
Secure discovery of genetic relatives across large-scale and distributed genomic data sets
- Matthew M. Hong1,5,
- David Froelicher1,2,5,
- Ricky Magner2,
- Victoria Popic2,
- Bonnie Berger1,2,3 and
- Hyunghoon Cho4
- 1Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA;
- 2Broad Institute of the Massachusetts Institute of Technology and Harvard, Cambridge, Massachusetts 02142, USA;
- 3Department of Mathematics, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA;
- 4Department of Biomedical Informatics and Data Science, Yale University, New Haven, Connecticut 06510, USA
-
↵5 These authors contributed equally to this work.
Abstract
Finding relatives within a study cohort is a necessary step in many genomic studies. However, when the cohort is distributed across multiple entities subject to data-sharing restrictions, performing this step often becomes infeasible. Developing a privacy-preserving solution for this task is challenging owing to the burden of estimating kinship between all the pairs of individuals across data sets. We introduce SF-Relate, a practical and secure federated algorithm for identifying genetic relatives across data silos. SF-Relate vastly reduces the number of individual pairs to compare while maintaining accurate detection through a novel locality-sensitive hashing (LSH) approach. We assign individuals who are likely to be related together into buckets and then test relationships only between individuals in matching buckets across parties. To this end, we construct an effective hash function that captures identity-by-descent (IBD) segments in genetic sequences, which, along with a new bucketing strategy, enable accurate and practical private relative detection. To guarantee privacy, we introduce an efficient algorithm based on multiparty homomorphic encryption (MHE) to allow data holders to cooperatively compute the relatedness coefficients between individuals and to further classify their degrees of relatedness, all without sharing any private data. We demonstrate the accuracy and practical runtimes of SF-Relate on the UK Biobank and All of Us data sets. On a data set of 200,000 individuals split between two parties, SF-Relate detects 97% of third-degree or closer relatives within 15 h of runtime. Our work enables secure identification of relatives across large-scale genomic data sets.
Collaborative studies that aim to jointly analyze genomic data from multiple parties are essential for increasing the sample sizes to enhance the discovery of biomedical insights. However, when sharing individual-level genetic data is not feasible owing to privacy concerns (e.g., Erlich et al. 2018), the range of joint analyses that can be performed is severely limited. As a result, many existing collaborations have relied on simplified analysis pipelines in which some key analysis steps, such as cohort identification, quality-control procedures, and correction for confounding factors (e.g., population structure), are performed independently by each party on their respective data sets without considering the pooled data. This presents a key barrier to realizing the full potential of collaborative genomics research.
An important analysis task that is commonly omitted in collaborative studies is the identification of genetic relatives across isolated data sets. Identifying and excluding close relatives within a study cohort is a standard step in many genetic analyses (e.g., genome-wide association studies [GWAS]) (Anderson et al. 2010), because the presence of relatives can introduce bias and confounding that undermine the accuracy of study results (Devlin and Roeder 1999; Newman et al. 2001; Voight and Pritchard 2005; Astle and Balding 2009; Kang et al. 2010; Shibata et al. 2013; Bhatia et al. 2016; Hellwege et al. 2017; Young et al. 2019). For large-scale biobanks, a substantial portion of study participants may be biologically related; an estimated 32.3% of the individuals in the UK Biobank (UKB) data set (Bycroft et al. 2018) have a third-degree or closer relative in the same data set. Thus, controlling for relatedness can have a major impact on the size and composition of the analysis cohort and thereby can affect the final analysis results. Removal of duplicate individuals across data sets is a special case of detecting relatives, which our work also addresses.
There are several key hurdles to identifying related individuals across data sets. Unlike other analysis tasks that derive aggregate-level insights from the pooled data, such as association tests, finding relatives is an inherently sensitive task, directly operating at the level of individuals. Consequently, most existing approaches for cross-site analysis, for example, meta-analysis or federated learning, cannot be applied in our setting, as they rely on sharing aggregate-level data between the parties. Furthermore, despite the growing literature on cryptography-based secure computation algorithms for biomedicine (Cho et al. 2018, 2022; Blatt et al. 2020; Froelicher et al. 2021b), which allow joint computation without sharing private data between parties, to our knowledge no practical solution exists for relative detection. This is mainly because standard tools for evaluating kinship require all pairs of individuals between two data sets to be compared (Manichaikul et al. 2010; Chang et al. 2015; Conomos et al. 2016) or involve complex combinatorial operations (e.g., string matching) (Gusev et al. 2009; Naseri et al. 2019; Shemirani et al. 2021), which incurs an overwhelming cost when implemented using cryptographic operations.
In this work, we introduce SF-Relate, a scalable and privacy-preserving solution for identifying relatives across distributed data sets, as illustrated in Figure 1. Our novel approach entails each party locally assigning their samples to buckets via a type of locality-sensitive hashing (LSH) (Indyk and Motwani 1998) and then securely estimating kinship only between samples that end up in the same bucket across parties. We devise new hashing and bucketing strategies aimed at effectively distinguishing relatives from nonrelatives while minimizing the number of sample pairs that need to be compared. Furthermore, to securely estimate kinship coefficients for these sample pairs, we introduce a secure approach based on homomorphic encryption, a cryptographic technique that allows direct computation on encrypted data. We combine this approach with our efficient distributed computation techniques to minimize the overhead of cryptographic operations. Overall, our study provides the first practical demonstration of secure relative identification across large-scale genomic data sets, including hundreds of thousands of individuals, with a strong, formal notion of privacy protection.
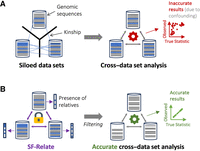
SF-Relate overview. (A) When genetic relatives across data sets cannot be identified owing to restricted data sharing, joint studies can suffer from bias and confounding. (B) SF-Relate allows parties to securely identify and correct for cross–data set relatives to enhance downstream analysis.
Results
Overview of SF-Relate
SF-Relate enables multiple parties to detect cross-site relatives in their joint data set without having to share any sensitive information (Fig. 1). The input data set for each party includes phased haplotype sequences from individuals within that party's cohort. We consider the parties to be honest-but-curious, meaning that they follow our analysis protocol faithfully but might try to infer information about other parties’ data sets based on what they observe individually during the protocol execution. Based on this model, SF-Relate guarantees end-to-end confidentiality for each party's input data set, protecting it from other parties in the protocol. During the protocol, any data exchanged between the parties are encrypted in a manner that requires the participation of all parties for decryption, thus ensuring a high level of protection. This approach allows parties to disclose only the information they agree to reveal, such as the final output.
To efficiently scale to large data sets, SF-Relate follows a two-step approach (Methods). In Step 1, Hashing and Bucketing, each party locally evaluates a series of hash functions on each individual's haplotype sequences to assign the individual to buckets across a collection of hash tables, such that related individuals are more likely to be assigned to the same bucket index. For this purpose, we devised a novel encoding scheme that splits and subsamples genotypes into k-SNPs (similar to k-mers, but noncontiguous; single-nucleotide polymorphism [SNP]), such that the similarity between k-SNPs reflects extended runs of identical genotypes, typically indicative of relatedness. We then leverage LSH to derive bucket indices from the k-SNPs. To capitalize on the fact that related samples will likely be assigned to the same buckets multiple times, SF-Relate merges buckets with the same indices across multiple hash tables (produced by different subchromosomes) and then filters every bucket down to a single element, thus minimizing the number of costly kinship evaluations. We refer to this as a microbucketing strategy. Despite the extreme level of filtering applied to each bucket during this process, our strategy enables accurate detection of relatives. At the end of this process, each party obtains a single hash table with size-one buckets, effectively an ordered list of samples.
In Step 2, Secure Kinship Evaluation, the parties securely perform element-wise comparisons between their ordered lists of samples from step 1. Each comparison involves evaluating a standard estimator of the kinship coefficient, KING (Manichaikul et al. 2010). To calculate the estimator without revealing private information between the parties, we employ multiparty homomorphic encryption (MHE) (Mouchet et al. 2021). Data encrypted under MHE can be directly used in computation without needing to be decrypted first, and decryption requires the cooperation of all parties. To minimize the computational overhead of MHE, SF-Relate uses sketching techniques on input haplotypes to reduce data dimensionality before performing kinship computations. Furthermore, our protocol is optimized to maximize the use of operations on local, nonencrypted data, which is more efficient than operations on encrypted data. Finally, the encrypted results are compared to relatedness thresholds and aggregated for each individual, providing each party with an indicator that reflects the presence of a close relative in the other data set.
We detail our algorithms and novel techniques in the Methods.
Data sets and evaluation settings
To evaluate SF-Relate, we obtained three genomic data sets of varying sizes, including a data set of 20,000 samples (individuals) with 1 million SNPs from the All of Us Research Program (AoU) (All of Us Research Program Investigators 2019) and two data sets from the UKB (Bycroft et al. 2018) including 100,000 and 200,000 samples, respectively, both with 650,000 SNPs. The two UKB data sets were uniformly sampled from the full UKB release v3 (n = 488, 377), and the AoU data set comprises the first 20,000 individuals in the All of Us release v5 (n = 98, 590). We then evenly split each data set into two parts to emulate a cross–data set analysis involving two parties. We compute the ground truth by evaluating all pairwise kinship coefficients using the KING approach (see Methods) (Manichaikul et al. 2010) in plaintexts on a set of ancestry-agnostic SNPs, as in UKB's pipeline (Bycroft et al. 2018). We provide further details on data set preparation in the Methods.
We evaluated the accuracy of our method in detecting close relatives between two data sets using the recall and precision metrics. Recall represents the fraction of samples with a close relative in the other data set (as determined by the baseline KING method given a threshold) that SF-Relate successfully identifies. Precision represents the fraction of samples identified by SF-Relate as having a close relative in the other data set that actually have such a relationship according to the baseline method. For evaluation of computational costs, we measured the elapsed wall-clock time and the total number of bytes sent from one party to another (given the symmetry of SF-Relate's computation) for runtime and communication costs, respectively. In addition, we monitored the peak memory usage of SF-Relate.
Although not required by our method, all of our experiments were performed using virtual machines (VMs) on the Google Cloud Platform (GCP). This represents a setting where parties use a cloud service provider to access high-performance computing resources that may not be readily available in the local environment. Furthermore, many biobank data sets, including AoU and UKB, are now hosted on cloud-native environments for data analysis. For UKB, we used two VMs (one for each party) with 128 virtual CPUs (vCPUs) and 856 GB of memory (n2-highmem-128) colocated in the same zone in GCP. For AoU, we emulated the two parties in a single VM with 96 vCPUs and 624 GB memory owing to the constraints of the provided data analysis platform. Supplemental Table S1 summarizes all symbols and default parameters.
SF-Relate accurately and efficiently detects close relatives between large-scale data sets
We summarize our results on the AoU and UKB data sets in Tables 1 and 2. Across all three data sets (AoU-20K, UKB-100K, and UKB-200K), SF-Relate obtains near-perfect recall and precision (both exceeding 97% in all cases) for detecting the presence of third-degree or closer relationships between two parties. Calculating the recall separately for each relatedness degree from zero (monozygotic twins) to third, we observe that most missing relationships are for the third degree; SF-Relate finds all existing relationships up to the second degree in all three data sets, with the exception of the second degree in UKB-200K, for which it missed two out of 1711 individuals with a relative (99.9% recall). The recall metric for third-degree relationships remains high, >94% for all three data sets. Note that the more distant the relationship, the more difficult it is to detect, because the identity-by-descent (IBD) segments become more scattered and reduced in quantity, which in turn results in a lower rate of surviving the filtering step in microbucketing. In UKB-200K, we observed that a small fraction (5%) of third-degree relatives, missed by SF-Relate, correspond to those with kinship coefficients near the fourth-degree threshold (Fig. 2), suggesting that some of them may not be real third-degree relationships considering the stochastic nature of the kinship estimator.

SF-Relate achieves higher accuracy for samples with closer kinship and enables a trade-off between accuracy and runtime. (A) We plot the distribution of kinship coefficients (KING) stratified by the (closest) relatedness degree of the relative pairs and by whether they were detected by SF-Relate as related. Misclassifications by SF-Relate are concentrated around kinship thresholds for different relatedness degrees, indicated by vertical dashed lines. (B) We vary the subsampling ratio (s) and the table ratio (τ) parameters in SF-Relate and report the resulting precision and recall for different relatedness degrees. For precision, only the overall metric for detecting third-degree or closer relatives is shown. By default, s = 0.7 and τ = 128. These parameters determine the trade-off between the runtime and accuracy of SF-Relate.
SF-Relate achieves near-perfect accuracy for identifying close relatives in the UK Biobank and All of Us data sets
SF-Relate scales efficiently to large data sets
Furthermore, SF-Relate consistently achieves high detection accuracy across a variety of populations with distinct ancestry backgrounds. SF-Relate maintains a recall rate >98% for a data set comprising individuals of African ancestry (Supplemental Table S2). SF-Relate largely remains effective in multiancestry data sets, achieving a recall >80% across all subpopulations (Supplemental Table S3). Ancestry groups with the lowest recall (Indian and Other with 84.4% and 82.4%, respectively) are associated with small sample counts, suggesting that the slight reduction in recall may be caused by sampling noise. Taken together, these results demonstrate SF-Relate's accurate relative detection performance across a range of data sets, which is achieved without revealing any private information between the two parties owing to SF-Relate's use of secure computation techniques when jointly analyzing the two data sets.
Despite the overhead of cryptographic protocols for secure computation, the runtime of SF-Relate remains practical for all three data sets, resulting in 5.8, 7.3, and 14.5 h of runtime for AoU-20K, UKB-100K, and UKB-200K, respectively. We note that the doubling of runtime from UKB-100K to UKB-200K reflects the linear scaling of SF-Relate in the number of individuals in the data set, because these data sets were analyzed using the same computing environment, unlike AoU. More precisely, the computational cost of the MHE calculation of pairwise kinship coefficients, which is the main computational bottleneck of SF-Relate, grows linearly in both the number of SNPs after sketching and the size of the hash table. Although both parameters can be adjusted by the user, to maintain accurate performance, these parameters need to be linearly scaled with the total number of SNPs and individuals in the original input data set, respectively. We provide a systematic evaluation of the runtime scaling of SF-Relate in Supplemental Figure S1.
The observed communication costs on the order of tens of terabytes (e.g., 93.2 TB for UKB-200K) are not small, but our results demonstrate that transferring such large amounts of data does not lead to impractical runtimes. In addition, we note that >99% of the communication bandwidth is owing to the exchange of encrypted hash tables, including the sketched haplotypes, which are 16 times larger than the unencrypted data with our cryptographic parameters. We note that the hash tables can, in principle, be transferred in a single round of communication. Therefore, we expect the impact of communication on runtime to be minimal even in a wide-area network (WAN) setting with high communication latency (round-trip delay).
Memory usage of SF-Relate can be easily adjusted to meet existing resource limits. SF-Relate computes the pairwise kinship coefficients in blocks, that is, in a streaming fashion over the encrypted genotype vectors. This approach allows users to control memory usage by setting the block size and the number of parallel processes. By default, SF-Relate configures these parameters to prioritize throughput while maintaining practical memory usage that can be supported by high-performance servers (e.g., 550 GB for 20 parallel processes for UKB-200K).
We highlight that without the hashing and bucketing strategy we introduced in SF-Relate, it would not be feasible to securely detect relatives between data sets by all-pairwise computation of the kinship coefficient (see all-pairwise columns in Table 2). Even with our efficient MHE implementation of the kinship calculation over the sketched haplotypes, performing all-pairwise comparisons for the UKB-200K data set is estimated to take 1.3 years based on the same computational setting. On the contrary, SF-Relate obtains practical runtimes by reducing the number of candidate individual pairs to test without compromising accuracy through our novel use of LSH hash tables. SF-Relate makes only 1.28%, 0.26%, and 0.13% of pairwise comparisons compared with the total number of individual pairs between the two data sets for AoU-20K, UKB-100K, and UKB-200K, respectively (Table 1). This drastically reduces not only the runtime but also the communication costs; for example, our MHE computation would require 65 PB of communication without our hashing techniques (Table 2).
Other cryptographic frameworks, such as secure multiparty computation (MPC), could be used to securely compute kinship coefficients instead of MHE. For comparison, we implemented an alternative MPC solution based on prior work on GWAS (Cho et al. 2018). We found that the MPC approach is consistently eight times slower than SF-Relate for varying data set sizes, resulting in an estimated runtime of ∼5 days for UKB-200K, compared with 14.5 h for SF-Relate (Supplemental Fig. S2). We ascribe this difference to the greater communication burden of MPC and the advantage of MHE in efficiently distributing the workload to leverage local operations on nonencrypted data. Moreover, MPC introduces a noncolluding third party for efficiency (Cho et al. 2018), which weakens the security model.
Navigating SF-Relate's accuracy–runtime tradeoffs and parametrization
The runtime and accuracy of SF-Relate are primarily influenced by the hash table size N = τ · n, determined by the data set size n and table ratio (τ; Methods), and the sample size for comparison, determined by the subsampling ratio (s) and the number of SNPs (Methods). As shown in Figure 2, increasing s improves the overall recall and precision, whereas increasing τ enables the detection of more distant relationships, also increasing overall recall. However, the runtime depends linearly on both s and τ, highlighting the trade-off between SF-Relate's accuracy and runtime. We expect the optimal trade-off to depend on the application setting.
For example, if users want to focus on identifying relatives up to the second degree within the UKB-200K data set, they could set the table ratio τ to 64 and the subsampling rate s to 0.7, instead of τ = 128 and s = 0.7 in our experiments (Methods). This results in a twofold improvement with respect to our experiments owing to halving of the hash table size. Even in this scenario, users would maintain an effective detection rate of >95% for individuals with relationships closer than the second degree (Fig. 2B). Alternatively, if users want to achieve perfect accuracy, they can increase s and τ. Increasing s from 0.7 to one (i.e., no sketching), improved SF-Relate's overall recall on UKB-200K from 97.0% to 98.7% and precision from 98.5% to 99.9% (Table 1; Supplemental Fig. S3). The runtime increased from 14 h to 21 h. Furthermore, by doubling the table ratio τ, SF-Relate achieves perfect accuracy for relations up to the third degree, while doubling its runtime. Overall, SF-Relate's recall remains consistently high, >95%, across a wide range of parameters and only starts to decrease when the parameters significantly deviate from the default setting (Supplemental Table S4).
To choose suitable values for s and τ in practice, we recommend that users first determine the farthest relationships they wish to detect and an acceptable level of recall. Using Figure 2, they can then determine the required s and hash table size, N = τn. The expected runtime can be estimated by considering the linear relationship between these parameters and SF-Relate's runtime. To select the Hashing and Bucketing parameters, the users may initially opt for our recommended parameters (Supplemental Table S1), which consistently achieve accurate and efficient performance across all our data sets. Optionally, users can conduct a local assessment using their own data set to verify and fine-tune the chosen parameters, which involves examining the number of shared genomic segments between local relatives and their matching probabilities (as depicted in Supplemental Fig. S4) and estimating the detection rate among local relatives.
SF-Relate's IBD-based hashing strategy improves detection accuracy over the KING estimator
SF-Relate leverages a secure implementation of the KING formula (Manichaikul et al. 2010) to estimate relatedness (Methods). Nevertheless, we observed that SF-Relate can sometimes lead to even more accurate identification of relatives than KING. This is because of SF-Relate's IBD-based approach to bucketing the samples, which helps filter out spurious pairs of samples that are identified by KING as being related that in fact do not share any long IBD segments. We confirmed this in our comparisons with PC-Relate (Conomos et al. 2016) and RAFFI (Naseri et al. 2021), recent methods for kinship detection designed to improve upon KING's accuracy by correcting for population structure and incorporating IBD segment detection, respectively.
We evaluated all methods on a subset of 20,000 samples from the UKB-200K, distributed across two parties. As expected, they produce highly similar results for up to third-degree relatives (Fig. 3; Supplemental Table S5; Supplemental Fig. S5). However, for detecting fourth-degree relatives, standard KING erroneously identified numerous individuals as being related owing to the presence of an outlier sample that is detected as related to thousands of samples in the other data set; SF-Relate, akin to the more advanced tools PC-Relate and RAFFI, successfully avoids these errors. Similar outlier-related issues regarding KING have been noted in UKB's official report on relatedness inference (see supplemental material in the work of Bycroft et al. 2018). In Figure 3, we visualize sequence similarity between four pairs of haplotypes involving the outlier sample, compared with typical fourth-degree relative pairs identified by PC-Relate. We observe light yellow bands of high sequence similarity regions exclusively in PC-Relate's pairs, which signifies real IBD segments. This suggests that SF-Relate's bucketing approach based on IBD segments can effectively distinguish outlier pairs from real ones, thus leading to more accurate detection of relatives.

SF-Relate excludes spurious fourth-degree relatives detected by KING. (A) We show confusion matrices assessing the relatedness degree classification accuracy of KING (left) and SF-Relate (right), comparing with the output of PC-Relate as the ground truth. SF-Relate is performed in plaintext (i.e., without MHE), focusing on the evaluation of the bucket assignment. Unlike SF-Relate, KING classifies many unrelated samples as fourth-degree relatives. Most of these pairs involve the same outlier sample, which has many spurious relationships. (B) We verify that pairs involving the outlier do not exhibit IBD sharing patterns (left), evident in fourth-degree pairs from PC-Relate (right). Four example pairs are shown for both cases. For each pair, we compute the Hamming similarity between the two samples of genomic segments across the genome. Bright yellow bands represent likely IBD segments. The locations of the bands are randomly permuted to obscure their positions.
SF-Relate supports alternative output settings
In SF-Relate's default setting, each party learns only whether each local individual has a close relative in the other party's data set. SF-Relate offers an option to output kinship computation results with more detailed granularity, summarizing them at various levels to address a range of analysis needs. Alternative outputs include the closest relatedness degree for each individual, the maximum kinship coefficient for each individual (discretized), and the full list of computed kinship coefficients. The SF-Relate output remains accurate in all settings: The individual kinship coefficients computed by SF-Relate exhibit a small average absolute error of 5.8 × 10−4 compared with KING (Supplemental Fig. S6). The closest degree reported for each individual matched with the ground truth for 99.9% of the individuals (Supplemental Fig. S7). Calculating the maximum kinship coefficient, discretized using the smallest bin width of 0.016, SF-Relate accurately assigned >85% of the samples to the correct bins, and >99.9% were within one bin of the true output (Supplemental Fig. S7).
A case study: SF-Relate reduces false positives in GWASs
We show that SF-Relate can be used to improve the accuracy of downstream studies without requiring the sharing of sensitive information. We illustrate this using the GWAS workflow and demonstrate SF-Relate's effectiveness in mitigating confounding from cryptic relatedness by enabling parties to detect and remove relatives from their joint data set prior to conducting the GWAS. We simulate a multisite GWAS using a subset of 100,000 samples from white British participants in UKB, distributed geographically among six parties (Methods) (Supplemental Table S6). Fifty percent of these samples have at least one relative of the third degree or less in the data set. We then simulate 100 phenotypes and perform a linear regression-based GWAS with the top principal component as a covariate (Methods). On average, using a nominal significance cut-off P-value of 0.05, SF-Relate removes 2.60% of the falsely identified loci (false positives) with a drop-in false-positive rate (FPR) from 5.14% to 5.01%, compared with when relatives are not removed from the data set (Fig. 4). When parties independently remove local relatives, 18% of the remaining samples still have relatives in the joint data set, and 2.03% of false positives are removed with a FPR drop from 5.14% to 5.04%. The one-sided Mann–Whitney U test P-value that SF-Relate produces lower FPRs across all phenotypes compared with the local removal of relatives is 1.25 × 10−5. Thus, SF-Relate significantly mitigates confounding, producing a FPR near the nominal cutoff P-value of 0.05, comparable to centrally coordinated sample removal (Fig. 4).
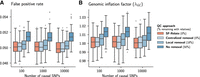
SF-Relate reduces false positives in multisite GWASs. We vary the number of causal SNPs used in simulating the phenotypes, and compare four quality-control (QC) approaches for excluding related individuals: (1) centralized removal (nonprivate), all relatives are removed from the pooled data set; (2) SF-Relate, relatives are removed using our secure approach; (3) local removal, each party filters relatives from its local data set independently; and (4) no removal, no relatives removed. Initially, 50% of samples have relatives, and local removal results in 18% of remaining samples still having a relative in the joint data set. We plot the fraction of significant loci (P-value < 0.05) on even numbered chromosomes that are designed to be noncausal in the simulation (A), and the genome inflation factor λGC in B. The filled boxes represent interquantile ranges of statistics across 100 simulated phenotypes. Although local removal of relatives helps reduce the confounding to some extent, SF-Relate significantly mitigates confounding, comparable to centrally coordinated sample removal.
Discussion
We presented SF-Relate, a secure federated algorithm for identifying close relatives between isolated genomic data sets. Using a novel strategy for hashing and bucketing individuals to capture shared IBD segments between relatives, SF-Relate achieves near-perfect detection while maintaining a practical runtime (i.e., less than a day) even on a large data set including 200,000 individuals. We expect SF-Relate to be a useful tool for the growing networks of collaborating institutions, which currently lack the tools to jointly perform a variety of genetic analyses without sharing data. To facilitate the use of our method, we provide an automated deployment workflow for SF-Relate on the sfkit web server (Mendelsohn et al. 2023), which streamlines the collaborative execution of secure federated tools similar to SF-Relate.
There are several directions that we would like to pursue in future research. First, although SF-Relate identifies relatives based on the standard KING-robust estimator (Manichaikul et al. 2010), there are other approaches that may provide more robust estimation, especially for more distant relatives beyond the third degree, in terms of both correcting for population structure (e.g., PC-Relate) (Conomos et al. 2016) and detecting IBD segments to allow a more direct calculation of the proportion of IBD sharing (e.g., RAFFI) (Naseri et al. 2021). Although we have demonstrated that our method can often mirror the behavior of these advanced methods (Fig. 3; Supplemental Table S5; Supplemental Fig. S5), directly implementing these approaches may be more effective for identifying distant relatives. Integrating our approach with a recently proposed secure federated algorithm for principal component analysis (Froelicher et al. 2023) may help to address the former. For the latter, we posit that an extension of our hashing strategy to quantify the rate of collision, which represents the sharing of a short IBD segment between individuals, may be possible.
Extending SF-Relate to accommodate a broader range of scenarios represents another key direction for future work. Although we have shown that SF-Relate can be used by consortia with large genomic data sets, developing a more efficient strategy than the pairwise execution of SF-Relate would be beneficial when a large number of parties are involved. Additionally, enabling the detection of relatives for a single query individual within a large, potentially distributed, database would be useful for personalized services that help individuals find lost biological relatives (e.g., MyHeritage). There is also a need to facilitate similarity computations for other data types, including medical records. In any of these scenarios, it would be meaningful to further explore potential information leakage in the output and devise strategies to mitigate any remaining risk. Overall, our work offers technical insights that are broadly applicable to discovering relations across siloed data sets.
Methods
Overview of the cross–data set kinship estimation problem
We consider the setting in which multiple parties (e.g., researchers from different institutions) wish to identify genetically related individuals between their private data sets while protecting the confidentiality of their data (Fig. 1). The goal of the parties is to use this information to facilitate downstream collaborative analysis by excluding duplicate or related individuals from the analysis to minimize bias in statistical analyses.
Without loss of generality, we focus on the setting with two parties, each holding a data set including phased haplotypes of n individuals over m genetic variants, such as SNPs. The desired output for each party is a list of individuals in their private data set who have at least one “close” relative in the other party's data set with respect to some relatedness threshold. When there are more than two parties, each pair of parties executes our protocol. We mainly consider the detection of third-degree or closer relatives, which are the closest relations most commonly used in genomic studies and data releases (e.g., UKB) (Bycroft et al. 2018). We assume each input data set to be locally phased by each party (i.e., each individual's genome is represented as two haplotype sequences), which is crucial for capturing identity-by-descent (IBD) sharing patterns, as we describe later.
We consider a threat model in which the parties are honest-but-curious; that is, they faithfully follow the protocol as specified but might attempt to infer private information about the other parties’ data sets based on the data observed during the process. Given this model, we aim to provide formal privacy guarantees for each party's input data set, ensuring that no information is revealed to other parties except for what can be gleaned from each party's respective output and global parameters of the problem (e.g., data set dimensions and security parameters).
Estimation of kinship coefficients
The kinship coefficient ϕ between a pair of individuals is defined as the probability that a pair of randomly sampled alleles is identical by descent (IBD), namely, when the pair of alleles is identical owing to genetic inheritance rather than by chance. For example, because human genomes are diploid, a direct descendant inherits exactly one set of chromosomes from each parent; in this case, the kinship coefficient between an individual and his or her parent is, in principle, 0.5 · 0.5 = 0.25.
Existing approaches for estimating the kinship coefficients typically fall into one of two classes: distance-based methods, which use a notion of distance between two genotype vectors that often incorporate information about minor allele frequencies (MAFs) (Manichaikul et al. 2010; Conomos et al. 2016), and IBD-segment-based methods, which first identify long shared segments between individuals that are likely owing to IBD (Naseri et al. 2019; Nait Saada et al. 2020; Shemirani et al. 2021) and estimate kinship based on the extent of these shared segments. In a benchmarking study, Ramstetter et al. (2017) showed that both approaches achieve high accuracy for up to third-degree relationships, whereas agreement becomes weaker for more distant relationships, which we also observe between PC-Relate and KING (Fig. 3). Although IBD-segment-based methods generally offer a more accurate estimation of kinship by analyzing IBD sharing patterns, distance-based approaches represent an efficient alternative that does not involve costly string matching, which often leads to substantially higher runtime (e.g., days vs. minutes) (Ramstetter et al. 2017). As a result, distance-based methods have been more commonly applied to large data sets (Bycroft et al. 2018). Recently proposed methods (e.g., RaPID) (Naseri et al. 2019) introduce hashing techniques to improve the scalability of IBD segment detection, which in some cases exceed the efficiency of distance-based methods owing to the quadratic scaling of pairwise distance calculation. However, IBD segment finding methods are still combinatorial in nature and cannot be efficiently implemented using existing secure computation techniques. In our work, we adopt a distance-based approach to minimize the computational overhead associated with secure computation but simultaneously exploit IBD sharing patterns to improve the scalability of our approach.
Our work addresses the problem of applying the widely adopted distance-based method for kinship estimation, the KING-robust
estimator (referred to as KING in what follows for simplicity) (Manichaikul et al. 2010), to find relationships between two data sets. This method is implemented in several standard genomic analysis toolkits,
such as Hail (https://github.com/hail-is/hail) and PLINK (Chang et al. 2015), and recently, the UKB released relatedness data for individuals in the data set using this estimator (Bycroft et al. 2018). KING estimates the kinship coefficient between two genotype vectors x and y ∈ {0, 1, 2}m using the following formula: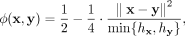 (1)
where each element in x and y represents the minor allele dosage of a genetic variant, and hx and hy represent the fraction of heterozygous loci in each vector (i.e., the heterozygosity of the individual).
(1)
where each element in x and y represents the minor allele dosage of a genetic variant, and hx and hy represent the fraction of heterozygous loci in each vector (i.e., the heterozygosity of the individual).
Existing cross–data set approaches and their limitations
Wang et al. (2022) proposed a homomorphic encryption method for identifying genetic relationships across parties, but their approach requires kinship computation for all pairs of samples, which does not scale to large data sets. Other previous approaches (Hormozdiari et al. 2014; Glusman et al. 2017; Robinson and Glusman 2018; Dervishi et al. 2023) rely on sharing a limited amount of processed data between parties to find related samples, which sacrifices both privacy and accuracy to some extent. For instance, Dervishi et al. (2023) introduced a solution in which the parties reveal a subset of SNPs in a shuffled order for their respective samples to estimate the kinship coefficients. Robinson and Glusman (2018) and Glusman et al. (2017) proposed to compare “fingerprints” obtained by applying a random projection to genomic samples to infer relatedness. He et al. (2014) and Hormozdiari et al. (2014) used error-correcting codes and fuzzy encryption to compare genotype vectors such that the comparison result can be decoded only if the two vectors are similar enough. These solutions require comparison between all pairs of samples between data sets, and the processing of genotype vectors into limited representations that can be shared leads to loss of precision.
In a recent competition organized by the iDASH Workshop 2023 (http://www.humangenomeprivacy.org/2023/), identifying the presence of relatives in encrypted data sets was posed as one of the challenge tasks. The challenge considered a setting that is different from our work. It involved a client-server scenario with small data sets (e.g., 2000 individuals and 16,000 variants) and restricted any data preprocessing for evaluation purposes, thus limiting the applicability of the proposed solutions in a real use-case scenario. The best-performing solutions to the challenge generally followed a similar approach adapting the work of Homer et al. (2008). In this approach, the presence of a relative in a data set is inferred based on a statistic evaluating whether the individual's genotype vector is closer to the allele frequencies in the data set than to background frequencies computed in a reference population. We show in Supplemental Figure S8 that computing this statistic securely does not lead to accurate results in realistic settings, involving complete genomes and many individuals (e.g., tens of thousands or more).
A novel approach to securely detect relatives across large-scale and distributed data sets
We developed SF-Relate to allow secure and efficient detection of relatives between large-scale data sets by drastically reducing the number of kinship computations, from quadratic to linear, while preserving accuracy. For a graphical overview of SF-Relate's workflow, see Supplemental Figure S9. To achieve this solution, SF-Relate draws ideas from both distance-based and IBD-segment-based kinship estimation methods. It first identifies pairs of samples to be compared between parties using a LSH function (Indyk and Motwani 1998), which we adapt to ensure that both (or all) parties assign individuals with shared IBD segments to the same bucket with a higher probability than for unrelated individuals. SF-Relate sets the capacity of each bucket to one, discarding duplicate hits, which we refer to as a microbucketing strategy. As we demonstrate, this novel approach is key to minimizing the number of comparisons while maintaining accuracy. Next, SF-Relate securely estimates and thresholds the kinship coefficients between pairs of samples in buckets with the same index across parties and then aggregates the results per sample using our secure implementation of a distance-based kinship estimator (KING-robust) (Manichaikul et al. 2010). To perform these operations while keeping each party's data confidential from other parties, we develop efficient two-party computation protocols based on MHE techniques (Mouchet et al. 2021). We additionally incorporate sketching techniques to further reduce the computational cost of MHE computations. In the following, we provide details of each step of our algorithm.
Step 1: hashing and bucketing
In this step, each party locally evaluates a series of hash functions on each individual's haplotype sequences to assign the individual to buckets across a collection of hash tables, such that related individuals are more likely to be assigned to the same bucket index. Only individuals in the same bucket across parties are compared in a later step. Note that the hash functions are synchronized among the parties based on a pseudorandom number generator with a shared seed, which is obtained prior to the execution of SF-Relate via a standard key exchange protocol (e.g., Diffie and Hellman 1976).
LSH to capture IBD segments
SF-Relate assigns individuals to buckets using a LSH (Indyk and Motwani 1998). LSH functions map similar items to the same value more frequently based on a similarity notion. Specifically, the Hamming LSH, which relies on the Hamming similarity (defined as the number of equal coordinates between vectors), projects a vector onto one random coordinate and uses its value as the output. In our setting, applying the Hamming LSH (or other LSH methods, such as MinHash) (Broder 1997) directly to each sample, encoded as a genotype vector, for bucket assignment would not work in practice. This is because the difference between related and unrelated individuals, with respect to the distance between samples’ encodings, is too small for LSH to distinguish; the average relative Hamming distance between third-degree relatives and unrelated individuals in UKB is 22% and 23%, respectively. Hence, SF-Relate applies an encoding scheme that results in highly similar Hamming vectors for related samples. This encoding captures the biological signal of IBD distributions and can be seen as a variant of the encoding in the method of Shemirani et al. (2021). It applies LSH on split chromosomes, exploiting the key insight that IBD segments are unevenly distributed on the genome. The subchromosomes are further divided into a short string of genotypes (similar to k-mers) to extract long runs of identical genotypes that are unlikely by chance.
The first three steps of our hashing approach encode IBD segments: First, in splitting, haplotypes are divided into genomic segments of fixed-length in genetic distance (centi-Morgans). Second, in subsampling, each segment is randomly projected down to a fixed number of SNPs, in which the SNPs are sampled with probability proportional to their MAF. This reduces the impact of genotyping errors and rare variants on hashing and unifies the SNP density across different segments and data sets, inspired by work by Naseri et al. (2019). Third, in k-merization, subsampled genotypes for several contiguous segments are concatenated to form a k-SNP (akin to k-shingles in the work of Shemirani et al. 2021), namely, a sequence of genotypes for k SNPs, which helps to identify matches of long genomic segments. Through these steps, we encode each sample as a list of subchromosome k-SNP vectors. In fact, we confirmed that related pairs share significantly more Hamming-similar subchromosomes under this encoding scheme (Supplemental Fig. S4).
The final step of hashing applies LSH on the subchromosome vectors to obtain the actual bucket index. To utilize the raw probability gap between non-IBD segments and IBD-segments produced by LSH (such as the gap between 0.5 and 1.0 in Supplemental Fig. S4) we need to amplify it. For this, we choose a concatenation parameter ℓ and define the bucket index as the FNV-1 hash (https://datatracker.ietf.org/doc/draft-eastlake-fnv/25/) on the concatenation of the outputs of ℓ independent Hamming LSH applied to the subchromosome vector. This in turn boosts the gap by raising it to the power of ℓ. An alternative (as in the work of Shemirani et al. 2021) would be to apply the LSH function MinHash (Broder 1997), which captures the set-based Jaccard similarity. Nevertheless, Hamming similarity is more natural for IBD detection, as IBD segments are, by definition, matching k-SNPs at the same genetic positions, whereas the Jaccard similarity discards positional information of the two sets of k-SNPs. Indeed, Hamming similarity detects more highly similar subchromosomes (Supplemental Fig. S10). The final output of this procedure is a list of hash tables, each consisting of buckets storing sample IDs.
Microbucketing strategy
To prevent leakage of private information, the parties need to compare the samples in corresponding buckets without exposing any additional information about their data sets, such as the distribution of nonempty buckets and their sizes. This requires that the buckets created in the previous step be padded to a fixed size by adding dummy samples. However, to keep the sample identities hidden, all pairs of samples in a bucket between the parties need to be compared, which leads to both quadratic scaling of comparisons with the bucket size and a large amount of wasted computation involving dummy samples. To address this issue, SF-Relate introduces a microbucketing strategy, which merges buckets across multiple hash tables (produced by different subchromosomes) and then filters every bucket down to a single element. Dummy samples are added only at the end to pad the empty bucket to size one. This effectively transforms the parties’ local bucket assignments into an ordered list of samples to be securely compared against the corresponding list obtained by the other party in an element-wise fashion. This approach avoids the quadratic scaling while minimizing the addition of dummy samples owing to the merging of buckets (i.e., a bucket is filled if at least one sample is assigned to it in one of the hash tables). Despite the extreme level of filtering applied to each bucket during this process, our strategy enables efficient and accurate detection of relatives.
SF-Relate chooses a table size parameter N and a bucket size parameter C. It aligns the different hash tables and merges buckets with the same remainder modulo N into one. This ensures the number of buckets is, at most, N. In practice, on a data set with n local samples, N is determined by first choosing a table ratio τ and letting N = τn (for a table of the main symbols and their default values, see Supplemental Table S1). After this merge, buckets with more than C samples are filtered until C samples remain. In this filtering step, samples from buckets built by a smaller subchromosome index are given higher preference, but otherwise, a uniformly random filtering is performed. The preference toward smaller subchromosome indices is to ensure samples in corresponding buckets in the hash tables more likely originate from the same hash table. The parties then repeat the entire hashing step locally L times (each with new randomness) until 99% of the buckets are full. Only at the end, dummy samples are inserted to ensure a constant bucket size.
On the realistic data sets UKB-200K, we evaluate the best bucket capacity C, by keeping the number of comparisons NC2 fixed and checking the fraction of related pairs that appear in at least one corresponding bucket after microbucketing. In practice, the minimal capacity C = 1 gives the highest recall. This is because related samples share many rare IBD segments (many of which is unique) that cause them to end up in a small-sized bucket (Supplemental Fig. S4). When this happens, both samples remain in the corresponding bucket with high probability (as there is no competition within that bucket). The average fraction of shared unique IBD segments is not high for the third degree (20/352), but given that our goal is to use the list of buckets to trigger subsequent kinship computations (which operates on the entire genome) and not identifying all IBD segments between pairs (unlike iLASH and RaPID) (Naseri et al. 2019; Shemirani et al. 2021), as long as one of the many small-sized buckets owing to rare IBD segments survives microbucketing, the pair of relatives would be discovered. This explains the effectiveness of microbucketing with a restricted microcapacity, C. In fact, for the rest of the paper, we always set C = 1.
In sum, each party in SF-Relate obtains a single hash table with N buckets (each with C = 1 samples), and microbucketing ensures the hash tables are highly utilized. The parties hence perform the C2N = N secure kinship computation in step 2, secure kinship evaluation.
Step 2: secure kinship evaluation
In this step, the parties perform element-wise comparisons between their ordered list of samples (representing elements in a merged hash table with size-one buckets), which were obtained in step 1. They first jointly evaluate the kinship coefficient for each pair (MHE-Phase 1) before aggregating the results to obtain an indicator for each individual, reflecting the presence of a close relative in the other data set (MHE-Phase 2).
Review of MHE
To compute on encrypted data in a secure manner, SF-Relate builds upon MHE (Froelicher et al. 2021a; Mouchet et al. 2021), extending the CKKS scheme (Cheon et al. 2017). CKKS encodes a vector of real number values in a single ciphertext and is well suited for calculations in which a small amount of noise can be tolerated. Like other HE schemes, it provides operations for addition, multiplication, and rotation (i.e., permutation of elements in a vector) of encrypted values in a ciphertext while providing the single-instruction, multiple-data (SIMD) property. Operations involving plaintext (unencrypted) data are substantially more computationally efficient; for example, ciphertext-plaintext multiplication is seven times faster than ciphertext–ciphertext multiplication based on our parameter setting.
MHE extends the CKKS scheme to the setting with multiple parties by splitting the decryption key into random shares (through a technique called secret sharing) and then distributing the shares among the parties. In this setting, all parties must cooperate to decrypt any ciphertext in SF-Relate, thus ensuring that only the final results of the protocol are revealed to each party. Other cryptographic keys involved in the system, including the encryption key and the evaluation keys, are jointly held by all parties, allowing the parties to locally encrypt data and perform homomorphic computations. A key advantage of MHE is that some costly cryptographic operations, such as the bootstrapping that is required to “refresh” a ciphertext after a certain number of multiplications, can be replaced by efficient interactive protocols (Froelicher et al. 2021a; Mouchet et al. 2021).
In the following, we describe our efficient MHE-based secure computation protocols for kinship evaluation between two parties (MHE-Phases 1 and 2) based on the bucketed samples from step 1. We assume that the MHE scheme has been initialized with the required cryptographic keys, which the parties generate via a collective key generation protocol (Mouchet et al. 2021), implemented in our base cryptographic library (https://github.com/tuneinsight/lattigo). We exploit the properties of MHE to minimize the cryptographic overhead of our computation protocols by maximizing the use of locally available plaintext data and balancing the workload between the parties. Although we focus on the two-party setting, our protocols naturally extend to settings with more than two parties, because we can execute the protocols between all pairs of parties and then aggregate the results.
MHE-Phase 1: secure computation of kinship coefficients
Given a list of N samples on both sides, where each sample is associated with a vector of M SNPs, the parties collaborate to calculate kinship coefficients for the N pairs of samples between them and compare each to a threshold. The desired comparison test given a threshold θ is 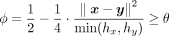 . For efficient evaluation under encryption, we can rewrite it as
. For efficient evaluation under encryption, we can rewrite it as 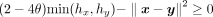 , thus avoiding the division operation. The comparison test passes when both hx and hy satisfies it, so we compute
, thus avoiding the division operation. The comparison test passes when both hx and hy satisfies it, so we compute
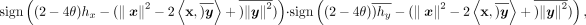 which evaluates to one if the coefficient is above the threshold and zero otherwise. Boxed terms represent encrypted data,
and
which evaluates to one if the coefficient is above the threshold and zero otherwise. Boxed terms represent encrypted data,
and 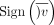 is the indicator function for v ≥ 0. Note that we assign the evaluation of this expression to the party that holds x (party 1) and have the other party (party 2) transfer the encrypted y for computation. This allows most operations to be performed efficiently using the plaintext x. We observe that hy (the number of heterozygous in the genotype) and
is the indicator function for v ≥ 0. Note that we assign the evaluation of this expression to the party that holds x (party 1) and have the other party (party 2) transfer the encrypted y for computation. This allows most operations to be performed efficiently using the plaintext x. We observe that hy (the number of heterozygous in the genotype) and 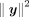 can be computed locally by party 2 before encryption. Hence, the most expensive operation is the inner product , which requires
a plaintext–ciphertext multiplication between vectors of size M and a summation of elements of the resulting vector. We use a polynomial approximation of the sign function to homomorphically
evaluate Sign(·). The cost of this function, despite requiring homomorphic evaluation of a high-degree polynomial, is dwarfed
by the cost of computing the inner products. Finally, in addition to the SIMD property of MHE operations, we process batches
of coefficients in parallel and evenly distribute the workload between parties by alternating their roles across batches with
respect to who holds the plaintext vector x. Our protocol is provided in Supplemental Note S1.
can be computed locally by party 2 before encryption. Hence, the most expensive operation is the inner product , which requires
a plaintext–ciphertext multiplication between vectors of size M and a summation of elements of the resulting vector. We use a polynomial approximation of the sign function to homomorphically
evaluate Sign(·). The cost of this function, despite requiring homomorphic evaluation of a high-degree polynomial, is dwarfed
by the cost of computing the inner products. Finally, in addition to the SIMD property of MHE operations, we process batches
of coefficients in parallel and evenly distribute the workload between parties by alternating their roles across batches with
respect to who holds the plaintext vector x. Our protocol is provided in Supplemental Note S1.
MHE-Phase 2: secure aggregation of results for individual samples
Next, the parties aggregate the comparison results for each individual to compute a single binary indicator representing the presence of a relative. For this, they first perform a linear scan over the comparison results, which selects the results corresponding to the same individual and masks the rest. The selected results are then accumulated, after which another sign test is performed to obtain a binary value as desired, hiding the number of identified relationships as a result. At the end of the protocol, the parties decrypt the vectors and each obtains a list of indicators, determining whether each sample has at least one close relative in the other data set; this is the only information that is revealed to each party. Note that the complexity of this step depends only on the number of comparisons and individuals, whereas MHE-Phase 1 also scales with the number of SNPs, which is typically large (e.g., more than 500,000). We provide our protocol for this step in Supplemental Note S1.
Accelerating kinship computation using sketching
To further reduce the cost of secure kinship evaluation, SF-Relate first reduces the size of each sample through sketching. In particular, given the subsampling ratio parameter 0 < s ≤ 1, SF-Relate randomly chooses an s fraction of SNPs to use for kinship evaluation. This provides a natural approximation for the KING kinship estimator, which includes the squared Euclidean distance between two genotype vectors, which can be estimated using a random subset of coordinates in an unbiased manner (see Supplemental Fig. S11). Our results show that this approach enables a meaningful trade-off between accuracy and efficiency; a minor loss in precision introduced by sketching allows us to obtain a substantial reduction in computational cost while maintaining near-perfect detection accuracy.
Alternative output modes
By default, SF-Relate computes a list of indicators representing whether each sample has at least one close relative in the other data sets. SF-Relate also supports securely computing other types of output, including the closest relatedness degree for each individual, the maximum kinship for each individual (discretized), and the full list of computed kinship coefficients.
For outputting all kinship coefficients, we replace the final sign test computations with the computation of the kinship coefficients
in MHE-Phase 1 (Supplemental Note S1). That is, the parties homomorphically compute 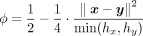 . To speed up the computation, they precompute the values
. To speed up the computation, they precompute the values 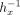 and
and 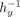 locally in plaintext, which allows replacing the division with a multiplication by
locally in plaintext, which allows replacing the division with a multiplication by 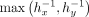 , which can be more efficiently computed.
, which can be more efficiently computed.
For both the closest relatedness degree and the maximum kinship coefficient, SF-Relate computes multiple comparison tests with respect to a series of kinship thresholds and then executes MHE-Phase 2 in parallel to accumulate the resulting comparison results. The decryption of these results reveals the largest threshold at which the comparison test succeeded. Based on this information, the parties can determine the closest degree or the maximum kinship as desired.
Note that, in any of these settings, the final results can also be kept in encrypted form and utilized in subsequent analysis steps without revealing the results of relative detection. Additionally, the complexity of MHE-Phase 1 is constant across all output modes with N plaintext–ciphertext multiplications between vectors of length M, whereas the complexity of MHE-Phase 2 increases linearly with the number of thresholds t. The default setting of SF-Relate corresponds to t = 1, whereas the version that reveals all coefficients corresponds to t = 0.
Data sets
We utilize three data sets sampled from prominent genomic data consortia: UKB and AoU. Data access application to genotypes and haplotypes from the UK Biobank can be submitted at https://www.ukbiobank.ac.uk/. The AoU controlled tier data set v5 is available through the controlled tier of the AoU researcher workbench. Application to access the AoU data set can be submitted at https://www.researchallofus.org/register/.
For our purpose, we specifically extract two data sets from UKB, one comprising 100,000 samples (UKB-100K) and the other comprising 200,000 samples (UKB-200K). In both cases, we randomly split the data sets among two sites. From AoU, we extracted a data set of 20,000 samples. Because of the smaller size of this data set and because we want to avoid a highly imbalanced distribution, we first split the individuals with close relations on the two sites and then randomly split the set of unrelated individuals across the two sites. On the UKB data set, we use phased autosomal haplotypes officially released in UKB v3 as input to the hashing (Methods), whereas for AoU, we phased a batch of 20,000 samples from AoU using Eagle 2 (Loh et al. 2016). We observe that independently phasing the data at each site does not affect the accuracy of SF-Relate (Supplemental Table S2).
Ground-truth preparation
To compute the ground truth of related individuals in our data sets, we follow the approach proposed in the UKB documentation (Bycroft et al. 2018). We first filter the SNPs based on their implications in population structure, before computing the kinship using the KING approach (Manichaikul et al. 2010). To determine the set of SNPs to retain, we conduct a PCA on a publicly available data set (i.e., 1000 Genomes) using the intersection of loci with our data set. Utilizing a reference data set ensures that our method is not tailored to the processed data set and effectively generalizes to other data sets. We then exclude SNPs that exhibit high PC loadings in the top three PCs, using a threshold set at the 75th percentile of these loadings. This strategy enables us to filter out SNPs with heavy loadings while retaining sufficient ancestry-agnostic autosomal SNPs for kinship inference.
Applying this approach to the UKB data sets results in selecting 90,000 SNPs, upon which the KING estimator predominantly identifies the same related pairs as those in UKB's relatedness release. The ground-truth relatedness degrees in our experiments are based on these KING coefficients, utilizing the recommended thresholds 2−d−1.5 for degree d (Manichaikul et al. 2010).
Kinship estimation using alternative nonsecure methods
Even though SF-Relate builds upon the KING estimator (Manichaikul et al. 2010), it outperforms it owing to its novel approach in preselecting individuals likely to be related, utilizing an encoding and
hashing scheme specialized to capture IBD signals. To showcase this, we compare SF-Relate and the standard KING estimator
alongside two advanced relative-detection tools: PC-Relate (Conomos et al. 2016) and RAFFI (Naseri et al. 2021). For PC-Relate, we rely on the hail implementation (https://github.com/hail-is/hail) and consider only biallelic variants from the UKB SNP panel to compute all pairwise coefficients. We set the MAF and the
number of PCs to 0.1 and 10, respectively, and remove variants with a missing rate >5%. Additionally, we perform ld-pruning
on the SNP set, reducing from 600,000 to 300,000 variants using parameters r2 = 0.05 and 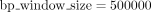 . For kinship estimation with RAFFI, we first run the IBD-finding tool RaPID with the parameters −r3 − s1 − d5 − w3, followed
by executing RAFFI (v0.1) for kinship estimation. However, we observed that the initial segment of length 10 cM on Chromosome
15 is only covered by 16 bp in the UKB data set, resulting in excessive candidate IBD segment pairs processed by RaPID. As
the 10 cM sharing has a tiny effect on the overall kinship coefficient, we removed these base pairs when running RaPID on
UKB.
. For kinship estimation with RAFFI, we first run the IBD-finding tool RaPID with the parameters −r3 − s1 − d5 − w3, followed
by executing RAFFI (v0.1) for kinship estimation. However, we observed that the initial segment of length 10 cM on Chromosome
15 is only covered by 16 bp in the UKB data set, resulting in excessive candidate IBD segment pairs processed by RaPID. As
the 10 cM sharing has a tiny effect on the overall kinship coefficient, we removed these base pairs when running RaPID on
UKB.
Phenotype simulation for the GWAS case study
To evaluate the effectiveness of SF-Relate in mitigating the confounding effects of cryptic relatedness for GWAS, we simulate
a GWAS study on a subset of 100,000 samples from UKB using simulated phenotypes, following the methodology proposed in REGENIE
(Mbatchou et al. 2021) as described next. We select a set of random variants from odd chromosomes to serve as causal variants, reserving the even
chromosomes to assess the level of false-positive associations. We exclude extremely rare SNPs (minor allele count < 5), before
randomly selecting P SNPs located in odd-numbered chromosomes, for P ∈ {100, 1000, 10000}. These selected SNPs are designated as causal. For each causal SNP, we sample its effect size βj with the constraint that the total variance (i.e., narrow-sense heritability) is h2 = 0.2. We then use a linear model with top principal component correction to simulate the phenotype Yi for each individual i as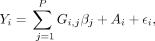 where Gi,j is the standardized genotype of individual i at SNP j, Ai is the first PC score of individual i (scaled to have variance 0.05), and
where Gi,j is the standardized genotype of individual i at SNP j, Ai is the first PC score of individual i (scaled to have variance 0.05), and  is a Gaussian noise variable with variance 0.75, representing environmental effects.
is a Gaussian noise variable with variance 0.75, representing environmental effects.
Software availability
Our open-source implementation of SF-Relate, including a script to create an example data set from the public 1000 Genomes phase 3 data set (Byrska-Bishop et al. 2022), is available at GitHub (https://github.com/froelich/sf-relate) and as Supplemental Code. Additionally, SF-Relate can be conveniently executed through sfkit (Mendelsohn et al. 2023; https://sfkit.org), a web server for secure collaborative genomic studies.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was accepted for an oral presentation at RECOMB 2024, where it received the “Best Student/Young Scientist Paper Award.” We thank Manaswitha Edupalli and Matthew Mosca for their help in processing the biobank data sets and Simon Mendelsohn for integrating SF-Relate into the sfkit web server. Additionally, we thank the RECOMB, Genome Research, and other reviewers for their helpful suggestions. This work is supported by National Institutes of Health (NIH) R01 HG010959 and U01 CA250554 (to B.B.), NIH DP5 OD029574 and RM1 HG011558 (to H.C.), and the Broad Institute Schmidt fellowship (to V.P.). Part of this work was completed while H.C. was at the Broad Institute. The All of Us Research Program is supported by the National Institutes of Health, Office of the Director; Regional Medical Centers 1 OT2 OD026549, 1 OT2 OD026554, 1 OT2 OD026557, 1 OT2 OD026556, 1 OT2 OD026550, 1 OT2 OD026552, 1 OT2 OD026553, 1 OT2 OD026548, 1 OT2 OD026551, and 1 OT2 OD026555; IAA # AOD 16037; Federally Qualified Health Centers HHSN 263201600085U; Data and Research Center 5 U2C OD023196; Biobank 1 U24 OD023121; The Participant Center U24 OD023176; Participant Technology Systems Center 1 U24 OD023163; Communications and Engagement 3 OT2 OD023205 and 3 OT2 OD023206; and Community Partners 1 OT2 OD025277, 3 OT2 OD025315, 1 OT2 OD025337, and 1 OT2 OD025276. In addition, the All of Us Research Program would not be possible without the partnership of its participants.
Author contributions: All authors developed the method. M.M.H., D.F., and H.C. implemented the software and performed experiments. M.M.H. and D.F. wrote the initial draft of the manuscript. All authors analyzed the results and edited the manuscript. V.P., B.B., and H.C. guided the work.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279057.124.
-
Freely available online through the Genome Research Open Access option.
- Received February 16, 2024.
- Accepted July 31, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








