance cutoff without much impact on the quality of the resampled panels. By default, ProxyTyper selects these “recombination loci” (Fig. 2A) by starting from the first variant on the panel and finding the next variant that is at least ΔΔg (set to 0.001 cM by default) away from the current recombination locus. Although resampling is being performed, ProxyTyper only evaluates recombinations on the recombination loci. Even using a small distance cutoff, we observe a large computational improvement in sampling. Of note, Beagle (Browning et al. 2018) uses a similar genetic distance cutoff while running its imputation HMM, which partially justifies this approach.
Typed variant augmentation
Typed variant augmentation aims to randomize the number of typed variants. Rather than introducing random variants, ProxyTyper reuses variants and augments them within the vicinity of the original variant.
Given the typed variant i, ProxyTyper selects a random position (in genomic coordinates) between the typed variant at 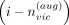 and typed variant at
and typed variant at 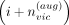 and augments a new proxy-typed variant. The genotype values of the augmented typed variant are (by default) copied from the
original typed variant's genotype vector. Users can also choose to set the genotypes to zero. To randomize the augmented typed
variant set, the augmentation is performed with a certain probability tuned by a user-defined value p(aug) ∈ [0, 1], which is, by default, set to 0.99. At each typed variant, a randomly generated value in [0, 1] is compared to p(aug), and augmentation is performed if the random value is smaller than p(aug). Augmentation can also be performed recursively to increase the number of typed variants in the panels arbitrarily. Augmentation
does not modify the panels’ untyped variants (if there are any).
and augments a new proxy-typed variant. The genotype values of the augmented typed variant are (by default) copied from the
original typed variant's genotype vector. Users can also choose to set the genotypes to zero. To randomize the augmented typed
variant set, the augmentation is performed with a certain probability tuned by a user-defined value p(aug) ∈ [0, 1], which is, by default, set to 0.99. At each typed variant, a randomly generated value in [0, 1] is compared to p(aug), and augmentation is performed if the random value is smaller than p(aug). Augmentation can also be performed recursively to increase the number of typed variants in the panels arbitrarily. Augmentation
does not modify the panels’ untyped variants (if there are any).
modulo-2 hashing of typed variant alleles
At the typed variant i of haplotype j, ProxyTyper calculates a hash of the alleles for the surrounding typed variants, 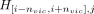 , as the proxy-allele of the variant. To increase hash complexity (nonlinearity), the hash includes second- and third-order
interaction terms and a bias term:
, as the proxy-allele of the variant. To increase hash complexity (nonlinearity), the hash includes second- and third-order
interaction terms and a bias term: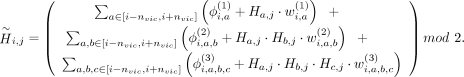
The three rows correspond to first-, second- and third-degree interactions of variants at a, b, and c for variants in (2nvic + 1) vicinity of variant i. 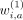 ,
, 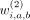 , and
, and 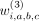 are the binary weights (specific to variant i) that add the corresponding component into hash.
are the binary weights (specific to variant i) that add the corresponding component into hash. 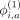 ,
, 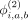 , and
, and 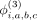 are the binary bias offsets that effectively flip the contribution of each component's effect on the final hash. The overall
hash is calculated by summing all components using modulo-2 arithmetic so that the final proxy-allele,
are the binary bias offsets that effectively flip the contribution of each component's effect on the final hash. The overall
hash is calculated by summing all components using modulo-2 arithmetic so that the final proxy-allele, 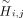 , is a binary value.
, is a binary value.
Recombination-dependent selection of vicinity variants
For variant i, the bias terms are selected randomly for each component using a Bernoulli distribution; that is, 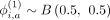 . Weight parameters tune the contribution of variants in the vicinity of the variant i, that is, [i − nvic, i + nvic], to the proxy allele of i. In certain cases, the vicinity may expand a large genetic distance. To ensure that the hashes are calculated in uniform
genetic vicinities, the selection of weights is performed using a genetic distance-dependent manner:
. Weight parameters tune the contribution of variants in the vicinity of the variant i, that is, [i − nvic, i + nvic], to the proxy allele of i. In certain cases, the vicinity may expand a large genetic distance. To ensure that the hashes are calculated in uniform
genetic vicinities, the selection of weights is performed using a genetic distance-dependent manner: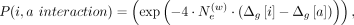 where
where 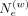 tunes the strength of interaction between pairs of variants dependent on their genetic distance, that is, (Δg[i] − Δg[a]). Low
tunes the strength of interaction between pairs of variants dependent on their genetic distance, that is, (Δg[i] − Δg[a]). Low 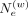 enables a more relaxed selection of weights and more involving variants i and a that are far from each other. This probability is used for preselecting the variants used for hashing the allele for the
variant i. In general, setting
enables a more relaxed selection of weights and more involving variants i and a that are far from each other. This probability is used for preselecting the variants used for hashing the allele for the
variant i. In general, setting 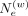 too high (above 1000) corresponds to focusing on the immediate vicinity of the variant i, that is, using the variant itself for hashing.
too high (above 1000) corresponds to focusing on the immediate vicinity of the variant i, that is, using the variant itself for hashing.
Random selection of weights
For each weight in the hash calculation, that is, 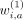 ,
, 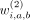 ,
, 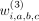 , we use a Bernoulli random variable:
, we use a Bernoulli random variable: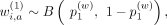
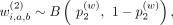
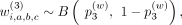 where
where 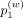 ,
, 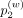 , and
, and 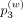 denote weight probability for first-, second-, and third-order weights, which are input arguments.
denote weight probability for first-, second-, and third-order weights, which are input arguments.
Overall, the variant selection and weight selection steps are performed independently for each variant and are parallelized. The final weights of the model include variant-specific weights and biases stored in a binary file. This file is the hashing “key” and should not be shared with entities other than the collaborating sites. ProxyTyper saves a summary file containing the number of hashing parameters at each vicinity for each typed variant.
Typed and untyped variant permutation on sliding windows
Given the typed (or proxy-untyped) variants [k, l], we permute the untyped variants’ indices [k, l] (Fig. 2E):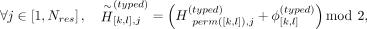 where
where 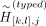 denotes the shuffled allele vector for typed variants for variants at indices [k, l] on haplotype j, and perm([k, l]) denotes a random permutation of indices k, k + 1, …, l − 1, l.
denotes the shuffled allele vector for typed variants for variants at indices [k, l] on haplotype j, and perm([k, l]) denotes a random permutation of indices k, k + 1, …, l − 1, l. 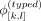 denotes a random binary bias that randomly flips the alternate alleles of variants in [k, l]. Each entry in the bias vector is selected with a Bernoulli variable, that is, B(0.5, 0, 5). To systematically protect the typed variants, ProxyTyper performs permutations recursively on sliding windows
of length
denotes a random binary bias that randomly flips the alternate alleles of variants in [k, l]. Each entry in the bias vector is selected with a Bernoulli variable, that is, B(0.5, 0, 5). To systematically protect the typed variants, ProxyTyper performs permutations recursively on sliding windows
of length 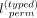 . A smaller
. A smaller 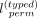 better preserves the ordering of the variants. The permutation and bias additions do not require the panel to be phased.
better preserves the ordering of the variants. The permutation and bias additions do not require the panel to be phased.
Generation of proxy-untyped variants by allele partitioning
Although permutation obfuscates variant positions, it precisely preserves the allele frequencies of the untyped variants.
ProxyTyper generates proxy-untyped variants by a mechanism called allele partitioning. Given an untyped variant, the basic
idea of partitioning is randomly splitting the alleles among two new proxy variants such that the union of the proxy variants
exactly recapitulates the alleles in the original variant (Fig. 2D):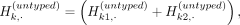 where we treat the alleles of kth untyped variant as a binary vector across haplotypes. This vector is equal to the summation of two proxy-untyped variant
allele vectors,
where we treat the alleles of kth untyped variant as a binary vector across haplotypes. This vector is equal to the summation of two proxy-untyped variant
allele vectors, 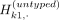 and
and 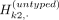 , which denote the (binary) allele vectors of the proxy-untyped variants k1 and k2. The above equation refers to partitioning the haplotypes that harbor alternative alleles for untyped variant k. For each original untyped variant in the reference panel, ProxyTyper generates two untyped proxy variants by randomly partitioning
the haplotypes that harbor alternate allele for the untyped variant. The original untyped variant is no longer included in
the proxy-untyped reference panel, and it has been replaced with two proxy-untyped variants, randomly placed between the nearest
tag variants on the left and right. After partitioning, the proxy-untyped alleles always have smaller allele frequencies than
the original variant.
, which denote the (binary) allele vectors of the proxy-untyped variants k1 and k2. The above equation refers to partitioning the haplotypes that harbor alternative alleles for untyped variant k. For each original untyped variant in the reference panel, ProxyTyper generates two untyped proxy variants by randomly partitioning
the haplotypes that harbor alternate allele for the untyped variant. The original untyped variant is no longer included in
the proxy-untyped reference panel, and it has been replaced with two proxy-untyped variants, randomly placed between the nearest
tag variants on the left and right. After partitioning, the proxy-untyped alleles always have smaller allele frequencies than
the original variant.
The motivation for using this partitioning as a mechanism is that it obfuscates the alleles of the original untyped variant
k by partitioning its alleles into two proxy variants. Although the original variant is not in the reference panel, the proxy
variants preserve the information needed to reconstruct the imputed allelic probabilities of the original variant. For example,
given a kth untyped variant, the imputed probability of allele 1 is equal to the summation of the HMM probabilities of all haplotypes
that harbor allele 1 on this variant: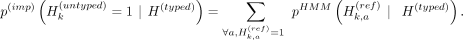 p(imp) denotes the imputed alternate allele probability of the kth variant by Beagle, and
p(imp) denotes the imputed alternate allele probability of the kth variant by Beagle, and 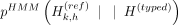 denotes the imputation HMM's forward-backward state probability for haplotype h at variant k, where the hth haplotype of the reference panel harbors an alternate allele for the variant k.
denotes the imputation HMM's forward-backward state probability for haplotype h at variant k, where the hth haplotype of the reference panel harbors an alternate allele for the variant k.
When we partition the kth variant's alleles into two proxy-untyped variants (k1 and k2), we effectively partition the haplotypes that harbor an alternate allele for variant k to these two variants: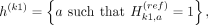
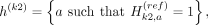
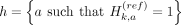 and
and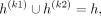
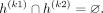
The last equations hold by the partitioning of the alleles such that 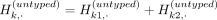 . The partitioning of haplotypes allows us to reconstruct the imputed alternate allele probability for the variant k in terms of the alternate allele probabilities assigned to k1 and k2. We first decompose the haplotypes in the equation for alternate allele probability at the variant k:
. The partitioning of haplotypes allows us to reconstruct the imputed alternate allele probability for the variant k in terms of the alternate allele probabilities assigned to k1 and k2. We first decompose the haplotypes in the equation for alternate allele probability at the variant k: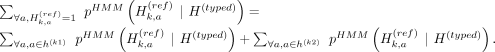
Given the panel with proxy-untyped variants k1 and k2, we can approximate the HMM haplotype state probabilities using the imputation performed with panel proxy-untyped: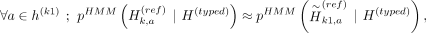
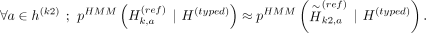
These equations hold approximately because of two reasons: First, the state probability at haplotype a of imputation HMM at untyped variants relies only on the closest typed variants on the left and right vicinity of the untyped variant. This is satisfied because k1 and k2 are constrained to be between the same typed variants as k (Fig. 2D). Second, k1 and k2 are not exactly at the same genetic position as k. Because of the linear interpolation that Beagle uses, the posterior probabilities assigned by Beagle to k1 and k2 will not be exactly the same as k, but the difference should be practically very small because k1 and k2 are placed very close to k while k is being partitioned.
The left-hand side of the equations refers to the Beagle HMM probability for the haplotype a when we perform imputation using typed variants as input and the original reference panel. The right-hand side indicates
the same probability when we use the untyped reference panel with proxy variants k1 and k2, which are new untyped variants that do not exist in the original panel. Replacing the right-hand side of the equations
with the above, we get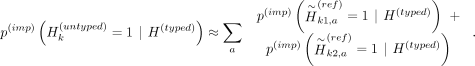
This equation indicates that we can use the reference panel with proxy-untyped variants 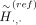 , obtain the imputed alternate allele probabilities (using Beagle) for the proxy-untyped variants at k1 and k2, and finally map them back to the original untyped variant k by summing their probabilities. ProxyTyper further obfuscates the proxy-untyped variants by randomly flipping their alleles
with 50% probability. This operation does not accrue any accuracy penalty because it also effectively flips the probability
of alternate alleles. Furthermore, allele flipping helps obfuscate the possible linkage between the original and proxy-untyped
variants because the proxy-untyped variants can have a higher frequency compared with the original variant. If a proxy-untyped
variant is flipped, ProxyTyper takes this into consideration by subtracting it from one before mapping it back to the original
untyped variant.
, obtain the imputed alternate allele probabilities (using Beagle) for the proxy-untyped variants at k1 and k2, and finally map them back to the original untyped variant k by summing their probabilities. ProxyTyper further obfuscates the proxy-untyped variants by randomly flipping their alleles
with 50% probability. This operation does not accrue any accuracy penalty because it also effectively flips the probability
of alternate alleles. Furthermore, allele flipping helps obfuscate the possible linkage between the original and proxy-untyped
variants because the proxy-untyped variants can have a higher frequency compared with the original variant. If a proxy-untyped
variant is flipped, ProxyTyper takes this into consideration by subtracting it from one before mapping it back to the original
untyped variant.
Currently, ProxyTyper can partition each untyped variant into two proxy-untyped variants, which doubles the number of untyped variants in the proxy panel. The alleles of each proxy-untyped variant are independently flipped. The partitioning information is stored in one file (kept secret at the reference site) that describes which proxy variants correspond to which original variants and describes the allele-flipping states of each proxy variant. Note that each untyped proxy variant is placed to a random position between the surrounding tag variants, similar to the permutation mechanism.
Anonymization of genetic maps and variant coordinates
After sampling of haplotypes and hashing of alleles, ProxyTyper first maps variant coordinates (including typed and untyped
variants) to a uniform range. This is set simply to 100 megabases by default. It should be noted that the variant coordinates
are only used for sorting variants and do not change imputation accuracy because genetic maps are provided. After coordinate
anonymization, the genetic maps are censored to release the genetic distances for only the typed variants. For this, ProxyTyper
first extracts the original cumulative genetic distances for only the typed variants. Next, a Gaussian noise is added to the
genetic distances: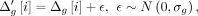 where σg is a user-defined standard deviation of genetic distance noise. Next,
where σg is a user-defined standard deviation of genetic distance noise. Next, 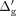 are sorted over all typed variants and finally assigned as the anonymized coordinates:
are sorted over all typed variants and finally assigned as the anonymized coordinates: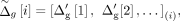 where
where 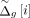 denotes the anonymized genetic distance for the typed variant i, and
denotes the anonymized genetic distance for the typed variant i, and 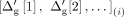 denotes the ith element in the sorted sequence of noisy genetic distances.
denotes the ith element in the sorted sequence of noisy genetic distances.
Imputation protocols, benchmarking, and imputation accuracy estimation
The four benchmarked imputation protocols are implemented using pipelines of calls to implement mechanisms in bash command line scripts. The protocols are run on single computers without explicitly simulating the network traffic. The accuracy of imputed genotypes was measured using the genotype level R2 by calculating the square of the Pearson correlation coefficient between the known and imputed genotype vector for each variant. The time and memory requirements of each protocol were measured using “/usr/bin/time” under Linux.
This study made use of data associated with a number of projects. The 1000 Genomes Project Phase 3 genotype data were downloaded from https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/.
GTEx project genotype data was accessed under terms of restricted access and can be obtained by application to the NCBI database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap/) accession number phs000424.
HRC data sets were accessed under terms of restricted access from the European Genome-Phenome Archive (EGA; https://ega-archive.org/) with the study identifier EGAD00001002729.
Software availability
All software developed in this study are available at GitHub (https://github.com/harmancilab/ProxyTyper) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We acknowledge funding from UTHealth startup funds and National Human Genome Research Institute grant R01HG012604 for A.H.
Author contributions: D.Z., X.J., and A.H. conceived the idea for building proxy panels. A.H. and D.Z. developed the conceptual ideas for implementations and also implemented the software. A.H. ran the computational experiments and performed the analysis. D.Z., A.H., and X.J. wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278934.124.
-
Freely available online through the Genome Research Open Access option.
- Received January 2, 2024.
- Accepted January 6, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








