
High-quality sika deer omics data and integrative analysis reveal genic and cellular regulation of antler regeneration
- Zihe Li1,11,
- Ziyu Xu2,3,11,
- Lei Zhu4,5,11,
- Tao Qin1,11,
- Jinrui Ma1,11,
- Zhanying Feng2,6,11,
- Huishan Yue1,
- Qing Guan7,
- Botong Zhou1,
- Ge Han1,
- Guokun Zhang8,
- Chunyi Li8,
- Shuaijun Jia4,5,
- Qiang Qiu1,
- Dingjun Hao4,5,
- Yong Wang2,3,9,10 and
- Wen Wang1,7
- 1New Cornerstone Science Laboratory, Shaanxi Key Laboratory of Qinling Ecological Intelligent Monitoring and Protection, School of Ecology and Environment, Northwestern Polytechnical University, Xi'an 710072, China;
- 2CEMS, NCMIS, HCMS, MADIS, Academy of Mathematics and Systems Science, Chinese Academy of Sciences, Beijing 100190, China;
- 3School of Mathematics, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Beijing 100049, China;
- 4Department of Spine Surgery, Honghui Hospital, Xi'an Jiaotong University, Xi'an, Shaanxi 710054, China;
- 5Shaanxi Key Laboratory of Spine Bionic Treatment, Xi'an, Shaanxi 710054, China;
- 6Department of Statistics, Department of Biomedical Data Science, Bio-X Program, Stanford University, Stanford, California 94305, USA;
- 7Key Laboratory of Genetic Evolution & Animal Models, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650223, China;
- 8Institute of Antler Science and Product Technology, Changchun Sci-Tech University, 130600 Changchun, China;
- 9Key Laboratory of Systems Health Science of Zhejiang Province, School of Life Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou, 310024, China;
- 10Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming 650223, China
-
↵11 These authors contributed equally to this work.
Abstract
The antler is the only organ that can fully regenerate annually in mammals. However, the regulatory pattern and mechanism of gene expression and cell differentiation during this process remain largely unknown. Here, we obtain comprehensive assembly and gene annotation of the sika deer (Cervus nippon) genome. We construct, together with large-scale chromatin accessibility and gene expression data, gene regulatory networks involved in antler regeneration, identifying four transcription factors, MYC, KLF4, NFE2L2, and JDP2, with high regulatory activity across the whole regeneration process. Comparative studies and luciferase reporter assay suggest the MYC expression driven by a cervid-specific regulatory element might be important for antler regenerative ability. We further develop a model called combinatorial TF Oriented Program (cTOP), which integrates single-cell data with bulk regulatory networks and find PRDM1, FOSL1, BACH1, and NFATC1 as potential pivotal factors in antler stem cell activation and osteogenic differentiation. Additionally, we uncover interactions within and between cell programs and pathways during the regeneration process. These findings provide insights into the gene and cell regulatory mechanisms of antler regeneration, particularly in stem cell activation and differentiation.
The cyclic renewal of deer antlers, the only known mammal organ that can fully regenerate annually, has long captivated numerous biologists (Li et al. 2014). Each spring, deer shed their hard antlers, and the pedicle scar heals as new bone and cartilage regenerate from the pedicle periosteum (PP). During late spring and early summer, antlers grow and calcify over 3 to 4 months at a rate of 2.75 cm per day, potentially reaching a weight of up to 15 kg by the end of the growth season (Price et al. 2005; Landete-Castillejos et al. 2019). In autumn, antlers rapidly calcify and shed their enveloping velvet skin, leaving an exposed bony surface. The cycle concludes with the casting of the antlers in spring, initiating a new regeneration phase. Researchers have discovered that antler regeneration is driven by stem cells located in the antler PP (Wang et al. 2019a). Casting of the PP results in failure of regeneration. Several somatic stem cell markers such as NT5E, THY1, and ENG have been found expressed in antler stem cells (Dong et al. 2020). Some researchers found embryonic stem cell factors such as MYC, KLF4, and POU5F1 expressed in the regenerative antler stem cells (Dąbrowska et al. 2016), whereas a single-cell transcriptome study suggested that these factors were specific to antlerogenic stem cells (Ba et al. 2022). Our recent work further unveiled the cell atlas of antler regeneration and elucidated the vital role of antler blastema progenitor cells (ABPCs) differentiated from PRRX1+ mesenchymal stem cells (PMCs) in the bone regeneration of sika deer antler (Qin et al. 2023). This study also demonstrated the involvement of early developmental pathways, including the WNT, TGFB, and FGF pathways, during antler regeneration. Despite these advancements, a comprehensive understanding of the complex gene and cell regulatory dynamics in deer antler regeneration remains elusive.
Gene regulatory networks (GRNs) constructed from transcriptomic and epigenomic data have been widely used to explain complex biological processes such as development and regeneration (Johnston et al. 2021; Jimenez et al. 2022). Recent studies on retinal regeneration have identified key GRNs and regulatory factors, such as NFIA/B, which constrain mammalian retinal regeneration (Hoang et al. 2020). However, most of these studies are limited to model species because of the lack of necessary data resources, including high-quality genomic data in nonmodel organisms. The quality of genomic data significantly impacts multiomic studies (Chisanga et al. 2022). Among Cervidae, the most comprehensive genomic data so far is for red deer (Cervus elaphus) (Pemberton et al. 2021), sequenced and assembled by the Darwin Tree of Life Project and annotated using the latest RefSeq pipelines (Pruitt et al. 2014; Blaxter 2022). However, antler regeneration related data are limited and difficult to obtain for red deer in contrast to sika deer (Cervus nippon), as many herds of sika deer are reared for antler medicine production in China (McCullough et al. 2009). Therefore, the sika deer could serve as a valuable model for mammalian regeneration research owing to the ease of sampling. Several sika deer genome assemblies derived from short-read or long-read sequencing are available (Chen et al. 2019; Han et al. 2022; Qin et al. 2023; Xing et al. 2023). Most of these studies focused on evolutionary analysis, revealing the genetic basis of unique traits such as high-tannin diet adaptation, rapid antler growth, and cancer resistance in sika deer. These resources have been utilized in transcriptomic research on the deer antler (Zhang et al. 2022). However, comprehensive genomic data, including high-quality genome assembly and annotation for sika deer, remain lacking. This gap, combined with the challenges of determining regulatory relationships between regulatory elements (REs) and target genes (TGs) using standard ATAC-seq and histone ChIP-seq methods, have greatly hindered efforts to unravel the gene and cell regulatory mechanisms underlying antler regeneration.
Recently, we developed paired gene expression and chromatin accessibility (PECA2) to more accurately infer regulatory relationships by integrating paired chromatin accessibility data and gene expression data in the mouse and humans (Duren et al. 2017). However, this approach relies on extensive paired chromatin accessibility and gene expression data, which are currently unavailable for deer species. In this work, we aim to generate comprehensive omics data for sika deer, including genomic, transcriptomic, and chromatin accessibility data. By leveraging high-quality data and newly developed methods, we seek to gain insights into the multiscaled regulatory dynamics of antler regeneration.
Results
Genome sequencing, assembly, and annotation for sika deer
We first conducted flow cytometry to estimate the sika deer genome size as 3.5 Gb (Supplemental Fig. S1A–C). Next, we employed multiple sequencing technologies to achieve a high-quality and highly contiguous genome assembly (Fig. 1A). Initially, we obtained 91.52 Gb (31×) of Pacific Biosciences (PacBio) HiFi reads and 360 Gb (120×) of Hi-C BGI reads (Supplemental Table S1) from a male sika deer. We utilized Hifiasm (Cheng et al. 2022) with these HiFi and Hi-C data, as well as Oxford Nanopore Technologies (ONT) long reads (N50 > 38 kb) from another deer in a previous study (Qin et al. 2023), to generate well-phased contig assemblies. Finally, we obtained two haplotype assemblies with sizes of 3.1 Gb and 3.0 Gb, respectively. We then employed a two-step haplotype-aware scaffolding strategy to finely phase and scaffold chromosomes for each haplotype using the Hi-C data (for details, see Methods). Finally, we anchored the contigs to 66 phased pseudochromosomes (32 × 2 + X, Y) (Fig. 1B; Supplemental Table S2). Compared with previous assemblies for sika deer (Han et al. 2022; Xing et al. 2023), our assembled haplotype sizes were much larger and closer to the estimated size. The majority of the additional assembly originated from satellite-rich (47.48%) heterochromatin sequences (Supplemental Table S2). The euchromatic contigs scaffolded to pseudochromosomes have a total size of ∼2.6 Gb, similar to other ruminant assemblies. Additionally, we found that the X and Y Chromosomes in the previous genome assembly lacked the attachment of the pseudoautosomal region (PAR) (Supplemental Fig. S1D,E). In our assembly, the PAR region was fully attached to the X and Y Chromosomes and showed good collinearity with those of other mammals (Supplemental Fig. S1F). Compared with the cow, the distal end of PAR in sika deer extended to SHROOM2, which is more similar to the pig (Liu et al. 2019a). Seventeen chromosomes contain no more than three contigs, indicating high continuity. Other quality metrics of our assembly further demonstrated the high quality of our data (Supplemental Table S2).
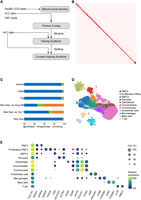
Assembly and annotation of the sika deer genome. (A) Workflow of phased genome assembly of sika deer. (B) Hi-C interaction heatmap of phased pseudochromosomes of the sika deer genome. (C) Bar plot of BUSCO evaluation of published genome annotations of sika deer and Cetartiodactyla species in RefSeq. (D) UMAP of cell atlas of antler regeneration based on our genome assembly and annotation. (E) Dot plot of cell marker gene expression profiles in each cell type. Dot size represents the proportion of cells expressing gene in a cell type.
We annotated 42.80% of repetitive sequences in the genome of sika deer. Notably, the proportion of satellite sequences (9.96%) was much higher compared with the previous (0.07%–1.53%) (Supplemental Table S3) sika deer genome assemblies (Han et al. 2022; Xing et al. 2023). Utilizing a range of methods, including RNA-seq and Iso-Seq transcriptome data (Supplemental Table S4), gene projection based on genome alignment, protein homology annotation, and de novo annotation, we annotated a total of 22,119 protein-coding genes, including 13 mitochondrial genes. The number of genes annotated was comparable to the RefSeq gene annotation of red deer. Functional annotations were obtained for 20,715 of these genes. The BUSCO completeness score of 99.5% is comparable with all previous sika deer annotations (Han et al. 2022; Qin et al. 2023; Xing et al. 2023) and annotations from published Cetartiodactyla reference genomes such as those of cattle, vaquita, and pig (Fig. 1C; Warr et al. 2020; Morin et al. 2021; Talenti et al. 2022). Moreover, we identified 35 genes in our annotation that spanned >1 Mb, a rarity in previous annotations owing to the splitting strategy. Finally, by integrating short-read and long-read transcriptomic data, we annotated 5′ UTR for 14,979 genes and 3′ UTR for 15,760 genes.
By utilizing our assembly and annotation to reanalyze previous single-cell data, we found a substantial increase in the number of detected cells and genes compared with our earlier version (Supplemental Fig. S2; Supplemental Table S5; Qin et al. 2023). Furthermore, the integration of single-cell quality control using mitochondrial gene data resulted in a more precise cell atlas (Fig. 1D,E). During our quality-control process, fibroblast populations with unknown functions identified in our previous study (Qin et al. 2023) were excluded owing to their association with low-quality cells characterized by high mitochondrial gene content and low read counts. We also successfully annotated T cells and mast cells for the first time in the context of antler regeneration. Furthermore, we discovered a proliferative subgroup of PMC (pPMC) that abundantly appeared at 5 days after casting (dac5), expressing both TNN (ABPCs marker) and ACTA2 (pericytes marker). This suggests that these cells are activated stem cells with the potential for both osteochondrogenesis and angiogenesis. In other words, the pPMCs may represent the intermediate stem cells from the resting cell to the osteochondrogenesis ABPCs and angiogenesis pericytes.
Chromatin accessibility landscapes and gene regulation networks for organs of sika deer
To construct regulatory networks involved in antler regeneration, we collected diverse samples to train the PECA2 model. In total, we performed RNA-seq and ATAC-seq sequencing for a total of 32 samples including two replicates for the lung, spleen, liver, adipose tissue, muscle, skin, rumen, and antler PP at four distinct stages of antler regeneration (Fig. 2A; Supplemental Tables S4, S6). The PP samples were the same as those used in previously published single-cell studies on antler regeneration, which encompassed critical stages of antler regeneration. The ATAC-seq samples achieved an average of 47,686,163 unique read pairs, with enrichment observed at transcription start site (TSS) (Supplemental Fig. S3). Overall, we identified 178,269 nonredundant open chromatin regions (OCRs) in these tissues. Among these, 41,142 OCRs were activated in the PP tissue, with 15,398 OCRs activated specifically in PP (Supplemental Fig. S4A). Compared with all OCRs, PP-specific OCRs were less abundant in promoter regions, with a larger proportion found in distal intergenic regions (Supplemental Fig. S4B). This suggests that PP-specific OCRs are more likely to function as distal elements during regeneration. When projected onto the red deer genome, 98.1% OCRs were fully mapped (Supplemental Fig. S4C), which is much higher than the ratio of synteny region (89.1%) among the global genomes between the two species, suggesting these identified OCRs are highly conserved and thus may have function. The 1808 OCRs that were absent or fragmented in red deer were considered specific to sika deer. Additionally, 4250 potential TGs of these 1808 sika deer-specific OCRs identified by Pearson's correlation analysis were enriched in immune-related functions (Supplemental Fig. S4D). Through hierarchical clustering and PCA analysis, we discovered notable distinctions between the PP and other tissue types, for both the ATAC-seq and RNA-seq data sets (Fig. 2B,C; Supplemental Fig. S5). Meanwhile, the skin and rumen, both of which have a similar stratified squamous epithelium composition (Pan et al. 2021), clustered together, indicating that our data accurately capture the similarities between these tissues in the sika deer.
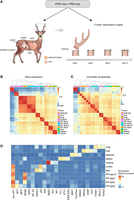
Chromatin accessibility and gene expression landscape for different organs of sika deer. (A) Schematic drawing of the study design covering primary organs and key stages of antler regeneration and collecting paired expression and chromatin accessibility data. (B,C) Hierarchical clustering heatmap of gene expression and chromatin accessibility of sika deer organs. (D) Hierarchical clustering heatmap of a TF-binding motif match profile showing a similar motif profile among self-renewable tissues.
To identify regulators driving the variance of chromatin accessibility, we conducted transcription factor (TF) motif enrichment analysis on the peaks from each tissue (Fig. 2D). We identified the TF motifs with the most significant enrichment for each organ and found that these TFs played important roles in the corresponding organs of model organisms, such as HNF4A in the liver (Radi et al. 2023) and MEF2s in the muscle (Taylor and Hughes 2017). The regulatory pattern of PP tissues was similar to that of skin and rumen, contrasting with the results of ATAC-seq and RNA-seq analysis. This similarity was mainly attributed to the binding of AP-1s, KLFs, and CNCs, which are TF families involved in cell proliferation and stimulus response. Meanwhile, the specific motif enrichment of the RUNX family in PP tissue indicates a strong osteogenic potential of PP tissues, which differs from epithelial tissues.
To establish accurate TF-RE and RE-TG regulatory relationships, we used PECA2 to model the regulatory network. These tissue-specific networks contain an average of 6190 TGs and 318 TFs. The TGs of PP-specific REs were enriched in antler-specific genes identified in a previous study (Fisher's exact test, P-value = 1.64 × 10−16) (Wang et al. 2019b), suggesting our model can identify important RE function in gene co-option for antler regeneration. We performed Kleinberg's HITS analysis (Kleinberg 2000) to calculate hub scores, which represent significance of TFs in network. We used Enrichr with “ARCHS4 tissue” database (Lachmann et al. 2018; Xie et al. 2021) to perform enrichment analysis with the top 50 TFs with the highest hub scores for each organ. The results for most major organs matched with corresponding human organs (Supplemental Table S7), indicating that our network could accurately identify the important TFs that contribute to tissue specificity. Tissue enrichment of PP tissues in four stages matched with the omentum, an organ with strong regenerative potential (Di Nicola 2019), suggesting that tissues with strong regenerative ability share similar TF regulatory patterns.
Hub TF dynamics in antler regeneration
Hierarchical clustering and TF-binding motif analysis of multiomic data revealed two main stages of deer antler regeneration (Figs. 2C,D, 3A). The first stage, encompassing dac0 and dac2, involved stem cell activity or early development (NFYs, SPs) and stimulus response (CREBs, CNCs, ZNF189, MITF, AP-1s, and sMAFs) as the main biological processes. From dac5 onward, TF families related to development (RUNXs, NFATs, TEADs, MEF2s) predominated, suggesting that stem cells had activated and initiated developmental processes such as chondrogenesis. These findings align with a previous study on dac5 PP ectopic ossification and confirm that dac5 is a critical turning point for antler regeneration (Qin et al. 2023). We then selected the hub TFs (those with the top 10 highest hub scores) of GRNs at each stage to identify the key TFs in regeneration (Fig. 3B). Similar to the TF motif patterns, some hub TFs are shared with other tissues, such as KLF4 in lung and skin (Supplemental Table S8). Notably, KLF4, JDP2, MYC, and NFE2L2 are hub TFs across all four stages, suggesting they serve as core TFs throughout the entire regeneration process. The hub TFs identified in the early regeneration stages were primarily associated with stimulus response (RXRA, RXRB, NR3C1), whereas those appearing later were mostly related to specific developmental processes such as chondrogenesis (RUNX2, SP7) and angiogenesis (SP3). We also identified neural crest-related TFs (TFAP2A, TEAD2), which supports the earlier hypothesis that antlers originate from the neural crest (Wang et al. 2019b).
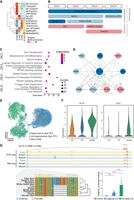
Hub TFs in antler regeneration. (A) Hierarchical clustering heatmap of the top 5 enriched motifs for each stage in antler regeneration. (B) Function and time course schema of top 10 TFs for each stage, suggesting four core TFs and the dynamics from cell stemness and stimulus response to development. (C) Functional enrichment of shared target genes of hub TFs in each antler regeneration stage. (D) Hierarchical structure of 10 hub TFs subnetwork in antler regeneration at dac5 with a similar pattern with time course schema. (E) UMAP projection of PMC cell lineage from antler and mouse digit tips distinguishes PMC of antler and mouse. Dash circle highlights the shared activated stem cell in antler and regenerative digit tip. (F) Higher KLF4 and MYC expression in PMC of deer antler compared with mouse digit tips. (G) Genome track of an antler-specific element near MYC (top), sequence alignment (bottom left), and luciferase assay (bottom right) suggest cervid-specific insertion has significantly increased the expression of MYC in the antler.
We observed a high degree of overlap among the regulons of hub TFs across all stages (Supplemental Fig. S6). Similarly, the expression profiles of hub TFs in the cellular atlas of antler regeneration reveal that most hub TFs are broadly expressed across various cell types, with only a few exceptions (Supplemental Fig. S7). For example, SP7 is specifically expressed in activated stem cells and osteoblasts. These findings suggest that the identified hub TFs may function throughout the entire antler regeneration process through combinatorial coordination.
To further elucidate the stage dynamics of the antler regeneration process, we conducted a functional enrichment analysis on the shared TGs of these hub TFs (Fig. 3C). The PI3K-AKT signaling pathway and collagen expression-related terms, such as extracellular matrix organization, were enriched across all four periods. During the dac0 period, there was notable enrichment in mesenchymal cell differentiation, indicating an early stem cell response to antler casting. Both the dac0 and dac2 periods exhibited similarities, with enrichment in terms associated with immune and stress responses, including the TNF signaling pathway, antigen processing and presentation of exogenous peptide antigen, and wound healing, indicating a response to wound exposure following antler casting. Significantly, both periods also showed enrichment in osteoclast differentiation, aligning with previous studies that suggest osteoclasts drive antler casting (Goss et al. 1992). In the dac5 period, when regenerative ABPCs appeared, enrichment was observed in developmental functions such as skeletal system development and embryonic organ development. Additionally, the emergence of the Relaxin signaling pathway, known for its role in inhibiting inflammation and fibrosis (Valkovic et al. 2019), marked the transition from an immune response to the initiation of the regenerative process during this period. The subsequent dac10 period showed enrichment in collagen and skeleton development-related terms. In summary, our networks provide a detailed explanation of the antler regeneration process, illustrating the development from antler stem cells to different tissues activating at dac5, driven by the immune response to antler casting.
As regeneration developmental process activated at dac5, we extracted the subnetwork of hub TFs at dac5 (Fig. 3D). This subnetwork has a clear hierarchical structure and consists of the time course pattern. The hub TFs related to stem cell and stimulus response in the initial stages are in the upstream of the subnetwork, whereas the hub TFs related to developmental processes are in the downstream from the subnetwork. Among the four core factors, MYC is located at the top of the subnetwork and KLF4 has the most regulatory connections in the subnetwork, indicating a crucial role in generation activation of these two Yamanaka stem cell factors. Sika deer antler stem cells have shown higher stemness and proliferation stability (more than 20 passages) than regular mesenchymal stem cells (Binato et al. 2013; Seo et al. 2014). Moreover, compared with mouse digit tip regeneration, a deer antler can regenerate many more times (Dolan et al. 2022). By comparing the stem cells from deer antler PP, a mouse regenerative digit tip (P3), and a nonregenerative digit tip (P2) (Fig. 3E), we found different quiescent stem cells from the antler and P3 differentiated into similar activated stem cells to initiate regeneration (Supplemental Fig. S8A,B). Specifically, KLF4 shows differential expression between the antler and P3 (P-value <1 × 10−300) by Wilcoxon analysis, whereas MYC is specifically expressed in the deer antler (Fig. 3F). Comparative analysis between human long bone and calvaria bone gene expression data (He et al. 2021) showed the different expression is not dominated by the difference between endochondral ossification (P2 and antler) and intramembranous ossification (P3) (Supplemental Fig. S8C), suggesting that the high expression levels of these two factors may contribute to the strong stemness of deer antler stem cells. Additionally, we examined the evolutionary status of their network-related REs and found a deer-specific open regulatory element (Chr 14: 13,194,714–13,195,323) with a deer-specific insertion upstream of MYC (Fig. 3G). This element contains a 7 bp deer-specific insertion in a conserved region, and our ATAC-seq data for different organs showed that it was exclusively open in PP tissue. Our dual luciferase reporter experiment demonstrated that following the cervid-specific insertion, this element exhibits a more significant enhancer effect than the wild-type RE with the pGL4.23 minimal promoter as a control (Fig. 3G). These findings suggest that the emergence of this stronger RE has led to the recruitment of MYC in deer antler stem cells, contributing to their strong stemness.
cTOP modeling uncovers regulation of cellular programs in antler regeneration
Although the hub TFs and their regulons have revealed the dynamics of gene regulatory profiles during antler regeneration, the regulation of cellular program dynamics remains unclear. To address this, we coupled our TF combinatorial regulatory modules identified from bulk GRNs with single-cell expression profiles to explore the cell subpopulation heterogeneity. We developed a model called the combinatorial TF oriented program (cTOP; for details, see Methods), which extracts cellular programs including combinatorial TF module and their specific TGs from single-cell RNA-seq data (Fig. 4A).
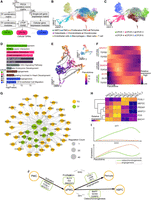
cTOP models cell programs in antler regeneration. (A) cTOP couples cell expression modules and TF combinatory modules under the PECA regulatory network to model cellular regulatory programs. (B,C) UMAP of cell types and highest CPCR in cell atlas at dac5. (D) Functional enrichment for TGs of each CPCR. (E) UMAP of pseudotime trajectory of stromal cells at dac5 by Monocle3. (F) cTOP-relative TFs (red) and genes (black) expression dynamics across the osteochondrogenesis trajectory. (G) Network of CPCR4 suggests PRDM1, FOSL1, and NFATC1 as the key factors for stem cell activation in antler regeneration. (H) Expression dynamics of TFs in CPCR4 during antler regeneration suggest their early function in antler regeneration. (I) Expression dynamic across differentiation of SP7 and SIX2 showing their program specific expression. (J) Schematic regulatory model of cellular programs in antler stem cell differentiation. Black arrows represent differentiation, and gray dashed arrows represent regulation.
We used the cTOP model to analyze the single-cell data at PP dac5, a stage when regeneration has been activated. In total, we identified six cellular programs mediated by TF combinatorial regulations (CPCRs). Uniform manifold approximation and projection (UMAP) dimensionality reduction revealed that the cell types assigned by the traditional clustering method (Fig. 4B) closely matched with the cellular program assignments based on the highest CPCR score (Fig. 4C), except for CPCR3, which was consistent with proliferating cells across different cell types. The strong correlation between functional enrichment (Fig. 4D) and cell classification demonstrated the accuracy and sensitivity of our method in extracting cellular programs. Furthermore, our findings revealed that cell types were not always defined by individual CPCRs but rather by different compositions of multiple CPCRs. For instance, CPCR2 was detected in nearly all cell types (Supplemental Fig. S9), indicating the involvement of immune responses triggered by antler casting in various biological processes during antler regeneration. Therefore, in our model, cell types are determined by the composition of CPCRs, and cell differentiation can be described as the dynamic interplay of CPCRs. We also applied the cTOP model to liver data. Our model successfully identified metabolism, immune, and angiogenesis programs in the relevant cell types (Supplemental Fig. S10A–E). Notably, the programs associated with endothelial cells in both PP and liver tissues shared nine TFs (AR, ID2, GATA2, KLF4, MAFK, MAFG, and PPARA) and 55 TGs tightly related to angiogenesis (Supplemental Fig. S10F; Lasorella et al. 2005; Torres-Estay et al. 2015; Sangwung et al. 2017; Dong et al. 2020), suggesting that the cTOP model is robust in identifying similar regulatory programs across different tissues.
Deer antler regeneration is a stem cell–based epimorphic process (Wang et al. 2019a). Within the stromal cell lineage that includes PMCs, pericytes, ABPCs, and osteoblasts, we identified five CPCR cell programs: CPCR1 (osteochondrogenesis), CPCR2 (stimulus response), CPCR3 (cell proliferation), CPCR4 (mesenchymal stem cell phenotype), and CPCR5 (pericyte-related angiogenesis). Pseudotime trajectories demonstrate that mesenchymal stem cells differentiate into pericytes and ABPCs through proliferative PMCs (Fig. 4E; Supplemental Fig. S11A–D). Throughout this process, the activity of CPCR4 gradually decreases, whereas CPCR3 is activated. Finally, the dominant program transitions into CPCR1 and CPCR5, respectively (Fig. 4F; Supplemental Fig. S11E,F).
At the beginning of differentiation, we found that CPCR4 functioned more as a PMC activation program rather than a PMC resting program, as inferred from the cell annotation. The TFs in this subnetwork include TF families NFATCs and TEADs that emerge at dac5 by motif enrichment analysis and regulate the osteochondrogenic TFs (RUNX2, SOX9, and EN1) of the dominant program in ABPCs: CPCR1 (Fig. 4G). Core TFs in the CPCR4, including FOSL1, FOS, PRDM1, BACH1 and NFATC1, expressed higher at the first 2 days after casting, suggesting their early roles in regulating stem cell activation (Fig. 4H). TFs in the AP-1 family, including FOS and FOSL1, have been found activating stem cells in muscle regeneration, and NFACT1 is crucial for ensuring bone repair and regeneration in skeletal stem cells (Yu et al. 2022). These TFs may be key factors for stem cell activation by regulating genes in the WNT and Hedgehog signaling pathway.
We further found regulation identified between programs following stem cell activation (Fig. 4J). The proliferation program CPCR3 was regulated by KLF4, ID2/E2Fs, CREBs, and SMADs, as well as SP7 from CPCR1 and SIX2 from CPCR5. Expression of SIX2 and SP7 specifically increased along differentiation of angiogenesis and osteochondrogenesis, respectively (Fig. 4I). SP7 is a well-known master regulator for skeleton development, whereas SIX2 is important for cranial skeleton development from cranial neural crest stem cells (Liu et al. 2019b). Additionally, CPCR5, the pericyte-dominant program, also regulated the CPCR1, suggesting multifunctional roles for pericytes during antler regeneration. These findings indicate that TFs expressed by the differentiation programs can, in turn, regulate the stem cell proliferation program and that SIX2 and SP7 may determine the fate of stem cell differentiation through this regulatory mechanism.
We also found significant effects of the TGFB pathway on different cellular programs involved in antler regeneration. In a mouse heterotopic ossification model, high levels of TGFB1 induce the fibroblasts differentiating to chondrogenic progenitor cells (Sorkin et al. 2020). We observed a similar role of TGFB1 in antler osteochondrogenesis in contrast to TGFB2/3. The expression of TGFB2 and TGFB3 peaks at the onset of osteochondrogenic differentiation and then declines, whereas the expression of TGFB1 increases during this process (Fig. 4F). TGFB2 and TGFB3 are crucial for the maintenance of the valvular interstitial cell phenotype, a multipotent cell type in heart valves (Wang et al. 2021). Additionally, TGFB2 has been reported to inhibit osteogenesis of mesenchymal stem cells, contrasting with the role of TGFB1 (Li et al. 2022). These results suggest a critical role of TGFB2/3 in PMCs stemness maintenance by suppressing differentiation. Similar interplays were also found between TGFB pathway–related TFs like ID2 and SMADs in CPCR3. In airway basal stem cells, the TGFB-ID2 axis was reutilized to promote tissue regeneration, while the overexpression of ID2 led to a tumorigenic phenotype (Kiyokawa et al. 2021). SMADs are canonical downstream factors of the TGFB pathway. SMAD4, in combination with TGFB-activated SMAD2 regulates the DNA repair and cell cycle to inhibit tumorigenesis. We found these factors regulating proliferative genes like UBE2C and TOP2A together. The coordination between ID2/E2Fs and SMADs may keep the balance between regenerative ability and cancer suppression in antler generation.
In summary, our cTOP approach effectively modeled the cellular dynamics of the stem cell differentiation process in antler regeneration. By constructing the subnetworks of CPCRs, we have found key factors operating within specific cellular programs and the interactions between these programs.
Discussion
The multiomic approach has become an efficient method for resolving complex biological processes. However, the application of such methods in nonmodel species has been hindered by the lack of high-quality omics data. This study generated the high-quality and comprehensive genomic data of sika deer, making it suitable for subsequent multiomic studies. We observed that the genome size estimated for flow cytometry is larger than that from k-mer estimation as well as previous assembly based on technologies like ONT and high-throughput sequencing, a discrepancy also noted in other ruminant species such as cows and reindeer (Kent et al. 1988; Goss et al. 1992). Our analysis indicates that this gap is mainly owing to the failure of anchoring massive centromeric sequences, a characteristic feature of ruminant genomes. Although we have nearly completed assembling all the euchromatic regions of the sika deer genome, overcoming the large centromeric regions and achieving a complete telomere-to-telomere genome assembly for ruminant species remain a significant challenge. Fortunately, the large gaps in the ruminant centromeric regions contain few or no genes because almost all mammalian genes have been annotated for the sika deer, and therefore, they have limited impact on the downstream functional omics analyses.
Based on the high-quality sika deer genome, we were able to integrate large amounts of omics data including bulk RNA-seq, single-cell RNA-seq, and ATAC-seq data, of sika deer to reconstruct the tissue-specific GRNs and resolve cellular dynamics during antler regeneration. The results reveal that antler PP shared some key regulator, such as AP-1 and KLF4, with epithelial tissues. AP-1 complexes are prevalent in the regenerative elements of various species, including fruit flies, acoel worms, and bony fish, and are crucial for activating regenerative response enhancers in bony fish (Wang et al. 2020). KLF4 is essential for the homeostasis and self-renewal of epithelial cells in various tissues (Segre et al. 1999; Angel et al. 2001). The recruitment and combination of these TFs may underlie the regenerative potential of antler PP. Several hub TFs, including MYC, KLF4, NFE2L2, and JDP2, coordinated the regeneration process from wound healing toward skeletal development. The uncovered cellular regulation dynamics indicated that FOSL1, PRDM1, and NFATC1 might drive the stem cell activation and that balancing between the oncogenic factors ID2/E2Fs and anticancer factors SMADs in the TGFB pathway might contribute to the well-organized stem cell proliferation during antler regeneration (Fig. 5E). The proliferation program might also receive potential feedback regulation by SP7 and SIX2 from differentiation programs. These findings for the first time revealed the gene and cell regulatory mechanism of deer antler regeneration.
Antler progenitor stem cells have been characterized as PMCs with certain embryonic stem cell characteristics. The involvement of embryonic stem cell–related TFs like KLF4 and MYC in antler regeneration has been a subject of debate. Our study suggested the hub role of KLF4 and MYC in the regulatory networks of antler regeneration, which could not be easily identified through gene expression analysis alone. Cross-species comparisons highlight their high expression might contribute to the high regenerative capacity of antler stem cells. Along with the identification of cervid-specific REs and the discovery of stem cell phenotype maintenance by TGFB2/3, we have preliminarily elucidated the evolutionary and molecular mechanisms underlying stemness in deer antler stem cells, which may provide valuable insights for regenerative medicine studies.
In addition, we proposed the new method, cTOP, in this study to combine our scRNA-seq and bulk ATAC-seq and to decode cellular GRNs. Existing GRN inference methods are not ideally suited for sika deer data. Most methods, such as SCENIC+, GLUE, and scReg (Cao and Gao 2022; Duren et al. 2022; Bravo González-Blas et al. 2023) require paired scRNA-seq and scATAC-seq, which is expensive and very sensitive to the quality of library preparation. Other methods, such as SCENIC and GRNBoost (Aibar et al. 2017; Passemiers et al. 2022), construct a coarse network using only scRNA-seq. Compared with cTOP, scRNA-seq-only methods, which infer regulatory relationships primarily from coexpression at the single-cell level, may identify many false-positive regulations among TFs and TGs within a coexpression module. The approach of cTOP, decomposing TF modules from bulk GRNs and then coupling with single-cell expression profile, offers a cost-effective alternative to obtain highly confident cellular GRNs. Importantly, cTOP fully utilized all data generated in a nonmodel animal. In future, we will generalize the cTOP method to scRNA-seq-data-only cases and compare it with existing methods. Furthermore, cTOP can easily be used in model animal–based development and disease research with even better performance, given the fact that the original PECA2 method was developed with larger and more diverse paired expression and chromatin accessibility data from the mouse and human.
In conclusion, we have identified the key factors and pathways in stem cell activation, proliferation, and differentiation of antler regeneration through cellular GRN modeling based on our high-quality omics data. The gene regulatory mechanism underlying the strong regenerative capacity and delicate balance between regeneration and cancer suppression in cervids will provide new clues for both regeneration and cancer medicine studies.
Methods
Sample collection and genome sequencing
Four 2-year-old male sika deer (C. nippon) were used for sampling regenerating antler tissues on dac0, dac2, dac5, and dac10. Another 2-year-old male sika deer was sacrificed for sampling normal organs. Blood from four chickens, sika deer, and rats was collected to conduct flow cytometry for genome size estimation (for details, see Supplemental Methods).
Genomic DNA was extracted from liver using the standard cetyltrimethylammonium bromide method. For HiFi sequencing, SMRTbell library construction and sequencing were performed at Novogene or BerryGenomics following the official protocols of PacBio for preparing ∼20 kb to 544 kb SMRTbell libraries. For Hi-C sequencing, we followed the standard protocol described previously with minor modifications, using the same sample with HiFi sequencing (Belton et al. 2012) at Novogene. ATAC-seq was performed by standard protocol as previously reported (Buenrostro et al. 2013) at Novogene. Details of DNA and RNA library preparation are described in the Supplemental Methods, and statistics of all data collected for each bat are provided in Supplemental Tables S1 and S3.
Genome assembly
Hifiasm (v0.19.5) was used to assemble the HiFi reads with ONT reads we generated previously and with Hi-C reads from same individual to generate phased contigs. Then, the contigs of each haplotype were merged and scaffolded with Juicer (Durand et al. 2016) and 3DDNA (Dudchenko et al. 2017) to check switch error of phasing (most in sex chromosomes) (Supplemental Fig. S1D). Then, each haplotig was scaffolded separately to increase scaffolding accuracy with higher Hi-C contact resolution.
Mitochondrial genome was assembled and annotated using MitoHiFi (v3.2.1) (Uliano-Silva et al. 2023) with HiFi reads.
We assessed genome completeness and consensus quality value (QV) using Merqury (Rhie et al. 2020). In addition, we performed a BUSCO (Waterhouse et al. 2018) assessment of the genome sequences using the certa_odb10 database.
Genome annotation
RepeatMasker (Tarailo-Graovac and Chen 2009) was first used to detect and mask the repetitive region. Then, we integrated different evidence to predict genes. First, we used GeMoMa (v1.9) to do homology-based annotation with annotations of human, cattle, red deer, and yarkand deer from the NCBI or Ensembl as references. Second, we processed whole-genome alignment to these related species genomes with the UCSC chain/net pipeline (Kent et al. 2003) and projected gene annotations using TOGA (v1.1.7) (Kirilenko et al. 2023). Third, we used the transcriptome workflow in REAT (v0.6.1; https://github.com/EI-CoreBioinformatics/reat) to integrate short and long transcriptomic data and generate a highly confident gene model. De novo prediction was applied with BRAKER (v2.1.5) (Hoff et al. 2016) with the transcriptomic model as a hint. All evidence was integrated using MAKER (v3.01.03) (Campbell et al. 2014).
ATAC-seq data process
ATAC-seq reads were cleaned by fastp (v0.23.1) (Chen et al. 2018) and then were aligned to the reference genome using Bowtie 2 (Langmead and Salzberg 2012). These reads were then filtered for high quality (MAPQ ≥ 13); we also removed reads that were not properly paired and with PCR duplicates by Picard (v2.25.7; https://broadinstitute.github.io/picard/). All peak calling was performed with MACS2 (v2.1.0) (Zhang et al. 2008) using “–call-summits nomodel –shift −100 –extsize 200.” Motif enrichment was performed by HOMER (Heinz et al. 2010).
RNA-seq data process
Short RNA-seq reads were cleaned by fastp and then were aligned to the reference genome with HISAT2 (Kim et al. 2019). The mapped reads of each sample were assembled by StringTie (v1.3.3b) (Kovaka et al. 2019).
Iso-Seq reads were preprocessed by IsoSeq3 (v3.8.0; https://github.com/PacificBiosciences/IsoSeq). The FLNC reads from IsoSeq3 were mapped to genome with minimap2 (Li 2018) and processed with TAMA pipeline.
Single-cell RNA data process
10x Genomics single-cell RNA-seq data were obtained from our previous research (Qin et al. 2023). Cell Ranger (v6.1.1) was used to make prepare the reference and count the gene expression profile. Then, we used SCANPY (v1.1.10) (Wolf et al. 2018) to process the analysis. First, we use Scrublet (v0.2.3) (Wolock et al. 2019) to mark and remove doublets. Then, we further filtered out cells with less than 500 counts or with a percentage of mitochondrial counts exceeding 20%. After preprocessing, we used harmony (v0.0.9) to integrate data of all stages. We used the Leiden method to cluster cells and the Wilcoxon method to identify marker genes.
The pseudotime trajectory inference of the dac5 data was applied with Monocle3 (v.0.5.0) (Setty et al. 2019) and Palantir (v1.3.0) (Setty et al. 2019) separately.
Optimization model to identify TF combinatorial programs by cTOP model
cTOP is a method to infer TF combinatorial program from scRNA-seq data. In brief, a guidance TF-TG regulatory network is first constructed for a given context. Here, we use PECA2 model from paired bulk gene expression and chromatin accessibility to construct a TF-TG regulatory network. Second, a guidance TF combinatorial network is built from the TF-TG network by a connection specificity index (CSI) to represent the similarity of regulons between TFs. Then, we apply a nonnegative matrix factorization (NMF)–based optimization model to both the CSI matrix and the single-cell expression matrix for identifying TF combinatorial program and cell embeddings on TF programs. Three essential steps of cTOP are detailed below.
Constructing TF-TG regulatory network and estimating TF combinatorial effect
We use the PECA2 model to build a TF-TG regulatory network. PECA2 takes paired gene expression (bulk RNA-seq) and chromatin accessibility data (bulk ATAC-seq) as input, with replicant merged for each tissue. The prior data of PECA2, including TF-TG correlation and RE-TG interaction, is calculated from the data of multiple deer organs (Supplemental Table S5) for deer specific regulatory network. The output of PECA2 is a TF-TG regulatory strength matrix, denotated by R matrix, for M TFs and N TGs. Specifically, Rij, which is the i-th row and j-th column of TRS matrix, is the regulatory strength score of the i-th TF on j-th TG.
Then, we use the connection specificity index (CSI) (Bass et al. 2013) to assess the combinatorial effect of two TFs. For the i-th and j-th TFs, we use Ri and Rj to represent their regulatory strength scores across all the TGs. Then, we calculate the Pearson correlation of their regulatory
strength PCCij = PCC(Ri, Rj). Then, CSI score considers the specificity of Pearson correlation to evaluate the combinatorial effect of i-th and j-th TFs:
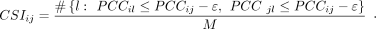 (1)
Here M was the total number of TFs. ε was a constant with a default value of 0.05. The sharp sign (#) denotes the number of elements
in a set. A high CSI score indicated that two TFs specifically regulated a group of TGs.
(1)
Here M was the total number of TFs. ε was a constant with a default value of 0.05. The sharp sign (#) denotes the number of elements
in a set. A high CSI score indicated that two TFs specifically regulated a group of TGs.
Identifying TF combinatorial program from scRNA-seq data
We use cTOP model to identify TF combinatorial program from single-cell RNA-seq data. There are three inputs of the cTOP model:
(1) the TF combinatorial network represented by above CSI matrix C, (2) the TF-TG regulatory network represented by above
trans-regulatory score matrix R, and (3) the single-cell gene expression matrix E. We expected the gene expression matrix E to be coupled with TF combinatorial programs through the regulatory network R. Formally, the cTOP optimization model is formulated as follows:
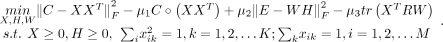 (2)
(2)
The cTop model has three components:
-
 : The first two terms are designed to detect TF modules from TF-TF combinatorial effect matrix. The first term decomposes
TF-TF combinatorial matrix for detecting TF combinatorial modules and the second term constrains the detected TF modules to
be TF combinations with large CSI scores. This component will output variable X, which is a M-by-K matrix to reveal the combinatorial effect of M TFs in K TF combinatorial programs. We used X to obtain TF modules of TF programs. Given the TFs’ combinatorial effect Xk = (X1k, X2k, …, XMk)T in the k-th combinatorial program, we computed the combinatorial effect of i-th TF and j-th TF in k-th TF programs:
: The first two terms are designed to detect TF modules from TF-TF combinatorial effect matrix. The first term decomposes
TF-TF combinatorial matrix for detecting TF combinatorial modules and the second term constrains the detected TF modules to
be TF combinations with large CSI scores. This component will output variable X, which is a M-by-K matrix to reveal the combinatorial effect of M TFs in K TF combinatorial programs. We used X to obtain TF modules of TF programs. Given the TFs’ combinatorial effect Xk = (X1k, X2k, …, XMk)T in the k-th combinatorial program, we computed the combinatorial effect of i-th TF and j-th TF in k-th TF programs:
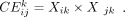 (3)
(3)
We assumed the combinatorial effect of TF pairs in each TF programs followed Gamma distribution. We used threshold P-value ≤0.01 to select TF pairs for k-th TF programs and the significant TF pairs formed the TF module of k-th TF programs.
-
 : the third term was to cluster and obtained gene expression programs for scRNA-seq. This component will output matrix W and H. W was a N-by-K matrix to represent the gene expression program, and each column of W indicated the mean expression of TGs regulated by the corresponding TF module. And, W was used to obtain the TGs of each TF program by gene expression. H was a K-by-c matrix to reveal assignment weights of c cells for K TF programs. We assigned each cell to a TF program with the largest assignment weight.
: the third term was to cluster and obtained gene expression programs for scRNA-seq. This component will output matrix W and H. W was a N-by-K matrix to represent the gene expression program, and each column of W indicated the mean expression of TGs regulated by the corresponding TF module. And, W was used to obtain the TGs of each TF program by gene expression. H was a K-by-c matrix to reveal assignment weights of c cells for K TF programs. We assigned each cell to a TF program with the largest assignment weight.
-
tr(XTRW): The last terms exerted specificity on TF program by coupling the TF modules with the gene expression programs through the regulatory network. This component gave constraints to TF modules: The TF modules should regulate TGs that have specific expression in certain cell types/states. This constraint enabled TF programs to utilize not only the specificity of TFs but also the specificity of REs-TGs and TF combinatorial effect to identify core TF combinations for cell type/states.
These three outputs (X, W, H) would be used for describing TF combinatorial programs. We modeled the k-th TF combinatorial program as follows:
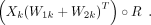 (4)
Here, Xk and Wk were the k-th column of X and W, respectively. The k-th TF combinatorial program was represented by the TF module defined by Xk with Equation 3. The TGs of k-th TF combinatorial program were given by W. The k-th cell state, which was cells governed by k-th TF combinatorial program, was defined by H.
(4)
Here, Xk and Wk were the k-th column of X and W, respectively. The k-th TF combinatorial program was represented by the TF module defined by Xk with Equation 3. The TGs of k-th TF combinatorial program were given by W. The k-th cell state, which was cells governed by k-th TF combinatorial program, was defined by H.
Annotating cell clusters with linear combinations of TF combinatorial programs
A cell cluster is a group of cells in scRNA-seq data. For a cell cluster, we suppose this cell cluster has n cells G = {g1, g2, … gn}. We represent this cell cluster as the TF combinatorial program with the averaged coefficients of all the cells in G, respectively:
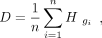 (5)
where
(5)
where 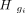 is the columns corresponding to the cell gi. Then, the TF combinatorial program combination coefficients of the given cell cluster will be D.
is the columns corresponding to the cell gi. Then, the TF combinatorial program combination coefficients of the given cell cluster will be D.
Model initiation, parameter selection, and optimization algorithm
To initiate our model, we first solved Components 1 and 2 of our model independently. These three components gave us the initiation of five variable: X0, W0, H0.
The initiation matrix of three variables enabled us to determine the hyperparameters in our model. There were three hyperparameters
in our model: μ1, μ2, μ3, and K. μ1, μ2, μ3, were parameters to balance the scale of five terms in our model, which could be determined by the initiation matrix:
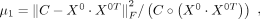 (6)
(6)
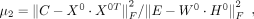 (7)
(7)
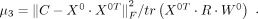 (8)
The hyperparameter K was the number of TF combinatorial programs, which was consistent with number of TF modules in C and number of cell type/states for single-cell data. K could be determined in two ways. First, if we had prior knowledge about the number of TF modules or cell types, we could
direct assign this number to K. Second, if we did not have biological insights about the data beforehand, we could try different K to find modules and compute modularity in the TF combinatorial effect matrix to select the best one. And, the number K could also be determined by a method similar to that in a previous study (Brunet et al. 2004).
(8)
The hyperparameter K was the number of TF combinatorial programs, which was consistent with number of TF modules in C and number of cell type/states for single-cell data. K could be determined in two ways. First, if we had prior knowledge about the number of TF modules or cell types, we could
direct assign this number to K. Second, if we did not have biological insights about the data beforehand, we could try different K to find modules and compute modularity in the TF combinatorial effect matrix to select the best one. And, the number K could also be determined by a method similar to that in a previous study (Brunet et al. 2004).
Starting from the initiation matrices and hyperparameters, we proposed a multiplicative update algorithm to solve the nonconvex
optimization problem of the cTOP model. We used Xij to represent the element of the i-th row and the j-th column in matrix X and used Wij and Hij as the corresponding elements in W and H. We adopted the following update roles and stopped the iteration when the relative error is <0.0001.
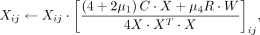 (9)
(9)
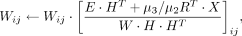 (10)
(10)
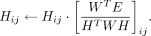 (11)
(11)
Cervid-specific structural variation identification
To check the structural variants in elements, we first used UCSC chain/net pipeline to generate whole-genome alignment to rein deer (HlranTar1). Then, we used liftOver to map elements from sika deer to rein deer. Then, we extracted a MAF file with rein deer as a reference from the HAL file generated by cactus in the Zoonomia project (Genereux et al. 2020). An in-house script called SV_caller was used to find structural variants following these criterions: (1) The identity of sequence for ingroup species in the SVs is not <90%, (2) the identity of sequence for all species 50 bp flank SVs is not <50%, and (3) the length of SV is >5 bp.
Comparative analysis of OCRs
To check the evolutionary conservation of OCRs, we first used UCSC chain/net pipeline to generate whole-genome alignment to red deer (mCerEla1.1). Then, we used liftOver to map elements from sika deer to red deer. Potential TGs were identified with a Pearson's correlation test. Functional enrichment was conducted with Enrichr.
Luciferase reporter assay
The dual luciferase reporter constructs engineered in this study were developed on the pGL4.23[luc2/minP] vector (Promega E8411) and the pGL4.74[hRluc/TK] vector (Promega E6911), which served as the base plasmid. The Dual-Glo Luciferase luminescent assay (Promega E1960) was carried out in accordance with the manufacturer's protocol with slight modifications. Detailed protocols for transient transfection and stable measurements are described here: After transient transfection in HEK-293T adherent cells, discard the cell culture medium and rinse the cells twice with PBS. Add 120 μL 1 × passive lysis buffer (Promega E194A) to each well to lyse the cells. After incubation for 10 min at room temperature, transfer lysates to 96-well flat-bottomed white polystyrene plates (Corning 3912). Using the automatic loading function of a multifunctional microplate reader (Synergy Neo2, BioTek), add 50 μL of firefly luminescence detection solution and 50 μL of sea cucumber luminescence detection solution accordingly to 2.5 μL of the sample to be tested in each well, and load the sample and detect the final luminescence value.
Data access
All raw sequencing data generated in this study have been submitted to the Genome Sequence Archive of China National Center for Bioinformation (GSA; https://ngdc.cncb.ac.cn/gsa) under accession number CRA018294 (single-cell RNA-seq data), CRA018238 (genomic data), and CRA015420 (RNA-seq and ATAC-seq data). The genome assembly generated in this study was submitted to the NCBI Genomes database (https://www.ncbi.nlm.nih.gov/home/genomes/) under accession number GCA_038088365.1. Genome assembly, gene annotation, and network files are available at Zenodo (https://zenodo.org/records/13298156). The source codes and sample data for cTOP and for the structural variation analyses are available as Supplemental Code and at GitHub (https://github.com/AMSSwanglab/cTOP and (https://github.com/lizihe21/SV_caller, respectively).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by two grants of National Natural Science Foundation of China (NNSFC, nos. 32030016 and 32220103005) and the New Cornerstone Investigator Program to W.W., as well as the National Key Research and Development Program of China (2022YFA1004800), Chinese Academy of Sciences Project for Young Scientists in Basic Research (YSBR-077), the NNSFC (12025107) to Y.W., and the NNSFC (32225009) to Q.Q.
Author contributions: W.W., Y.W., Q.Q., and D.J.H. conceived and designed the project. Z.H.L., Z.Y.F., W.W., and Y.W. drafted the manuscripts. Z.H.L., T.Q., B.T.Z., W.W., S.J.J., and L.Z. revised the manuscript. T.Q., G.K.Z., and C.Y.L. collected and prepared the samples and helped with the experiments. J.R.M., Q.G., and H.S.Y. designed and performed all experiments. Z.H.L., Z.Y.X., Z.Y.F., B.T.Z., L.Z., and G.H. performed the data analysis.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279448.124.
-
Freely available online through the Genome Research Open Access option.
- Received April 7, 2024.
- Accepted October 28, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








