
The chromatin landscape of the histone-possessing Bacteriovorax bacteria
- 1Department of Genetics, Stanford University, Stanford, California 94305, USA;
- 2Department of Computer Science, Stanford University, Stanford, California 94305, USA;
- 3Center for Personal Dynamic Regulomes, Stanford University, Stanford, California 94305, USA;
- 4Department of Applied Physics, Stanford University, Stanford, California 94305, USA;
- 5Arc Institute, Palo Alto, California 94304, USA
Abstract
Histone proteins have traditionally been thought to be restricted to eukaryotes and most archaea, with eukaryotic nucleosomal histones deriving from their archaeal ancestors. In contrast, bacteria lack histones as a rule. However, histone proteins have recently been identified in a few bacterial clades, most notably the phylum Bdellovibrionota, and these histones have been proposed to exhibit a range of divergent features compared with histones in archaea and eukaryotes. However, no functional genomic studies of the properties of Bdellovibrionota chromatin have been carried out. In this work, we map the landscape of chromatin accessibility, active transcription, and three-dimensional (3D) genome organization in a member of Bdellovibrionota (a Bacteriovorax strain). We find that, similar to what is observed in some archaea and in eukaryotes with compact genomes such as yeast, Bacteriovorax chromatin is characterized by preferential accessibility around promoter regions. Similar to eukaryotes, chromatin accessibility in Bacteriovorax positively correlates with gene expression. Mapping active transcription through single-strand DNA (ssDNA) profiling revealed that unlike in yeast, but similar to the state of mammalian and fly promoters, Bacteriovorax promoters exhibit very strong polymerase pausing. Finally, similar to that of other bacteria without histones, the Bacteriovorax genome exists in a 3D configuration organized by the parABS system along the axis defined by replication origin and termination regions. These results provide a foundation for understanding the chromatin biology of the unique Bdellovibrionota bacteria and the functional diversity in chromatin organization across the tree of life.
Histones and nucleosomal chromatin are one of the defining features of the broadest divisions of life on our planet. Nearly all eukaryotes (Marinov and Lynch 2016) package their chromatin around nucleosomal particles, composed of two dimerized tetramers of the four core histones H2A, H2B, H3, and H4, and histones are the most conserved proteins in their genomes (Postberg et al. 2010), especially when it comes to the key residues playing a vital role in the so-called histone code (and through it in all aspects of chromatin biology: transcription, gene regulation, DNA repair, DNA replication, mitosis and meiosis, and many others) (Jenuwein and Allis 2001), which are almost universally invariant across all eukaryotes.
Eukaryotic histones trace their ancestry to Archaea (Woese and Fox 1977), which is the half of the prokaryote diversity from within which eukaryotes appear to have emerged, and Archaea is where the informational processing systems of eukaryotes are thought to derive from (Lake et al. 1984; Lake 1988; Cox et al. 2008; Koonin 2010; Koonin and Yutin 2014; Spang et al. 2015). However, archaeal histones are quite different from those of eukaryotes. Although they all contain the same canonical histone fold domain (Sandman and Reeve 2006), consisting of three alpha helices (Arents et al. 1991) dimerizing with another histone molecule, archaeal histones, with some notable exceptions (Hocher and Warnecke 2024), only contain the histone fold domain (Henneman et al. 2018; Stevens et al. 2020), whereas those of eukaryotes have long tails (that are the primary sites of post-translational modifications). Another major difference is that unlike the eukaryotic octamer nucleosome, archaeal histone dimers can oligomerize into so-called hypernucleosomes (Maruyama et al. 2013; Mattiroli et al. 2017; Bowerman et al. 2021; Henneman et al. 2021), which are stacks of individual histone dimers, and can be of variable length.
Although bacteria (the other main branch of prokaryotes) do not have histones as a rule, recent phylogenomic and experimental studies have revealed that some bacteria in fact do possess histone proteins (Alva and Lupas 2019; Schwab et al. 2024). Most notably, in the bacterium Bdellovibrio bacteriovorus and other members of the Bdellovibrionota phylum, histones are both abundant and essential for viability components of the nucleoid (Hocher et al. 2023; Hu et al. 2024). Bdellovibrionota histones have been shown to bend DNA as dimers and in a sequence-independent manner (Hu et al. 2024), whereas others have suggested that they exhibit yet another major deviation from the conventional eukaryote state of nucleosomal architecture by binding to DNA headon to coat it, as opposed to binding to DNA by wrapping the DNA strand around histone proteins (Hocher et al. 2023).
These observations pose a long list of questions about the physical organization of the genomes of these histone-possessing bacteria, about the relationship between chromatin structure and gene expression, and about how such features compare to those of archaea. However, although archaea have received some experimental attention in the past decade (Ammar et al. 2012; Nalabothula et al. 2013; Badel et al. 2022; Marinov et al. 2023), these properties have not been assayed so far with functional genomic tools in the bacterial clades that have histones.
To begin addressing these outstanding issues, we have mapped chromatin accessibility using assay for transposase-accessible chromatin using sequencing (ATAC-seq) (Buenrostro et al. 2013), transcriptional activity using kethoxal-assisted single-stranded DNA sequencing (KAS-seq) (Wu et al. 2020), and three-dimensional (3D) genome organization using Hi-C (Lieberman-Aiden et al. 2009) in a member of the Bdellovibrionota phylum (a Bacteriovorax sp. ICPB 3264 [H-I A3.12] strain).
Results
Genome assembly and annotation of Bacteriovorax sp. ICPB 3264 [H-I A3.12]
We first set out to map the chromatin accessibility landscape in B. bacteriovorus using ATAC-seq, given that Bdellovibrio histones were recently structurally characterized (Hocher et al. 2023) and B. bacteriovorus is the most well studied Bdellovibrionota representative. However, a major technical challenge to the application of ATAC-seq to Bdellovibrio is presented by the unique lifestyle of these bacteria. Bdellovibrio is the most famous example of a predatory prokaryote (Stolp and Starr 1963; Stolp and Starr 1965; Lovering and Sockett 2021): It feeds by attaching onto the cell wall of Gram-negative bacteria, creating an opening into it and then inserting itself into the periplasmic space between the inner and outer bacterial membranes, breaking down the host cell subsequently undergoing polyploid division, and eventually lysing the remnants of the host. This means that B. bacteriovorus is usually grown together with another prey species, which are bacteria without histones.
This predatory lifestyle makes applying ATAC-seq to Bdellovibrio challenging as the basic principle behind ATAC-seq is the strong preference of the Tn5 transposase for open chromatin not protected by nucleosomes (Buenrostro et al. 2013). In mammalian cells, this can result in extreme overrepresentation of mitochondrial DNA sequences in final libraries if mitochondria have not been properly filtered out during the nuclei isolation procedure. In a mixture of bacteria without and with histones, if the latter's histones are strongly protecting DNA from Tn5 insertion, ATAC-seq libraries could easily consist almost entirely of fragments from the prey rather than Bdellovibrio.
It happens that prey-independent strains of Bdellovibrio are also available, allowing axenic growth in media free of other bacteria. We obtained “B. bacteriovorus” strain ICPB 3264 [H-I A3.12] from ATCC and carried out ATAC-seq on it. However, almost no reads from these libraries aligned to any of the previously sequenced Bdellovibrionota strains available in the NCBI databases (neither Bdellovibrio nor strains from Bacteriovorax, which is the other major division of the phylum).
We thus carried out de novo genome sequencing of the strain we worked with, using a combination of nanopore long reads and short Illumina reads (for details, see Methods). This resulted in a single contig of 4,148,738 bp (∼667× coverage) in length predicted to encode (for details, see Methods) 4127 protein-coding genes (Fig. 1A).

Genome assembly and annotation of Bacteriovorax sp. ICPB 3264 [H-I A3.12]. (A) Circos (Krzywinski et al. 2009) plot of the Bacteriovorax sp. ICPB 3264 [H-I A3.12] chromosome. Forward- and reverse-strand protein-coding genes are shown in red and blue, respectively, in the inner tile layer; ribosomal RNAs and tRNAs are shown in the outer tile layers. The red histogram shows the cumulative GC skew (Grigoriev 1998), with the inflection points corresponding to the replication origins and termini (Lobry 1996). The light blue histogram in the innermost circle shows the GC skew for each individual bin along the genome. (B) Place of Bdellovibrionota in the global prokaryote phlyogeny (obtained from Zhu et al. 2019 at https://biocore.github.io/wol/gallery/ranks/order.pdf) on the left and the maximum likelihood 16S rRNA tree (generated using RAxML-NG) (Kozlov et al. 2019) of members of the Bdellovibrionota phylum and the phylogenetic location of strain ICPB 3264 [H-I A3.12] on the right. Bdellovibrionota is highlighted with a red dot on the left, and the strain we worked with is highlighted with a red dot on the right. (C) Multiple sequence alignment of histone proteins identified in select Bacteriovorax and Bdellovibrio genomes. In contrast to the singlet histone folds in Bdellovibrio, the histone gene in Bacteriovorax sp. ICPB 3264 [H-I A3.12] is a histone doublet; the two histone folds are shown separately (top two rows).
Analysis of potential DNA modifications revealed the existence of 5-methylcytosine methylation in a CpGGCC context. This signature is likely associated with a restriction-modification (R-M) system (Tock and Dryden 2005) in Bacteriovorax.
We then established the precise phylogenetic positioning of the strain by analyzing available 16S rRNA sequences for Bdellovibrionota strains. The phylum Bdellovibrionota (Oren and Garrity 2021) includes three major clades/classes: Bacteriovoracia, Bdellovibrionia, and Oligoflexia. Bacteriovoracia includes the Bacteriovorax, Halobacteriovorax, and Peredibacter genera, whereas Bdellovibrionia features Bdellovibrio and Pseudobdellovibrio. Our phylogenetic analysis points to strain ICPB 3264 [H-I A3.12] belonging to the Bacteriovorax genus, being most closely related to Bacteriovorax sp. B1T4-F (Fig. 1B), even though it was originally labeled as a “Bdellovibrio” strain. We refer to it as Bacteriovorax sp. ICPB 3264 [H-I A3.12] or simply Bacteriovorax in the subsequent text.
Finally, we examined putative Bacteriovorax sp. ICPB 3264 [H-I A3.12] histone genes in order to confirm their presence in its genome, and we found one histone protein to be encoded in our assembly. Unlike Bdellovibrio histone genes, which encode histone singlets that later form dimers, in Bacteriovorax sp. ICPB 3264 [H-I A3.12], the histone gene encodes a histone doublet that presumably functions equivalently to a dimer of singlets on its own (Fig. 1C). This doublet is highly structurally similar to the histone dimer of B. bacteriovorus (Supplemental Fig. 1).
ATAC-seq reveals the accessible chromatin landscape in Bacteriovorax
To map the chromatin accessibility landscape in Bacteriovorax, we adapted the previously described bacterial ATAC (bac-ATAC) protocol (Melfi et al. 2021), which involves cross-linking with 1% formaldehyde and permeabilization of the cell wall with lysozyme (for details, see Methods).
In metazoans, ATAC-seq libraries display a clear nucleosomal signature, with a strong subnucleosomal peak (≤120 bp) followed by a robust mononucleosomal (∼ 150 bp) peak, a weaker dinucleosomal peak, and so on. In Bacteriovorax, we do not observe a nucleosomal protection pattern but only a single broad fragment peak centered between 100 bp and 200 bp (Fig. 2A). Thus, Bacteriovorax histones do not appear to confer the same arrayed local protection to DNA as eukaryotic ones.
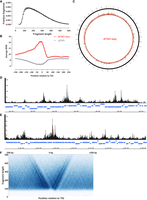
The accessible chromatin landscape in Bacteriovorax. (A) ATAC-seq fragment length distribution. (B) ATAC-seq metaprofile around annotated Bacteriovorax sp. ICPB 3264 [H-I A3.12] TSSs. (C) Circos plot of the global accessibility distribution along the Bacteriovorax sp. ICPB 3264 [H-I A3.12] chromosome. (D,E) Representative snapshots of local ATAC-seq signal distribution in Bacteriovorax (exponentially growing culture). (F) V-plot of ATAC-seq fragment distribution around annotated Bacteriovorax sp. ICPB 3264 [H-I A3.12] TSSs.
Accessibility is centered around the promoter regions between genes (Fig. 2B–E). We observe transcription start site (TSS) scores (indicating the level of enrichment over promoter regions relative to flanking regions; see Methods and previous detailed discussions) (Marinov and Shipony 2021) in the 1.35–1.40 neighborhood, which is comparable to what we had previously found in the archaeon Haloferax volcanii (Marinov et al. 2023) and is also to the lower end of what is usually seen in eukaryotes with highly compact genomes such as yeast (Supplemental Fig. 3; Shipony et al. 2020). In contrast, naked gDNA controls exhibit a slight depletion around TSSs (Fig. 2B). Manual inspection of ATAC-seq profiles along the genome confirms these global observations but also shows frequent cases of elevated accessibility extending into gene bodies (Fig. 2D,E).
We also note that a previous ATAC-seq study of conventional bacteria that do not possess histones (Caulobacter crescentus) reported the existence of broad domains of elevated and decreased accessibility spanning hundreds of kilobases (Melfi et al. 2021). This is not readily observed in Bacteriovorax (Fig. 2C), just as it is not observed in the archaeon H. volcanii.
Finally, we asked whether patterns of nucleosomal positioning are observed around promoters using V-plot analysis (Henikoff et al. 2011). We do not observe strongly positioned nucleosomes around its TSSs (Fig. 2F). However, this is with the major caveat that we do not yet have precise TSS annotations for Bacteriovorax; maps of RNA transcript 5′ ends will be needed to establish a reliable such set.
The ssDNA and active transcription landscape in Bacteriovorax
Next, we mapped the landscape of active transcription in Bacteriovorax using KAS-seq (Wu et al. 2020), which strongly and specifically enriches for ssDNA structures by labeling exposed guanine (G) bases with N3-kethoxal, followed by biotin labeling through a click reaction, fragmentation of DNA, and streptavidin pulldown (for details, see Methods). Although KAS-seq labels all ssDNA structures (G-quadruplexes, replication intermediates, etc.), the most abundant sources of ssDNA in the cell are transcriptional bubbles associated with RNA polymerases directly engaged with DNA, both paused and actively elongating (Wu et al. 2020).
As with ATAC-seq, we do not observe broad large-scale domains of active transcription along the Bacteriovorax chromosome in our KAS-seq data sets (Fig. 3A). The global landscape is largely uniform, punctuated by strong localized peaks.
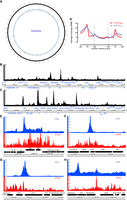
The ssDNA and active transcription landscape in Bacteriovorax. (A) Circos plot of the global KAS-seq signal distribution along the Bacteriovorax sp. ICPB 3264 [H-I A3.12] chromosome. (B,C) Local KAS-seq signal distribution in Bacteriovorax shows strongly localized peaks associated with promoter regions. (D) Metaplot of KAS-seq and ATAC-seq signal distribution along Bacteriovorax genes. (E) KAS-seq and ATAC-seq profiles around the ribosomal RNA locus of Bacteriovorax. (F) KAS-seq and ATAC-seq profiles around the heat shock protein GrpE2 gene. (G) KAS-seq and ATAC-seq profiles around the LuxR2 transcriptional activator gene. (H) KAS-seq and ATAC-seq profiles near the origin of replication of the Bacteriovorax chromosome.
Examination of local browser tracks (Fig. 3B,C) revealed these peaks to be associated with promoters, indicating the existence of very strong promoter pausing in Bacteriovorax. Polymerase pausing and its later controlled release are decisive steps in gene regulation in many eukaryotes (Core and Adelman 2019), having been first described in the fruit fly Drosophila melanogaster (Gilmour and Lis 1986; Rougvie and Lis 1988; Muse et al. 2007; Nechaev et al. 2010), and are also shown to be widespread in mammals (Kwak et al. 2013; Williams et al. 2015), most metazoans, and some plants are but absent in yeast and Arabidopsis thaliana (Chivu et al. 2023). Bacteria do exhibit sequence-dependent polymerase pausing, but this has been primarily reported in the context of transcription elongation and termination (Kassavetis and Chamberlin 1981; Winkler and Yanofsky 1981; Yanofsky 1981; Lau et al. 1983; Kireeva and Kashlev 2009; Kang et al. 2019), and Escherichia coli promoters are not characterized by the typical peaks associated with pausing in global run-on GRO-seq data sets (Chivu et al. 2023). The strong KAS-seq peaks we observe are not caused by transcription termination-associated pausing because they are also found in between divergent promoters (Fig. 3B,C).
We had previously observed promoter pausing using KAS-seq in the euryarchaeon H. volcanii (Marinov et al. 2023). Bacteriovorax promoters are even more accentuated in KAS-seq profiles than in Haloferax: The promoter KAS signal peaks at about 12 reads per million mapped reads (RPM) over Bacteriovorax promoters compared with a trough at about 10 RPM in intergenic space and about eight over gene bodies (Fig. 3B–D); in Haloferax, these values are about 11, about 10, and about eight RPM, respectively (Marinov et al. 2023).
The most strongly transcribed (as assessed by KAS-seq levels over the gene body) genes in Bacteriovorax are the two ribosomal RNAs (Fig. 3E). Notably, these do not exhibit a KAS-seq peak in their promoter (although a very strong such peak is located downstream from the ribosomal RNA operon). This region is also one of the most accessible over the gene body loci in the genome as measured by ATAC-seq; this points to an analogous situation to the one well known from yeast and other eukaryotes. Yeast rDNA exists in two states: a transcriptionally inactive chromatinized condition, on one hand, and a highly transcriptionally active, almost entirely devoid of histones state (Conconi et al. 1989; Merz et al. 2008; Shipony et al. 2020) on the other. These observations also contrast with those from ATAC-seq for C. crescentus, which does not possess histones and in whose genome ribosomal RNA clusters were reported to be one of a handful of highly transposase-inaccessible transcribed regions (HINTs) (Melfi et al. 2021).
Other genes in which particularly strong putative paused RNA polymerase peaks are observed include the GrpE2 heat shock protein (Fig. 3F), a LuxR2 transcriptional activator gene (Fig. 3G), the chaperone protein ClpB ATP-dependent unfoldase (Supplemental Fig. 2A), the RNA Polymerase Sigma Factor RpoH1 (Supplemental Fig. 2B), and others. For each of these proteins, especially the heat shock ones, it is plausible that polymerase pausing is a regulatory mechanism specifically designed to enable their very rapid activation, in a manner analogous to heat shock genes in Drosophila in which the phenomenon was originally described (Gilmour and Lis 1986; Rougvie and Lis 1988).
The strongest ATAC-seq peak in the genome is found in an unusually large intergenic space without any annotated genes located between replication-related genes and near the origin of replication (Fig. 3H). This locus exhibits only a modest ssDNA peak and may be associated with replication initiation.
Chromatin accessibility correlates with transcriptional activity in Bacteriovorax
Our next step was to characterize the relationship between chromatin accessibility and transcriptional activity in Bacteriovorax as well as their dynamics upon large-scale gene expression perturbations. Although genome-wide gene expression dynamics in these organisms have not been studied extensively previously, it has been reported that starvation of Bdellovibrio cells for 4 h in HEPES buffer results in major changes in gene expression (Dwidar et al. 2017).
We carried out ATAC-seq and KAS-seq in multiple replicates from the same cells and at the same time in both exponentially growing and HEPES-starved cultures and, indeed, observed large-scale changes in transcriptional activity and chromatin accessibility (Fig. 4A–C). Chromatin accessibility around promoters largely disappeared in starved cells, even though transcriptional activity was either unaffected at many promoters or shifted markedly in others. The overall properties of the active transcription landscape were similar to those of exponentially growing cells, except for a slight shift from promoters toward presumed elongation over gene bodies for a number of genes (Fig. 4C). The observed ATAC-seq patterns in HEPES-starved cells, namely, the disappearance of the open chromatin promoter signature, can be explained in three ways. It is possible (1) that starvation induces closure of promoters; (2) that it results in the opposite, histones are actively removed from chromatin in general; and (3) that we observe the effects of cell death and transposition into naked DNA. However, we replicated these results over multiple time courses, and the active transcription maps, which look largely the same in terms of general properties as those of exponentially growing cultures, were generated side by side, from the very same samples that chromatin accessibility was mapped in. Therefore, one of the first two explanations is likely to be correct, but which of the two is a question to be resolved by the subsequent studies.
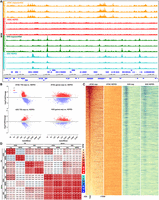
Relationship between chromatin accessibility and transcriptional activity in Bacteriovorax. (A) Starvation of Bacteriovorax cells induces changes in the chromatin accessibility and active transcription landscapes. Shown are three independent KAS-seq and ATAC-seq replicates for each condition. (B) Differential accessibility and KAS levels between exponentially growing and starved (HEPES) cells. (C) Global ATAC-seq and KAS-seq profiles over Bacteriovorax genes under exponentially growing and starvation conditions. (D) Correlation between ATAC-seq and KAS-seq signal over TSSs and gene bodies.
Previously, we found no correlation between chromatin accessibility and transcriptional activity in the archaeon H. volcanii (Marinov et al. 2023). In contrast, ATAC-seq signal over both promoter regions and gene bodies correlates positively with KAS signal, again over both promoter regions and gene bodies (Fig. 4D).
Frequent independent transcription initiation/regulation of individual genes within Bacteriovorax operons
Functionally related genes are often organized into operons in both bacteria and archaea (Price et al. 2006) that are transcribed together. However, evidence has accumulated over the years that certain genes within operons may be transcribed and regulated separately from the whole operon (Koide et al. 2009; Babski et al. 2016; Grüberger et al. 2019; Laass et al. 2019). KAS-seq and ATAC-seq data for the archaeon Haloferax also demonstrated this phenomenon on a genome-wide scale: Individual promoters, discernible by strong ATAC-seq and/or KAS-seq peaks inside operons, are frequently observed in that organism, as are obviously different KAS-seq levels over different genes within a single operon (Marinov et al. 2023).
We aimed to determine whether similar unexpected complexity in the functional organization of operons also exists in the genomes of bacteria with histone-based chromatin. To this end we identified cases of clear operons (i.e., colinear genes obviously belonging to the same functional group) and examined KAS-seq and ATAC-seq profiles in both conditions that we assayed (Fig. 5). We observed multiple cases of both internal KAS-seq and ATAC-seq peaks inside operons (e.g., Fig. 5B,E). Although internal KAS-seq peaks could be associated with polymerase pausing owing to regulation of transcriptional elongation, RNA processing, or cotranscriptional translation events, internal ATAC-seq peaks most likely correspond to independent promoters.
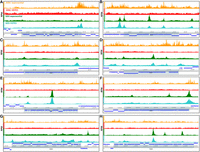
Chromatin accessibility and active transcription levels in Bacteriovorax operons. (A–H) Genome browser snapshots of ATAC-seq and KAS-seq levels in exponentially growing and starved (HEPES) cells for eight different Bacteriovorax operons. (A) NADH ubiquinone oxidoreductase, (B) UDP-N-acetylmuramic acid synthesis pathway enzymes, (C) NADH ubiquinone oxidoreductase (another operon), (D) DNA gyrase, (E) flagellum genes, (F) general secretion pathway genes, (G) flagellum genes (another operon), and (H) flagellum genes (another operon).
3D organization of the Bacteriovorax genome
Finally, we characterized the 3D organization of the Bacteriovorax genome. We employed a modification of the Hi-C assay, using multiple restriction four-cutter enzymes to digest cross-linked Bacteriovorax chromatin to improve resolution in its very small and compact genome (for details, see Methods).
Hi-C has been previously applied to study the physical organization of several bacterial and archaeal strains such as, originally, C. crescentus (Le et al. 2013; Yildirim and Feig 2018) and, later, multiple others (Marbouty et al. 2014, 2015, 2017, 2021; Trussart et al. 2017; Lioy et al. 2018; Böhm et al. 2020; Lamy-Besnier et al. 2023). These studies have revealed two main typical features of bacterial chromosomes. In Caulobacter and most other bacteria, Hi-C maps exhibit a prominent pattern of increased interactions perpendicular to the main diagonal and connecting the origin and the terminus of replication, reflecting a chromosome configuration driven by SMC/condensin complexes loaded at centromere-like parS sites near replication origins (Wang et al. 2017). The parABS system (Austin and Abeles 1983a,b; Abeles et al. 1985; Jalal and Le 2020) plays a major role in this process, with most bacteria having one or multiple highly conserved (Livny et al. 2007) parS sites near their origin of replication. The second common major feature are self-interacting chromosomal interaction domains (CIDs), areas of increased local contact frequency corresponding to plectonemes arising because of transcription-induced supercoiling (Le et al. 2013).
Hi-C maps of archaea differ considerably: Sulfolobus, which does not have histones, appears to possess three distinct origins of replication, resulting in a much more complicated chromosomal configuration, and does not display CIDs (Takemata et al. 2019), whereas Haloferax, which has histones (although it is not clear to what extent they package the genome), was reported to exhibit CIDs but not the perpendicular to the main diagonal feature typical to bacteria (Cockram et al. 2021).
In contrast, Bacteriovorax, although it does possess histones, exhibits a typical large-scale bacterial organization of its chromosome, with very strong cross-diagonal interactions connecting the replication origin and termination regions (Fig. 6A). However, we do not observe clear evidence for plectonemic CID-like structures in our maps. HEPES-starved cells exhibit decreased strength of the cross-diagonal feature (Fig. 6B), likely reflecting much lower replication activity.
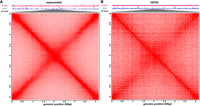
Three-dimensional organization of the Bacteriovorax sp. ICPB 3264 [H-I A3.12] chromosome. (A) Hi-C map of exponentially growing Bacteriovorax cells. (B) Hi-C map of starved (HEPES) Bacteriovorax cells.
Discussion
The existence of bacteria with histone-based chromatin represents one of the most surprising and exciting discoveries in chromatin biology in recent years, coming after the assumption that histones are restricted to archaea and eukaryotes had reigned for many decades. Our study, carried out in a novel Bacteriovorax sp. strain whose genome we assembled and annotated de novo, presents the first direct measurements of chromatin accessibility, the distribution of active transcription and ssDNA, and 3D genome conformation in such bacteria, and it allows us to chart some of the broadest trends in chromatin organization across the tree of life (Fig. 7; Supplemental Fig. 3).

Comparison of the chromatin landscape across the deep organismal diversity. Shown are the presence of histones, histone-based chromatin, the basic genomic properties (i.e., genome size, gene length, gene density/intergenic space size), and the extent of concentration of ATAC-seq signal around promoters (TSS score; for details, see Methods).
We find that Bacteriovorax sp. encodes a histone that is a doublet of two histone folds. Like the euryarchaeon Haloferax and similar to eukaryotes with nucleosomal chromatin, Bacteriovorax’s chromatin is characterized by preferentially accessible promoter regions. The relative level of promoter accessibility is comparable to that of Haloferax, is slightly lower than yeast, and is considerably lower than metazoans with much larger genomes. Likely this is a reflection of high transcriptional activity in extremely compacted genomes; that is, actively transcribed regions temporarily lose the protection from transposase insertion conferred by histones or other packaging proteins, and the density of actively engaged polymerase molecules is higher in such cases than it is in mammals with highly bloated genomes. We see some more direct evidence for this phenomenon in the case of the rDNA operon in Bacteriovorax, which is the most broadly accessible gene in the genome, similar to the situation with rDNA in yeast.
Common with eukaryotes but unlike the euryarchaeon Haloferax, Bacteriovorax exhibits positive correlation between chromatin accessibility and active transcription, suggesting that displacing histones from promoters might play a regulatory role in this organism. However, two observations complicate such an interpretation. First, we do not observe a protection signature in the ATAC-seq fragment length distribution analogous to what is seen in eukaryotes and also in some archaea with histone-based chromatin (Ofer et al. 2023); it is thus not entirely clear how exactly Bacteriovorax histones physically protect DNA and bind to it (somewhat different models have been proposed in the past year) (Hocher et al. 2023; Hu et al. 2024). Second, in starved cells we observe loss of ATAC-seq enrichment over promoters but not of actively transcribing polymerases. This observation can be interpreted in two ways: either promoters close upon strong stress signals, implying that Bacteriovorax histones are not strongly inhibitory to polymerase activity, or, alternatively, histones are lost from chromatin, and this loss does not strongly affect polymerase engagement with DNA, even though histones appear to be essential in these organisms. Single-molecule mapping of chromatin accessibility (Kelly et al. 2012; Krebs et al. 2017; Shipony et al. 2020) and examination of a much broader array of conditions can be expected to shed light on these questions.
We also observe evidence for very strong polymerase pausing over Bacteriovorax genes, in contrast to what has been reported for bacteria such as E. coli (Chivu et al. 2023) and similar to what is seen in many (though not all) eukaryotes. Regulation of productive transcriptional elongation might therefore be a key mechanism of modulating gene expression in bacteria with histone-based chromatin.
We also find evidence for independent regulation of and transcription initiation from internal promoters inside Bacteriovorax operons, similar to what is observed in the euryarchaeon Haloferax.
Finally, the Bacteriovorax genome exhibits 3D organization typical for bacteria, centered on the axis defined by the replication origin and terminus. However, it does not display clear plectonemic CIDs. This interesting observation might further illuminate the mechanisms of formation or lack of such domains. Strong plectonemic CIDs are rare in eukaryotes with one notable exception: dinoflagellates, which display the largest and most pronounced such domains of any organism assayed so far, likely thanks to their unique genomic organization featuring unidirectional gene arrays many hundreds of kilobases long (thus generating extreme levels of supercoiling stress) and the also unique-to-them loss of histones as a main packaging component; it has been thus hypothesized that the frequent and strong interactions between nucleosomes in other eukaryotes override the formation of plectonemes driven by transcription-induced supercoiling, but in dinoflagellates, that effect is unmasked (Marinov et al. 2021). Conversely, if analogous interactions are introduced in groups that normally do not feature histones and exhibit supercoiling domains, such as bacteria, it may be expected for CID-like domains to disappear. We do not observe clear CIDs in Bacteriovorax, but they are also not always robust in all bacteria and not seen in the non-histone-carrying crenarcheaote Sulfolobus while having been reported from Haloferax. However, more recent evidence points to Haloferax possessing histone genes but not actually having histone-based chromatin (Dulmage et al. 2015; Jevtić et al. 2019; Sakrikar and Schmid 2021), in which case some other chromatin protein must be responsible for the high protection from Tn5 insertion measured over its genome, and other chromatin proteins might also be actively shaping the physical genome of other archaeons and bacteria. Further expanding the coverage of physical genome mapping over the deep diversity in the tree of life can be expected to resolve these and many other questions.
Methods
Cell culture
We obtained the strain designated as B. bacteriovorus “ICPB 3264 [H-I A3.12]” from ATCC (https://www.atcc.org/products/25637). As discussed in the main text, subsequent analysis revealed that this strain does not in fact belong to the B. bacteriovorus clade but is instead a Bacteriovorax strain for which sequence data did not previously exist.
The cells were grown in ATCC medium 526 (10.0 g peptone, 3.0 g yeast extract, 1.0 L distilled water, autoclaved at 121°C) at 30°C, except where otherwise indicated for the HEPES starvation condition (in which case cells were centrifuged down, and the media was replaced with HEPES buffer supplemented with 2 mM CaCl2 and 4 mM MgCl2 as previously described) (Dwidar et al. 2017).
Genome sequencing
Genomic DNA (gDNA) was isolated using the NEB Monarch gDNA purification kit (T3010).
For Illumina sequencing, gDNA libraries were generated using Tn5 transposition as previously described (Marinov et al. 2023). Briefly, ∼200 ng DNA gDNA was used, with the volume increased to 22.5 μL using ultrapure H2O. Transposition was carried out with 22.5 μL gDNA, 25 μL 2 × TD buffer (20 mM Tris-HCl at pH 7.6, 10 mM MgCl2, 20% dimethyl formamide), and 2.5 μL Tn5, incubating for 15 min at 37°C. Transposed DNA was immediately purified using the QIAGEN MinElute PCR purification kit (28004), with the reaction stopped with 250 μL PB buffer, and elution in 10 μL EB buffer. Final library amplification was carried out by mixing the 10 μL eluate, 10 μL H2O, 2.5 μL i5 primer, 2.5 μL i7 primer, and 25 μL NEBNext high-fidelity 2× PCR master mix, using the following thermocycler program: 3 min at 72°C; 30 sec at 98°C; and 10 cycles of 10 sec at 98°C, 30 sec at 63°C, and 30 sec at 72°C. Final libraries were purified using the MinElute PCR purification kit.
Illumina libraries were sequenced in a 2 × 150 bp format using the Novogene sequencing service.
Nanopore sequencing was carried out using Flongle flowcells and the SQK-LSK114 library generation kit following the manufacturer's instructions.
Genome assembly
The guppy basecaller (version 6.4.6) was used for nanopore basecalling with the following settings: ‐‐flowcell FLO-FLG114 ‐‐kit SQK-LSK114 ‐‐recursive ‐‐trim_strategy none.
Genome assembly was generated using the SPAdes assembler (version 3.15.4) (Bankevich et al. 2012) in a hybrid nanopore/Illumina mode.
Genome annotation
Genome annotation was carried out using the RAST web server (Aziz et al. 2008). Ribosomal RNAs and other ncRNAs were annotated using Infernal (version 1.1.1) (Nawrocki and Eddy 2013) and the RFAM database (version 12.0) (Nawrocki et al. 2015) and RNAmmer (version 1.2) (Lagesen et al. 2007).
Sequence analysis
DNA/RNA and protein multiple sequence alignment was carried out using MUSCLE (Edgar 2004).
Ribosomal RNA phylogenetic trees were built using RAxML-NG (version 1.2.0) (Kozlov et al. 2019) with the following settings: ‐‐model GTR + G. All full-length or near-full-length 16S rRNA sequences for Bdellovibrionota strains are available from the NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) database.
Protein sequences were translated from the newly generated genome annotation and available genome annotations for other sequenced Bdellovibrionota strains, and then protein domains were annotated using HMMER3 (version 3.2.1) (Eddy 2011) and the Pfam database (version 32.0) (Finn et al. 2014).
Short-read 5-methylcytosine profiling
gDNA was sheared on a Covaris E220; converted into sequencing libraries following the EM-seq protocol, using the NEBNext enzymatic methyl-seq kit (NEB E7120L); and sequenced as 2 × 150mers on a NovaSeq X through Novogene.
Adapters were trimmed from sequencing reads using Trimmomatic (version 0.36) (Bolger et al. 2014). Trimmed reads were aligned against our de novo genome assembly using bwa-meth with default settings. Duplicate reads were removed using picard-tools (version 1.99) (http://broadinstitute.github.io/picard/). Methylation calls were extracted using MethylDackel (https://github.com/dpryan79/MethylDackel). Additional analyses were carried out using custom-written Python scripts (see Data access).
ATAC-seq experiments
ATAC-seq experiments were carried out as previously described (Melfi et al. 2021; Marinov et al. 2023).
Cells were fixed by adding 37% formaldehyde (Sigma-Aldrich) at a final concentration of 1% and incubating for 15 min at room temperature. Formaldehyde was then quenched using 2.5 M glycine at a final concentration of 0.25 M. Cells were subsequently centrifuged at 13,000 rpm for 1 min, washed once in 1× PBS, and centrifuged again at 13,000 rpm for 1 min. Lysis was then carried out by resuspending cells in 400 μL permeabilization buffer (33 mM Tris-HCl at pH 8.0, 20% sucrose, 25 μg/mL lysozyme) and by incubating 5 min at 30°C in a ThermoMixer (with shaking at 1000 rpm). Cells were again pelleted at 13,000 rpm for 1 min, resuspended in 50 μL transposition mix (25 μL 2× TD buffer, 2.5 μL Tn5, 22.5 μL ultrapure H2O), and incubated for 15 min at 37°C. The reaction was stopped with the addition of 150 μL IP elution buffer (1% SDS, 0.1 M NaHCO3) and 2 μL Proteinase K (Promega MC5005) and then incubated overnight at 65°C to reverse cross-links. DNA was isolated by adding an equal volume of 25:24:1 phenol:chloroform:isoamyl solution, vortexing and centrifuging for 3 min at 14,000 rpm, purifying the top aqueous phase using the MinElute PCR purification kit, and eluting in 10 μL EB buffer. Libraries were generated as described above.
ATAC-seq data processing
Demultipexed FASTQ files were mapped to the de novo Bacteriovorax sp. genome assembly as 2 × 36mers using Bowtie (version 1.0.1) (Langmead et al. 2009) with the following settings: -v 2 -k 2 -m 1 ‐‐best ‐‐strata. Duplicate reads were removed using picard-tools (version 1.99).
TSS scores were calculated as the ratio of ATAC signal in the region ±100 bp around TSSs versus the ATAC signal of the 100 bp regions centered at the two points ±2 kbp of the TSS as previously described (Marinov and Shipony 2021).
Peak calling was carried out using MACS2 (Feng et al. 2012) with the following settings: -g 4150000 -f BAM ‐‐to-large ‐‐keep-dup all ‐‐nomodel.
KAS-seq experiments
KAS-seq experiments were carried out following the previously published protocol (Wu et al. 2020) with some modifications in the sequencing library generation part (Marinov et al. 2023).
Briefly, a 500 mM N3-kethoxal solution was brought to 37°C and then added to 1 mL of culture at a final concentration of 5 μM. Cells were then incubated for 5 min at 30°C in a ThermoMixer at 1000 rpm.
Cells were then pelleted by centrifugation at 13,000 rpm for 1 min and resuspended in 100 μL 1× PBS buffer, and DNA was immediately isolated using the Monarch gDNA purification kit (NEB T3010S), with the modification that elution was carried out with 87.5 μL 25 mM K3BO3 solution (pH 7.0).
Biotin was clicked onto kethoxal-modified G's by mixing 87.5 μL DNA, 2.5 μL 20 mM DBCO-PEG4-biotin (Sigma-Aldrich 760749; DMSO solution), and 10 μL 10× PBS and incubating for 90 min at 37°C.
DNA was isolated using AMPure XP beads and eluted in 130 μL 25 mM K3BO3 (pH 7.0) and then sheared on a Covaris E220 for 120 sec down to ∼150–200 bp.
Libraries were built on beads using the NEBNext Ultra II DNA library prep kit (NEB E7645L). Biotin pull down was initiated by pipetting 20 μL Dynabeads MyOne streptavidin T1 beads (Thermo Fisher Scientific 65306) into DNA lo-bind tubes. Beads were separated on magnet, resuspended in 200 μL of 1× Tween washing buffer (TWB; 5 mM Tris-HCl at pH 7.5, 0.5 mM EDTA, 1 M NaCl, 0.05% Tween 20), and then separated on magnet again and resuspended in 300 μL of 2× binding buffer (BB; 10 mM Tris-HCl at pH 7.5, 1 mM EDTA, 2 M NaCl). The DNA (130 μL) was added together with 170 μL 0.1× TE buffer and incubated at RT on rotator for ≥15 min. Beads were separated on magnet, resuspended in 200 μL of 1× TWB, and incubated in a ThermoMixer for 2 min with shaking at 1000 rpm at 55°C. Beads were again separated on magnet, and the 200 μL 55°C TWB wash step was repeated. Beads were separated on magnet and resuspended in 50 μL 0.1× TE.
End repair was carried out by adding 7 μL NEB end repair buffer and 3 μL NEB end repair enzyme, incubating for 30 min at 20°C then for 30 min at 65°C.
End repair was followed by adaptor ligation by adding 2.5 μL NEB adaptor, 1 μL NEB ligation enhancer, and 30 μL NEB ligation mix; incubating for 20 min at 20°C; adding 3 μL USER enzyme; and incubating for 15 min at 37°C. Beads were separated on magnet, resuspended in 200 μL of 1× TWB, and then incubated in a ThermoMixer for 2 min with shaking at 1000 rpm at 55°C. Subsequently, beads were separated on magnet and resuspended in 100 μL of 0.1× TE, separated on magnet again, resuspended in 15 μL of 0.1× TE buffer, and transferred to PCR tubes.
Beads were then incubated for 10 min at 98°C, and libraries were amplified by adding 5 μL of i5 primer, 5 μL of i7 primer, and 25 μL of 2× Q5 Hot Start polymerase mix, using the following PCR program: 30 sec at 98°C, 15 cycles for 10 sec at 98°C, 30 sec at 65°C, and 30 sec at 72°C; and a final extension for 5 min at 72°C.
Beads were separated on magnet, and the final libraries were purified from the supernatant using 50 μL AMPure XP beads, eluting in 0.1× TE buffer.
KAS-seq data processing
Demultipexed FASTQ files were mapped to the de novo Bacteriovorax sp. genome assembly as 2 × 36mers using Bowtie (Langmead et al. 2009) with the following settings: -v 2 -k 2 -m 1 ‐‐best ‐‐strata. Duplicate reads were removed using picard-tools (version 1.99).
Differential accessibility/KAS-seq analysis
The analysis of differential chromatin accessibility as measured using ATAC-seq or enriched for KAS-seq signal was carried out using DESeq2 (Love et al. 2014). Read counts were calculated over promoters or gene bodies and used as input into DESeq2.
Hi-C experiments
Hi-C experiments were carried out as previously described (Marinov et al. 2021, 2022) with some modifications.
Cells were cross-linked using 37% formaldehyde (Sigma-Aldrich) at a final concentration of 1% for 15 min at room temperature. Formaldehyde was then quenched using 2.5 M glycine at a final concentration of 0.25 M. Cells were centrifuged at 13,000 rpm for 2 min, washed once in 1× PBS, and stored at −80°C.
Denaturation was carried out by resuspended cells in 50 μL of 0.5% SDS and incubating for 10 min at 62°C. SDS was quenched by adding 145 μL of H2O and 25 μL of 10% Triton X-100 and incubating for 15 min at 37°C.
Restriction digestion was carried out by adding 25 μL of 10× CutSmart buffer and 100 U of the MluCI restriction enzyme (NEB R0538) plus 100 U of the MboI enzyme (NEB R0147) and then by incubating for ≥2 h at 37°C in a ThermoMixer at 900 rpm. The reaction was then incubated for 20 min at 62°C in order to inactivate the restriction enzymes.
Fragment ends were filled in by adding 37.5 μL of 0.4 mM biotin-14-dATP (Thermo Fisher Scientific 19524-016); 1.5 μL each of 10 mM dCTP, dGTP, and dTTP; and 8 μL of 5U/μL DNA Polymerase I large (Klenow) fragment (NEB M0210). The reaction was the incubated in a ThermoMixer at 900 rpm for 45 min at 37°C.
Fragment end ligation was carried out by adding 663 μL H2O, 120 μL 10× NEB T4 DNA ligase buffer (NEB B0202), 100 μL of 10% Triton X-100, 12 μL of 10 mg/mL bovine serum albumin (100× BSA, NEB), 5 μL of 400 U/μL T4 DNA ligase (NEB M0202) and by incubating for ≥4 h with rotation at room temperature.
Cells were then pelleted by centrifugation at 13,000 rpm for 5 min. The pellet was resuspended in 200 μL elution buffer (1% SDS, 0.1 M NaHCO3); Proteinase K was added and incubated overnight at 65°C to reverse cross-links.
After addition of 600 μL 1 × TE buffer, DNA was sheared using a Covaris E220 instrument. DNA was then purified using the MinElute PCR purification kit (QIAGEN 28006), with elution in a total volume of 300 μL 1× EB buffer.
For streptavidin pulldown of biotin-labeled DNA, 150 μL of 10 mg/mL Dynabeads MyOne Streptavidin T1 beads (Life Technologies 65602) was separated on a magnetic stand and then washed with 180 μL of 1× Tween washing buffer (TWB; 5 mM Tris-HCl at pH 7.5, 0.5 mM EDTA, 1 M NaCl, 0.05% Tween 20). Beads were then resuspended in 300 μL of 2× binding buffer (10 mM Tris-HCl at pH 7.5, 1 mM EDTA, 2 M NaCl); the sonicated DNA was added; and beads were incubated for ≥15 min at room temperature on a rotator. After separation on a magnetic stand, the beads were twice washed with 180 μL of 1× TWB and heated in a ThermoMixer with shaking for 2 min at 55°C.
Final libraries were prepared on beads using the NEBNext Ultra II DNA library prep kit (NEB E7645). End repair was carried out by resuspending beads in 50 μL 1× EB buffer and adding 3 μL NEB ultra end repair enzyme and 7 μL NEB ultra end repair enzyme, followed by incubation for 30 min at 20°C and then for 30 min at 65°C.
Adapters were ligated to DNA fragments by adding 30 μL blunt ligation mix, 1 μL ligation enhancer, and 2.5 μL NEB adapter; incubating for 20 min at 20°C; adding 3 μL USER enzyme; and incubating for 15 min at 37°C.
Beads were then separated on a magnetic stand and washed with 180 μL TWB for 2 min at 55°C at 1000 rpm in a ThermoMixer. After separation on a magnetic stand, beads were washed in 100 μL 0.1 × TE buffer, resuspended in 16 μL 0.1 × TE buffer, and heated for 10 min at 98°C.
For PCR, 5 μL of each of the i5 and i7 NEB Next sequencing adapters were added together with 25 μL 2× NEB ultra PCR Mater Mix. PCR was carried out with a 98°C incubation for 30 sec; 12 cycles of 10 sec at 98°C, 630 sec at 5°C, and 1 min at 72°C; and incubation for 5 min at 72°C.
Beads were separated on a magnetic stand, and the supernatant was cleaned up using 1.8× AMPure XP beads.
Libraries were sequenced as 2 × 150mers on a Illumina NovaSeq X through Novogene.
Hi-C data processing
Hi-C sequencing reads were processed against the de novo Bacteriovorax sp. assembly using the Juicer pipeline for analyzing Hi-C data sets (version 1.6 of Juicer and version 2.13.07 of Juicer Tools) (Durand et al. 2016a) with default settings.
Hi-C matrices were visualized using Juicebox (Durand et al. 2016b).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE245010. Except where explicitly indicated otherwise, data were processed using custom-written Python scripts available at GitHub (https://github.com/georgimarinov/GeorgiScripts) and as Supplemental Code.
Competing interest statement
W.J.G. is a consultant and equity holder for 10x Genomics, Guardant Health, Quantapore, and Ultima Genomics; is cofounder of Protillion Biosciences; and is named on patents describing ATAC-seq. A.K. is a consulting Fellow with Illumina; a member of the SAB of OpenTargets (GSK), PatchBio, and SerImmune; and is a scientific cofounder of RavelBio.
Acknowledgments
We thank members of the Greenleaf and Kundaje laboratories for helpful discussions. This work was supported by National Institutes of Health grants (P50HG007735, RO1 HG008140, U19AI057266, and UM1HG009442 to W.J.G., 1UM1HG009436 to W.J.G. and A.K., 1DP2OD022870-01 and 1U01HG009431 to A.K.), the Rita Allen Foundation (to W.J.G.), the Baxter Foundation Faculty Scholar Grant, and the Human Frontiers Science Program grant RGY006S (to W.J.G.). W.J.G. is a Chan Zuckerberg Biohub investigator and acknowledges grants 2017-174468 and 2018-182817 from the Chan Zuckerberg Initiative.
Author contributions: G.K.M. conceptualized the study, carried out cell culture, and performed ATAC-seq, KAS-seq, and Hi-C experiments together with B.D.; analyzed the data; and wrote the manuscript, with input from all authors. B.D. performed EM-seq experiments. W.J.G. and A.K. supervised the study.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279418.124.
- Received March 29, 2024.
- Accepted November 19, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








