
Analyzing the large and complex SFARI autism cohort data using the Genotypes and Phenotypes in Families (GPF) platform
- Liubomir Chorbadjiev1,7,
- Murat Cokol2,7,
- Zohar Weinstein3,
- Kevin Shi4,
- Christopher Fleisch5,
- Nikolay Dimitrov1,
- Svetlin Mladenov1,
- Ivo Todorov1,
- Iordan Ivanov1,
- Simon Xu5,
- Steven Ford5,
- Yoon-ha Lee6,
- Boris Yamrom6,
- Steven Marks6,
- Adriana Munoz6,
- Alex Lash5,
- Natalia Volfovsky5 and
- Ivan Iossifov6
- 1SeqPipe Limited, Sofia 1000, Bulgaria;
- 2Rodop Biotechnology, 54050 Sakarya, Turkey;
- 3Atrius Health, Cambridge, Massachusetts 02139, USA;
- 4New York Genome Center, New York, New York 10013, USA;
- 5Simons Foundation, New York, New York 10010, USA;
- 6Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724, USA
-
↵7 These authors contributed equally to this work.
Abstract
The exploration of genotypic variants impacting phenotypes is a cornerstone in genetics research. The emergence of vast collections containing deeply genotyped and phenotyped families has made it possible to pursue the search for variants associated with complex diseases. However, managing these large-scale data sets requires specialized computational tools to organize and analyze the extensive data. Genotypes and Phenotypes in Families (GPF) is an open-source platform that manages genotypes and phenotypes derived from collections of families. GPF allows interactive exploration of genetic variants, enrichment analysis for de novo mutations, phenotype/genotype association tools, and secure data sharing. GPF is used to disseminate two family collection data sets, SSC and SPARK, for the study of autism, built by the Simons Foundation. The GPF instance at the Simons Foundation (GPF-SFARI) provides protected access to comprehensive genotypic and phenotypic data for SSC and SPARK. GPF-SFARI also provides public access to an extensive collection of de novo mutations from individuals with autism and related disorders and to gene-level statistics of the protected data sets characterizing the genes’ roles in autism. However, GPF is versatile and can manage genotypic data from other small or large family collections. Here, we highlight the primary features of GPF within the context of GPF-SFARI.
The substantial reduction in sequencing costs has made generating whole-exome or whole-genome sequences for large family collections feasible. This development allows for the direct observation and analysis of genetic variants across the entire frequency spectrum, spanning from common to rare and de novo mutations. In addition, large-scale sequencing in genetic studies provides researchers with the ability to pinpoint the causal variant itself rather than relying on genetic markers that merely point to a genomic region harboring these variants. Over the past ten years, numerous sequencing studies involving hundreds to thousands of families have significantly advanced our understanding of the genetic underpinnings of various phenotypes. Notably, this approach has yielded remarkable insights, particularly in the context of childhood disorders that have a substantial impact on an individual's reproductive ability, such as autism spectrum disorder (ASD) and congenital heart disorders (CHDs), in which de novo mutations were confirmed to play a pivotal causal role. However, the complexity and scale of data generated by sequencing large family collections surpass that of traditional genotyping studies. As a result, there is a pressing need to devise specialized methodologies and tools for the efficient management, analysis, and dissemination of this vast and intricate data set.
The development of genomic analysis tools is an actively evolving field. Platforms like Bravo (Taliun et al. 2021), Genebass (Karczewski et al. 2022), and the gnomAD browser (Gudmundsson et al. 2022) provide access to population variants from hundreds of thousands of individuals, including rich annotation features and phenotype associations in Genebass. However, these platforms are primarily designed for predefined data sets; lack the flexibility to create a fresh, customizable instance for personalized analysis; and do not directly support families. The seqr (Pais et al. 2022) platform focuses on the collaborative analysis and annotation of clinical variants and phenotypes in individuals or trios. However, installing seqr on personal computers or local clusters can be challenging, restricting the import of sensitive data. Furthermore, importing large data sets (e.g., hundreds of thousands of individuals) and managing genomic annotations are also difficult. Commercial platforms, such as DNAnexus (https://www.dnanexus.com) and the suite of Qiagen products (https://www.qiagen.com/us), offer flexibility and scalability, enabling users to create new instances for genomic data analysis. These platforms are well suited for large-scale genomic projects, offering robust infrastructure and a broader range of tools for data management, integration, and analysis. However, they often come with a substantial cost for large projects.
Genotypes and Phenotypes in Families (GPF) (https://iossifovlab.com/gpf) is an open-source platform that can handle millions of genetic variants and deep phenotypic data for individuals. GPF allows users to create custom instances, input family-based genotype and phenotype data, annotate variants, and perform comprehensive analysis through an intuitive graphical interface. Additionally, GPF promotes collaboration by enabling users to share projects, making it a unique tool for both personalized and large-scale genomic research.
We developed GPF to manage data related to the Simons Simplex Collection (SSC), a collection comprising about 2800 families, each with one child affected by autism (Fischbach and Lord 2010). The platform proved extensible and flexible enough to accommodate the larger Simons Foundation Powering Autism Research (SPARK) collection, a growing collection of about 200,000 individuals with autism and their families (Feliciano et al. 2019; Zhou et al. 2022), as well as other collections supported by Simons Foundation Autism Research Initiative (SFARI). Our GPF instance, GPF-SFARI (https://gpf.sfari.org), enables researchers to interactively conduct complex queries for variant selection and perform genotype–phenotype association and gene-set variant enrichment analyses using the SSC and SPARK data sets. Because of the sensitive nature of these data sets, users need permission to access the full extent of SSC and SPARK. Still, GPF-SFARI offers public access to summary statistics and analysis tools for all data sets, keeping confidential information about individuals and families secure. In addition, GPF-SFARI has two publicly accessible components for unregistered users: the “sequencing de novo” data set contains published de novo mutations for six developmental disorders in typically developing children; the “gene profiles” set contains summary statistics for all human genes, including the number of variants of various types in the SSC, SPARK, and “sequencing de novo” data sets, as well as gene properties relevant to autism. Although we developed GPF in the context of the SFARI collections, it is designed so that other researchers may create their own GPF instances and analyze their genotypic and phenotypic data using the provided tools and visualizations. We created extensive GPF documentation (https://iossifovlab.com/gpfuserdocs) to support such uses.
Here, we will describe the general features of GPF and the data sets within the GPF-SFARI instance. Then, we will describe the query, enrichment, and phenotype tools GPF provides in the context of GPF-SFARI. Finally, we discuss how users can use GPF to analyze and share their genotypic and phenotypic data.
Results
General features of GPF
The GPF system manages genotypes and phenotypic measures of individuals from collections of families. Genotypes of various types identified through different technologies (whole-exome, whole-genome, etc.) and genotyping tools are imported into the system. Separately, phenotypic measures are imported. The system then provides an intuitive interface for exploring, jointly analyzing, and securely sharing the genotypic and phenotypic data.
GPF is designed to accommodate a diverse range of family structures (Fig. 1A). The basic family type is the nuclear family, with two parents and their children. Nuclear families (especially the trios with two parents and one child) are the main family types used in the study of de novo mutations. GPF also supports complex multigenerational families like the ones frequently used in linkage analysis of Mendelian phenotypes. Additionally, to accommodate the needs of case-control studies, the system supports families composed of single individuals. GPF uses pedigree files to capture the family relationships and basic phenotypic information, such as gender, affected status, and the indication of the individual proband.

General features of GPF. (A) GPF supports various family structures represented as pedigree diagrams, in which parent–child relationships are shown with lines; males and females with squares and circles, respectively; and phenotypes with colors. In this panel, red indicates individuals with autism, and white indicates those without. Shown are a family with a single individual (left), three nuclear families (two parents and their children; middle), and a complex multigenerational family (right). Arrows indicate the probands. (B) GPF supports various genetic variant types represented relative to a reference genome, including substitutions, short insertions and deletions, copy-number variants, and tandem repeats (or microsatellites). (C) The gene view component shows the population (or summary) variants in a gene of interest. The gene view represents the genomic location of the variants on the x-axis in the context of the gene isoforms and the variant frequencies on the y-axis. Stars, triangles, and circles are likely-gene disruption (LGD), missense, and synonymous variants, respectively. Red squares indicate de novo variants. (D) The family variants view, organized into three sections, displays variants that segregate within families. The first section shows the family (the family ID/study column), the variant (the location/variant column), the segregation pattern (the pedigree column), and the variant's predicted effect on the protein-coding genes (the worst effect/genes column). In the segregation patterns, numbers indicate the alternative alleles segregating in the family. For example, an individual with “1” is heterozygous with a first alternative allele and a reference allele, and an individual with “1/1” is homozygous with two alternative alleles. The second section shows the GPF's extensive genomic annotations assigned to each variant. The third section shows relevant phenotypic measures of the probands or their family members.
GPF supports a wide range of variant types, each representing distinct genetic alterations (Fig. 1B). These variant types can be identified through various technologies. For example, single-nucleotide variants (SNVs), indels, and copy-number variants (CNVs) can be detected using methods such as whole-exome sequencing (WES) and whole-genome sequencing (WGS). Single-nucleotide polymorphisms (SNPs) and CNVs can also be identified through array hybridization.
GPF further categorizes alleles based on their inheritance patterns within a family. Mendelian alleles exhibit typical inheritance patterns from parents to offspring. De novo alleles arise in the child without being inherited from either parent. Omission refers to alleles that should logically be passed on to a child according to Mendelian principles but were not.
The easiest way to query variants in GPF is through its gene browser tool, which summarizes information about variants observed in a single gene within a selected data set. Here, population variants for a specific gene are visually represented through the gene view, in which variants are depicted as symbols on a scatter plot, providing information about their genomic locations (x-axis), frequencies (y-axis), and types (symbol shapes). Figure 1C shows the gene view for 185 alleles in CHD8 (as observed in the “SSC WES” data set in the GPF-SFARI; see below), a chromodomain helicase DNA-binding protein associated with autism (Bernier et al. 2014; Cotney et al. 2015). GPF's gene view is interactive, allowing users to select subsets of gene variants by type, location, or frequency and to view the families in which the selected variants segregate in the family variants view.
The family variants view offers insight into the segregation of genetic variants in families. Figure 1D illustrates this by presenting information on four family variants organized into three groups of columns. The first group outlines essential details about the variant, including its familial context, segregation pattern within the family, and a prediction of its impact on protein-coding genes. The second group shows in-depth genomic annotations assigned to each variant during import. These annotations include a range of data, such as variant frequencies in reference populations like gnomAD (Chen et al. 2024), conservation metrics like phyloP (Pollard et al. 2010) and phastCons (Siepel et al. 2005), pathogenicity scores like MPC (Samocha et al. 2017) and CADD (Kircher et al. 2014), and gene intolerance scores like RVIS (Petrovski et al. 2013) and pLI (Lek et al. 2016). The third group comprises phenotypic measures, highlighting GPF's capacity to integrate information about family members’ phenotypes and the segregation of the genetic variant within the family structure.
GPF organizes its data into genotype and phenotype studies. The genotype studies comprise a set of genotypes for individuals from a specific set of families. Similarly, phenotype studies consist of phenotypes from individuals from a set of families. Phenotypes in GPF are further structured into instruments or diagnostic instruments applied to the study's individuals. Each instrument includes a set of specific measures. A genotype study can be linked with a phenotype study, forming a cohesive connection between genetic and phenotypic information. Data sets, in turn, are composite groupings of studies and previously defined data sets. The studies grouped into data sets can contain similar data types for different sets of families or different data types for the same families. This flexible framework enables users to work on individual studies, multiple studies, or all studies in a GPF instance.
We will use the data sets in GPF-SFARI to describe and demonstrate the additional features of the GPF. These genotype and phenotype data sets are built from the largest autism family collections. In the following section, we provide an overview of SFARI and the GPF-SFARI data.
GPF at SFARI
SFARI aims to improve the understanding, diagnosis, and treatment of ASD by supporting and funding innovative scientific research. SFARI's two main initiatives are SSC (Fischbach and Lord 2010) and SPARK (Feliciano et al. 2019; Zhou et al. 2022), the most extensive collection of families with autism.
SSC encompasses roughly 2800 simplex families, each including a single child with autism, the affected child's parents, and, for most families, one or more unaffected siblings. SSC has comprehensive individual phenotypic profiles, especially for the affected probands. The phenotypes are based on the application of diagnostic instruments, including autism diagnostic tools like ADI-R, ADOS, measures of core features of autism and intellectual ability, and family history (Fischbach and Lord 2010). SFARI has funded several large projects to generate array hybridization genotypes and WES and WGS data from all SSC individuals. These data have been used by many research groups to transform the study of autism genetics, demonstrating the large contribution of de novo mutations to autism and establishing a process for identifying autism genes using de novo variants that has produced hundreds of high-confidence genes (Iossifov et al. 2015; Sanders et al. 2015; Turner et al. 2016; Yuen et al. 2017; Satterstrom et al. 2020). Following the success of SSC, SFARI initiated SPARK, a growing collection of self-registered families with autism, which currently has about 100,000 families or approximately 200,000 individuals (Feliciano et al. 2019; Zhou et al. 2022). SPARK includes both multiplex and simplex families, accommodating a wide range of family structures. SFARI has funded the generation of whole exomes from about 120,000 SPARK individuals at Regeneron and whole genomes from an additional about 30,000 individuals at the New York Genome Center (NYGC).
Through its data-sharing policy, SFARI requires researchers to return and deposit their results of genotyping and analysis of sequence data back to SFARI. These returned data include genotypes of various types—CNVs, SNVs, indels, microsatellites, etc.—both de novo and inherited. Part of SFARI's mission is to distribute these data to the scientific community. However, the data are large and complex, and most are sensitive and require protection through strict authorization processes. SFARI uses GPF to share the data, allowing outside (non-SFARI) researchers to explore and analyze the large and complex data sets of genotypes in deeply phenotyped families. The user can also use the genetic and phenotypic data in GPF to select materials distributed by SFARI, such as DNA, cells, or tissues from the individuals in the SSC and SPARK collections. We will refer to the GPF deployed at SFARI as GPF-SFARI (https://gpf.sfari.org/).
A subset of the data in GPF-SFARI is freely accessible to everyone. The rest comprises sensitive genotypic and phenotypic data that require a rigorous authorization process to grant access to qualified researchers. SFARI has implemented such a process through its SFARIBase infrastructure, which governs the access to the sensitive data within GPF and, generally, all the data and materials managed by SFARI. The close integration enables SFARIBase to automatically manage GPF's user accounts and permissions, reflecting the status of researchers’ requests and approvals for genotypic and phenotypic data sets, ensuring consistency and a seamless experience for researchers interacting with both systems. Importantly, although the individual genotypic and phenotypic data in SPARK and SSC are safeguarded, the users of GPF-SFARI can access aggregate and summary statistics for these data.
Figure 2A shows the major data components of the GPF-SFARI. The “sequencing de novo” component is a data set that contains freely accessible lists of de novo variants found in the SSC and SPARK collections, in other collections with autism, and in de novo variants identified in children with five additional developmental disorders (Methods) (see Fig. 2B). The two largest data sets contain the protected genotypic and phenotypic data related to the SSC and SPARK collections (Methods) (see Fig. 2C). The “gene profiles” component integrates freely accessible summary information of the data from the three components, organized by gene (Table 1). The gene profiles also contain external information relevant to studying the genes’ roles in autism.
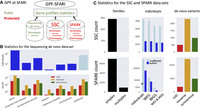
GPF-SFARI. (A) We organized the data in the GPF instance deployed at SFARI (GPF-SFARI) into three large data sets. Two data sets, SSC (Fischbach and Lord 2010) and SPARK (Feliciano et al. 2019; Zhou et al. 2022), are protected under a rigorous authorization process and comprise the de novo and transmitted genotypes and phenotypes from the two large collections of families with autism built by SFARI. The third data set, sequencing de novo, is a collection of publicly available de novo variants identified in individuals diagnosed with one of six disorders (autism, schizophrenia, developmental disorder, intellectual disability, congenital heart disease, and epilepsy) and in typically developing children labeled as unaffected. In addition, GPF-SFARI includes a publicly accessible gene profiles component (see Table 1), displaying autism-relevant information for each human gene, including variant counts across three data sets. (B) The panel shows the number of individuals (top) and the number of de novo coding variants (bottom) included in the sequencing de novo data set separately for each diagnosis. The bottom section displays the counts for three types of de novo coding variants: de novo likely-gene-disrupting (LGD) variants (blue), de novo missense variants (orange), and de novo synonymous variants (green). (C) The panel shows the numbers of families (left), individuals (middle), and de novo coding variants (right) in the SSC (top) and SPARK (bottom) data sets. The number of families is organized by type: simplex (one individual with autism) and multiplex (more than one individual with autism). Individuals are categorized by diagnosis: dark blue for affected (autism) and light blue for unaffected. We present the numbers of individuals with phenotypic data (individuals) and those with genotypes derived from whole-exome sequencing (WES), whole-genome sequencing (WGS), or both (WES & WGS). Finally, the numbers of de novo coding variants are presented separately by the variant effects: LGD, missense, or synonymous.
Gene profiles provide freely accessible summary statistics of the possibly protected data managed by GPF and additional relevant information organized by gene
GPF power tools
Genotype browser
GPF provides users with power tools for interacting with the data. One of these key tools is the genotype browser, which enables users to create and execute intricate queries for genetic variants. Users can search for variants based on the properties of the variants, the properties of the genes that harbor the variants, and the phenotypic properties of individuals carrying the variants (Supplemental Fig. 1). The variant properties that can be used in queries include the variant location, type, homozygous/heterozygous status, genes, predicted effects on the protein-coding genes they affect, and genomic scores such as phyloP, CADD, and MPC assigned to variants during import in GPF.
The properties of genes that can be included in a query are of two types: gene-set membership and gene scores. Gene sets are a handy abstraction in many biological contexts, including genes in a pathway, the targets of a transcription factor or RNA-binding protein, and genes containing a given protein domain. Relevant collections of gene sets can be imported into GPF. For example, in GPF-SFARI, we have imported the GO Ontology (The Gene Ontology Consortium 2000), MSigDB (Subramanian et al. 2005; Liberzon et al. 2011), sets of published lists of autism genes (autism gene sets), and gene sets implicated in autism etiology (relevant gene sets) (e.g., chromatin modifier targets and embryonically expressed genes). Each of the imported gene sets has a name, and users can select gene sets by name to formulate a query like “search for variants affecting genes involved in apoptosis (a pathway from MSigDB) or genes implicated in autism.” GPF provides two alternatives to selecting a predefined gene set: The users can provide their gene set as a list of genes, or they use the GPF genetic variants to define a gene set, such as the gene affected by de novo LGDs in a GPF data set. Gene scores are numerical values assigned to genes that describe properties such as gene length and degree of intolerance to damaging mutations. The relevant gene scores, obtained from external sources and imported into GPF, can be used to formulate queries like “find genetic variants that affect genes with RVIS intolerance score larger than 4.”
Finally, users can search for variants based on the phenotypic properties of the individuals carrying the variants. The phenotypic properties include diagnosis, gender, and the measures of instruments in the phenotypic studies.
Recessive variants can be identified by querying for homozygous variants. GPF currently does not support compound heterozygosity, a feature we plan to implement (see Discussion). Searching for de novo or ultrarare variants automatically implies a search for dominant-acting variants. GPF does not have explicit support for common dominant variants, but this can still be explored through postprocessing of the variants downloaded from GPF. The downloaded files conveniently include the genotypes for all individuals in the family, enabling users to analyze segregation patterns associated with dominant variants.
Figure 3A presents the results of a complex query through the genotype browser, highlighting four of the 102 de novo LGD variants in the SSC affected children within FMRP target genes. The FMRP targets are the genes that encode mRNAs targeted by the fragile X mental retardation protein (Darnell et al. 2011). Users may download the selected variants in a comma-separated file format for further analysis beyond the GPF platform. These downloaded files include the genotypes of all family members, along with a comprehensive set of annotation attributes for all variants.

Query and statistical tools at GPF. (A) The genotype browser enables users to construct complex queries that involve the properties of genetic variants, the properties of affected genes, and the phenotypic properties of carrier individuals (Supplemental Fig. 1). Here, we partially present the results of a query for de novo LGD variants that fall within the FMRP target gene set and occur in children diagnosed with autism (for a description of the columns, see Fig. 1D). (B) The enrichment tool (Supplemental Fig. 2) enables users to test for enrichment of de novo variants within a gene set. The enrichment tool results for de novo mutations are shown in children with autism and unaffected children from the autism-related subset of the sequencing de novo “SD autism” in the FMRP target gene set. The top table shows that among 21,795 individuals with autism, 3421 de novo LGD mutations are found (N). Of these, 586 fall in FMRP target genes (O), although we expect to see only 347 (E). This corresponds to an enrichment with a significant P-value (pV), indicated with a red background. Missense mutations are similarly enriched in FMRP target genes. However, synonymous mutations are not enriched (white background). The bottom table shows the same analysis for unaffected individuals, which finds no enrichment for LGD, missense, or synonymous mutations. (C) The phenotype browser allows users to explore the available phenotypic data associated with a data set and download the subsets of interest. The panel shows a part of the search results for phenotypic measures related to “communication” in the SSC data set. The results are presented in a table, with one row per measure. For each measure, the table includes histograms of its values, separately in groups of individuals by gender, role, and affected status. The zoomed-in view (a feature provided by GPF) of the histograms plot for the communications_standard measure from vineland_ii shows the number of individuals for which the measure is available for the affected probands and the unaffected siblings separately by males and females and the histograms of the four groups (male and female probands and male and female siblings). The histograms show that the affected probands have diminished vineland_ii communication scores. The result table also displays the scatter plots and regression lines for each of the four measures against the individuals’ ages at assessment and their IQs. (D) The phenotype tool enables users to test whether a given phenotypic measure (e.g., nonverbal IQ) differs between SSC children who carry a specified type of genetic variant (e.g., de novo LGDs) and those who do not have such variants (Supplemental Fig. 3). The panel illustrates the impact of four de novo variant types (LGD; missense; CNV+, or large duplications; and CNV−, or large deletions) in genes with a low pLI rank (less than 1000) on nonverbal IQ. For each de novo variant type, the mean and 95% confidence interval are plotted for four groups of individuals: males and females with and without a de novo variant in the selected genes. There is a significant decrease in nonverbal IQ for affected children with de novo LGDs or CNV− within the genes with a low pLI rank. There is no effect of CNV+ mutations, and only a marginal effect is observed for missense variants. We note that panels B and D are stylized versions of content from GPF-SFARI, with reduced white space for better clarity. Supplemental Figures 2 and 3, respectively, illustrate the result representation generated by GPF.
Enrichment tool
The enrichment tool allows the user to test if a given set of genes is affected by more or fewer de novo mutations in the children in the data set than expected. We and others have used such an approach to demonstrate a functional convergence of de novo mutations in autism. For example, prior studies show that damaging de novo mutations in autistic children target synaptic genes and genes encoding chromatin modifiers (Neale et al. 2012; O'Roak et al. 2012a; Iossifov et al. 2014). The approach has also been used to demonstrate that the de novo mutations in autism target similar genes as the de novo mutations in schizophrenia, intellectual disability, and epilepsy (Iossifov et al. 2014). Users can use the enrichment tool to explore their research-driven hypotheses using the extensive genetic data managed by GPF-SFARI. Importantly, because the enrichment tool uses aggregate measures of genotypic data (i.e., the number of mutations in a gene or gene set), it is available for protected data sets as well, without the need to register for data access.
To use the enrichment tool, a user must choose a set of genes either by selecting one of the gene sets that have already been loaded in GPF or by providing their own gene set. In addition, the user must select the data set that contains the de novo variants to be used in the enrichment analysis. Finally, the user must select among the background models that GPF uses to compute the expected number of de novo mutations within the given data set. GPF supports several background models, including mutability models based on sequence context (Samocha et al. 2014) and models based on gene length or the number of variants of no consequence (e.g., de novo or rare synonymous; see GPF documentation) (Supplemental Fig. 2).
Figure 3B shows an example output for the enrichment tool using the FMRP targets identified by Darnell et al. (2011) and the de novo variants from the autism-related subset of the sequencing de novo (SD autism), reproducing a result we previously reported (Iossifov et al. 2012). The de novo LGDs and missense mutations from autistic children are significantly enriched in FMRP genes. In contrast, synonymous mutations are not enriched, and none of these variant types are enriched in unaffected children. These observations suggest an etiological role of the FMRP gene set in autism.
Phenotype browser
The phenotypic data within GPF is organized by instruments, or diagnostic tools applied to the subset of the data set's individuals. Each instrument includes several measures. The value of a measure can be a number, for example, for measures of IQ or height, a yes or no answer to a question, or a free-text form. Using GPF, users can find the list of instruments in the data set, view all measures for each instrument, and search for measures based on their names and descriptions. GPF provides summary-level information for each measure, including the type of its values and histograms for those values, as well as a mechanism to download the values for each individual for one or more measures of interest.
The phenotype browser enables the exploration, search, and download of available phenotypic data across various data sets. Users can easily view the available instruments for a data set and the phenotypic measures associated with each instrument. They can also search for measures across all instruments using their names and descriptions. The phenotype browser shows the high-level statistics for each measure, including the number of individuals with values for the measure (or the number of individuals for which the instrument has been applied) and histograms of the measures’ values, separately for different roles (e.g., proband, mother, sibling), affected statuses, and genders. It can also provide correlations of all measures with a set of core phenotypic properties. For example, the phenotype browser for the SSC data set displays the correlations between all measures and the individual's age and IQ. Figure 3C shows the high-level statistics for four measures related to “communication” in the SSC data set. The phenotype browser can be used to get an overview of the phenotype instruments in both public and protected data sets. Registration is required to download individual phenotype data in protected data sets.
Phenotype tool
The phenotype tool enables users to explore associations between genotypes and phenotypes, harnessing the wealth of integrated data from both domains (Fig. 3D). Its operation is straightforward: Users select a specific phenotypic attribute and choose a type of genetic variant based on frequency and the effect on target genes. Subsequently, the tool assesses whether individuals carrying such a variant exhibit statistically significant differences in the chosen phenotypic attribute compared with individuals lacking that variant (Supplemental Fig. 3). This tool has played a pivotal role in our research and published findings. For instance, we showed that children with autism who harbor detrimental de novo mutations tend to display lower IQ levels (O'Roak et al. 2012b; Iossifov et al. 2014) and impaired motor skills in comparison to their counterparts without such variants (Bishop et al. 2017; Buja et al. 2018). Because the phenotype tool utilizes aggregate data measures (i.e., the number of individuals with a specific genotype or phenotype), it is available for both public and protected data sets, without requiring registration for data access.
GPF “at home”
GPF is an open-source project with comprehensive documentation (https://iossifovlab.com/gpfuserdocs) that others can easily install and use to manage their genotypic and phenotypic data. Users can seamlessly browse, analyze, and securely share their data with colleagues and the broader scientific community (Fig. 4).
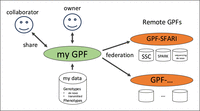
GPF at home. Users can instantiate their own GPF (my GPF) to manage and analyze their data. Through GPF, they may also share their data with other researchers. Finally, users may connect to other GPF instances (e.g., GPF-SFARI) using the federation feature and perform joint queries and analysis of their data and the data in the remote instances.
Resource requirements in GPF depend on the task, data set size, and number of genetic variant annotations. Small data sets (up to variants from exome sequencing of a couple of hundred families) can be handled on a modern laptop. In contrast, larger data sets, such as those from WGS of more than a handful of individuals or exome sequencing of thousands of samples, require high-performance computing clusters or large-scale hosts for efficient data import and analysis. For example, importing the SPARK Consortium iWGS v1.1 data set comprising about 13,000 individuals takes about a day on 300 cores of a computation cluster, and interactivity is ensured through hosting two GPF production instances on a cluster of three nodes, each equipped with 8 CPU cores, 32 GB of RAM, and 15 TB of disk space.
To help new users get started with GPF, we added a “getting started guide” to GPF's documentation. In the guide, we provided step-by-step instructions on how to install the GPF system, configure a new empty GPF instance, import genotypic and phenotypic data using both small mock example data sets and real data sets available as supplemental data in published manuscripts, and query the newly imported data. We also provided instructions on how to add custom genomic annotations, connect genotypic and phenotypic data, configure the genotype browser views, utilize the phenotype and enrichment tools on the newly imported data, and configure the gene profiles component that provides gene-level statistics for the data in the new GPF.
The “federated access” feature extends GPF's capabilities by allowing one GPF instance to connect with one or more additional GPF instances. Users can jointly analyze the data in multiple GPF instances through federated access. A typical scenario involves a user configuring a GPF instance to work with their private data while accessing a public GPF instance, such as the GPF-SFARI, which hosts relevant data sets and resources. Once the necessary authorization is obtained for remote GPF instance data, establishing federated access within the local GPF instance is straightforward. In the getting started guide, we provided simple instructions on connecting the new GPF instance with the public GPF-SFARI using the GPF's federated access feature and performing a simple joint analysis across the two GPF instances.
Finally, we added instructions on how to configure a permanent GPF instance that can be used to securely share data in the “publicly accessible GPF” section of the GPF instructions. We deployed the instance built by the end of the “getting started guide” as a publicly accessible GPF instance at https://demo.iossifovlab.com/gpf.
Discussion
Here, we described our platform, GPF, which offers tools for exploring and analyzing genetic and phenotypic variation in families. GPF accommodates the genetic variant and family pedigree types typically used in genetic analysis and enables users to browse and select variants using an extensive array of variant properties and phenotypic measures of the carrier individuals. The GPF power tools allow the user to conduct enrichment and phenotypic association analysis. As a flexible and scalable platform capable of handling genotypic and phenotypic data from millions of individuals, GPF can be applied to a large class of genetic study types.
We used GPF on the extensive autism genetic and phenotypic data collected by SFARI, corresponding to about 9000 and about 200,000 individuals in the SSC and SPARK collections. The resulting variant collections, as well as the associated phenotypic measures, are accessible through our GPF instance, GPF-SFARI. Although these two data sets are accessible only after registration, a third one, sequencing de novo, which includes de novo variants from studies of six developmental disorders, is publicly available at GPF-SFARI. GPF-SFARI also provides a freely accessible collection, gene profiles, of statistics for each gene regarding the number of genetic variants of different types in the three data sets without any connection to sensitive individual-level data. The powerful interactive query interface and the analysis tools of GPF enable researchers to use the rich data set of genotypic and phenotypic variation in GPF-SFARI easily and effectively.
SFARI support has been instrumental in the development of GPF. SFARI is committed to supporting the further maintenance and development of GPF. Additionally, as an open-source platform, GPF enables community-driven contributions, supporting its sustainability and further development. We are actively developing new features to enhance GPF's functionality and address its limitations. These include the ability to store phasing data, which will improve the understanding of haplotype structures and implement queries for compound heterozygosity. Similarly, we will implement a general approach to store genotype quality metrics and to query based on them. We are also working on the option to download VCF files, providing users with more flexibility to operate on common variants and to improve the interoperability of GPF. Additionally, we plan to integrate RNA sequencing data measures, such as RPKM, allele-specific expression, and splicing, to enable more comprehensive analysis of gene expression alongside genomic variants and phenotypes. To simplify the user experience, we are developing a web-based system for easier GPF instance management, reducing the need for technical expertise in setup. For example, we will allow the import of genotype-, phenotype-, and RNA-associated data and configure the genomic annotations and the user interface through the new management interface. Finally, the federation system will be expanded to enable users to interact with multiple GPF instances without needing to install their own, improving collaboration and accessibility.
Methods
Architecture and implementation
The structure of GPF comprises three stacked layers (Supplemental Fig. 4). The system's core is the data access environment (DAE) layer. The DAE is implemented in Python 3 and provides access to all underlying genotypic and phenotypic data, as well as all relevant genomic information (e.g., the reference genome, gene, and variant properties). It also includes the set of authorized users and their access privileges (represented as groups), as well as analysis tools.
The phenotypic and genotypic data are stored in phenotype and genotype storages, respectively. The phenotype storage is implemented as a relational data schema that can be managed by various relational database systems, like DuckDB (https://duckdb.org) or SQLite (https://www.sqlite.org). We based our genotype storage on the modern columnar database organization to enable interactive access even for a genotyping data set over millions of individuals. GPF's genotype storages can be deployed on different frameworks. The DuckDB (https://duckdb.org) genotype storage is suitable for small- to intermediate-sized data sets and requires no management. The Impala (https://impala.apache.org) genotype storage is a distributed server environment that can handle any data size and can be set up on an existing computational infrastructure. The BigQuery (https://cloud.google.com/bigquery) genotype storage operates on Google's cloud infrastructure, requires minimal management, and can handle any data size. The BigQuery's drawback is that it may prove expensive for systems with high loads.
The second layer in the GPF is the WDAE REST API. It is implemented using Python's Django framework (https://www.djangoproject.com). WDAE enables remote scripts to access the public or protected data and analysis tools provided by the DAE, as documented in the WDAE Interface section of the GPF documentation. The last layer is the GPF's user web interface. It is implemented on top of the WDAE layer, utilizing the Angular web framework. When publicly deployed, GPF uses HTTPS to ensure the encryption of input and output data streams, which is essential for secure data handling.
The three layers enable users to interact with GPF in different ways. The GPF web interface enables all users to access the data interactively. Everyone can also download the relevant data for further analysis, typically in a text table format. Users with a computational background can also create scripts to automate the interaction with the system. For example, if a researcher uses Python and has access to the computer at which GPF is deployed, they can create Python scripts to access the data through the DAE API. Alternatively, a researcher who prefers other programming languages or does not have access to the GPF's host can still create scripts accessing the GPF's data and tools through the WDAE interface.
Access management
We designed a flexible authorization system based on groups, in which both users and data sets are assigned to specific groups, allowing users to interact only with data sets within their assigned groups. We developed an intuitive management interface to control user and data set assignments to groups. The authorization system is flexible, supporting various access policies, ranging from freely accessible data to the SFARI's internal project-based access policies. The interface and the underlying authorization system are detailed in the user management section of the GPF documentation.
Annotation infrastructure
Genotypic data analyses rely on examining the genotypes in the context of the vast amounts of genomic data generated in recent decades. To facilitate joint analysis of multiple data sets, GPF's import process annotates all the variants uniformly across all imported studies with a user-specified list of genomic properties defined in an annotation pipeline (see the Annotation Infrastructure section of the GPF documentation). The source for these properties is one or more genomic resource repositories (GRRs). We have compiled a sizeable GRR and made it accessible at https://grr.iossifovlab.com. Our repository comprises publicly available resources including population frequencies from gnomAD (Gudmundsson et al. 2022); conservation scores like CADD (Kircher et al. 2014), phyloP (Pollard et al. 2010), and phastCons (Siepel et al. 2005); and missense scores like MPC (Samocha et al. 2017). Each resource within the repository is assigned a resource identifier. GPF users only need to specify the list of resource identifiers with which they wish to annotate the GPF instance variants. GPF's GRR infrastructure is flexible and allows users to configure one or more private GRRs, including other public or private resources, and annotate their variants with the additional resources. GRR supports standard formats for most resource types, such as FASTA for reference genomes, VCF for variant frequencies and properties, BED and bigWig for position scores, and simple columnar text files. The structure and maintenance of a GRR, along with detailed descriptions of acceptable file formats, are outlined in the “Genomic resources and resource repositories” section of the GPF documentation. We also prepared an extensive demo to help users get started with creating their own GRR (https://github.com/iossifovlab/mini_grr).
Importantly, GPF also has a new reannotation feature, which enables users to update the genomic annotations of all data without the computationally expensive process of reimporting them. When users update the GPF instance annotation pipeline, by adding or removing annotation attributes or upgrading the versions of some of the used resources, GPF will automatically and efficiently reannotate the data within the instance. This workflow is demonstrated in the Getting Started guide. The reannotation feature allows annotations to be kept up to date, even for large GPF instances.
The annotation pipeline used by GPF-SFARI is included in our public GRR as a resource with the ID “pipeline/GPF-SFARI_annotation.” The pipeline resource provides an extensive and comprehensive description of the genomic resources it utilizes. Specifically, we used hg38 as the genome and RefSeq as the gene model for variant effect annotations (e.g., missense, synonymous). The annotations used in GPF-SFARI include phyloP, phastCons, FitCons, Linsight, CADD, MPC, and gnomAD v2.1.1 liftover and v3 allele frequencies. Except for MPC, all the above cover both the coding and the noncoding genomic regions.
Import and import cluster
DAE supports importing data in standard file formats when appropriate, for example, pedigree (PED) files to describe families. When the standard formats were unavailable or could not meet our requirements, we defined our custom formats, like the file format for phenotypic data.
GPF can import genotypes from two main file formats: the standard VCF format (Danecek et al. 2011) and a simple list of variants typically used for importing de novo variants. The GPF's import functions are flexible and can directly handle a variety of input file configurations. For example, researchers can equally easily import a single VCF file with all of their data; multiple VCF files, each containing genotypes for a human chromosome; or multiple VCF files, each representing genotypes for a group of families.
GPF does not currently support dynamic or built-in quality filtering of variant/genotype data. Filtering is expected to be performed externally prior to importing data sets into GPF. Users can import multiple versions of the same data set with different filters applied, allowing for flexibility in the analysis. In GPF-SFARI, specific filters have been applied to some data sets, and the details of these filters are included in the corresponding data set descriptions.
The phenotypic data are imported from a set of simple files (one for each instrument) organized as a table with columns representing the instrument's measures and with lines representing the individual to which the instrument has been applied.
Importing a large data set in a GPF instance is slow, particularly if the user has requested many annotation properties. GPF allows users to configure an “import cluster” that they can use to parallelize and thus substantially speed up the import process. The “import cluster” can be configured to run on multiple cores of one host, a local computation cluster like clusters controlled by OGE (https://www.oracle.com/technetwork/oem/host-server-mgmt/twp-gridengine-overview-167117.pdf) or Slurm (https://slurm.schedmd.com/), or a cloud Kubernetes (https://kubernetes.io) cluster.
GPF-SFARI components
The “sequencing de novo” is a comprehensive resource that compiles lists of de novo substitutions and short indels associated with six distinct abnormal development disorders, as well as de novo variants found in typically developing children. The data set predominantly comprises data from published research articles, providing a valuable reference for genetic research. Additionally, it incorporates de novo variants obtained from the SPARK collection, some of which are yet to be formally published, expanding the breadth of genetic insights available for analysis. Figure 2B shows the number of children and de novo LGD, missense, and synonymous variants included in “sequencing de novo” for each disorder. Some of the studies included in “sequencing de novo” are based on WGS data and contain noncoding de novo variants. The “sequencing de novo” description in GPF-SFARI lists the publications used to build the data set.
SSC and its large sequence data have been around for a while and have been extensively studied. Many groups, including us, have applied different tools to generate genotypes of various types. These genotypes, aggregated and imported in GPF-SFARI, include de novo and transmitted substitutions and indels from whole-exome (Iossifov et al. 2012; Krumm et al. 2012; O'Roak et al. 2012a,b; Sanders et al. 2012; Iossifov et al. 2014) and whole-genome (Turner et al. 2016, 2017; An et al. 2018; Yoon et al. 2021) data sets, de novo CNVs called from hybridization array (Levy et al. 2011; Sanders et al. 2015) and from whole-genome data (Yoon et al. 2021), and microsatellites called from whole genomes (Mitra et al. 2021). The SSC data set in GPF-SFARI also contains the complete phenotypic data associated with SSC, comprising about 100 phenotypic instruments or about 10,000 individual measures.
SPARK is a more recent and growing collection, so its processing and analysis are ongoing and incomplete. To date, about 100,000 families, comprising around 200,000 individuals, have self-registered in SPARK; however, sequence data (whole exome and whole genome) have been generated from only a subset (see Fig. 2C). Genotypes have been generated and imported into GPF-SFARI for an even smaller subset (Feliciano et al. 2019; Zhou et al. 2022). The major group that produces genotypes from the WES data is the SPARK Consortium, which includes SFARI's bioinformatics team. The SPARK Consortium generates updates of the SPARK genotypes regularly, and currently, GPF-SFARI includes transmitted and de novo substitutions and indels from about 106,000 individuals. In addition, the SPARK data set contains about 400,000 and about 3,000,000 transmitted LGD and missense variants. NYGC performs genotyping from the whole-genome data in batches, and GPF-SFARI includes transmitted substitutions and indels from about 10,000 individuals from the first large batch of whole genomes.
Gene profiles provides a comprehensive list of variant counts with various effects for all genes, along with additional relevant gene properties (Table 1). For instance, within the “gene profiles” on GPF-SFARI, you can find counts of de novo likely gene-disrupting (LGD), missense, and synonymous variants, as well as the number of transmitted variants involving de novo LGD variants from both SSC and the continuously expanding SPARK collection. Furthermore, the tool indicates whether genes have been identified as autism genes in multiple publications and whether they belong to relevant gene sets, such as FMRP target genes, CHD8 targets, or chromatin modifiers. Additionally, GPF-SFARI's gene profiles presents various measures of gene intolerance, including RVIS (Petrovski et al. 2013) and pLI (Lek et al. 2016) scores.
Software availability
The source code for GPF is freely accessible at GitHub (https://github.com/iossifovlab/gpf). The system's latest release at the time of submission is available as Supplemental Code.
Data access
The sequencing de novo data set, as well as the gene profiles and aggregated statistics from the SSC and SPARK data sets, are publicly available. Individual genotypes and phenotypes from the SPARK and SSC data sets are available with permission from SFARI. Instructions on requesting permission are included on the GPF-SFARI's about page at https://gpf.sfari.org/hg38/about.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the Simons Center for Quantitative Biology at Cold Spring Harbor Laboratory; SFARI Grants SF497800, SF529232, SF677963, SF666590 (to I. Iossifov); and the Centers for Common Disease Genomics grant UM1 HG008901. The Centers for Common Disease Genomics are funded by the National Human Genome Research Institute and the National Heart, Lung, and Blood Institute.
Author contributions: L.C., M.C., C.F., and I. Iossifov designed the GPF; L.C., M.C., K.S., C.F., N.D., S. Mladenov, I.T., I. Ivanov, S.X., S.F., Y.L., B.Y., S. Marks, A.M., and I. Iossifov implemented the GPF; L.C., M.C., Z.W., A.L., N.V., and I. Iossifov wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280356.124.
-
Freely available online through the Genome Research Open Access option.
- Received December 13, 2024.
- Accepted July 31, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








