
Genetic complexity of killer-cell immunoglobulin-like receptor genes in human pangenome assemblies
- Tsung-Kai Hung1,
- Wan-Chi Liu2,
- Sheng-Kai Lai3,4,
- Hui-Wen Chuang1,
- Yi-Che Lee2,
- Hong-Ye Lin5,
- Chia-Lang Hsu1,6,
- Chien-Yu Chen5,
- Ya-Chien Yang2,7,
- Jacob Shujui Hsu1 and
- Pei-Lung Chen1,3,4,8,9
- 1Graduate Institute of Medical Genomics and Proteomics, College of Medicine, National Taiwan University, Taipei 100233, Taiwan;
- 2Department of Clinical Laboratory Sciences and Medical Biotechnology, College of Medicine, National Taiwan University, Taipei 100229, Taiwan;
- 3Department of Medical Genetics, National Taiwan University Hospital, Taipei 100229, Taiwan;
- 4Genome and Systems Biology Degree Program, Academia Sinica and National Taiwan University, Taipei 10617, Taiwan;
- 5Department of Biomechatronics Engineering, National Taiwan University, Taipei 10617, Taiwan;
- 6Department of Medical Research, National Taiwan University Hospital, Taipei 100229, Taiwan;
- 7Department of Laboratory Medicine, National Taiwan University Hospital, Taipei 100229, Taiwan;
- 8Graduate Institute of Clinical Medicine, College of Medicine, National Taiwan University, Taipei 100229, Taiwan;
- 9Division of Endocrinology and Metabolism, Department of Internal Medicine, National Taiwan University Hospital, Taipei 100229, Taiwan
Abstract
The killer-cell immunoglobulin-like receptor (KIR) gene complex, a highly polymorphic region of the human genome that encodes proteins involved in immune responses, poses strong challenges in genotyping owing to its remarkable genetic diversity and structural intricacy. Accurate analysis of KIR alleles, including their structural variations, is crucial for understanding their roles in various immune responses. Leveraging the high-quality genome assemblies from the Human Pangenome Reference Consortium (HPRC), we present a novel bioinformatic tool, the structural KIR annoTator (SKIRT), to investigate gene diversity and facilitate precise KIR allele analysis. In 47 HPRC-phased assemblies, SKIRT identifies a recurrent novel KIR2DS4/3DL1 fusion gene in the paternal haplotype of HG02630 and maternal haplotype of NA19240. Additionally, SKIRT accurately identifies eight structural variants and 15 novel nonsynonymous alleles, all of which are independently validated using short-read data or quantitative polymerase chain reaction. Our study has discovered a total of 570 novel alleles, among which eight haplotypes harbor at least one KIR gene duplication, six haplotypes have lost at least one framework gene, and 75 out of 94 haplotypes (79.8%) carry at least five novel alleles, thus confirming KIR genetic diversity. These findings are pivotal in providing insights into KIR gene diversity and serve as a solid foundation for understanding the functional consequences of KIR structural variations. High-resolution genome assemblies offer unprecedented opportunities to explore polymorphic regions that are challenging to investigate using short-read sequencing methods. The SKIRT pipeline emerges as a highly efficient tool, enabling the comprehensive detection of the complete spectrum of KIR alleles within human genome assemblies.
Killer-cell immunoglobulin-like receptors (KIRs), expressed primarily on the surface of natural killer (NK) cells and specific T lymphocytes, are epistatic for interacting with human leukocyte antigens (HLAs) on target cells to regulate host innate and adaptive immunity (Martin et al. 2002; Vilches and Parham 2002; Pollock et al. 2022). The KIR complex comprises 15 genes with diverse inhibitory and activating domains, although not all are essential for healthy individuals. KIR haplotypes exhibit extensive diversity through expansion and recombination (Shilling et al. 1998), with gene contents ranging from eight to 14 (Hsu et al. 2002). The Genome in a Bottle (GIAB) consortium excluded KIR regions from benchmarking (v4.2.1) owing to their copy number variability (Wagner et al. 2022), highlighting the complexity of accurate determination of KIR gene copy numbers (Béziat et al. 2013).
The biological and clinical significance of KIR genes is rooted in their involvement in immune-mediated diseases, hematopoietic stem cell and organ transplantation (Boudreau et al. 2017; Dizaji et al. 2021; Zamir et al. 2022), and associations with autoimmune diseases such as rheumatoid arthritis, psoriasis, type 1 diabetes, and reproductive health issues (Alexandrova et al. 2022; Feng et al. 2022). KIR genes are also implicated in infectious diseases, including human immunodeficiency virus (HIV) and hepatitis C virus (HCV) infections, in which specific KIR and HLA combinations influence disease progression and response to antiviral therapy (Khakoo and Carrington 2006; Boudreau et al. 2016; Boelen et al. 2018; Li et al. 2022). Furthermore, KIR gene diversity is linked to cancer susceptibility and progression, including leukemia, lymphoma, and melanoma (Pende et al. 2019). Clinical trials show promising results using lirilumab, an anti-KIR monoclonal antibody (mAb) that targets HLA-C and KIR2DL1/L2/L3, combined with anti-PDCD1/CD274 (commonly known as PD-1/PD-L1) immunotherapies for treating squamous cell carcinoma of the head and neck (SCCHN) and human papillomavirus (HPV)–positive cervical cancer (Hanna et al. 2022; Liu et al. 2022). Another phase I/II clinical trial of lirilumab combined with an anti-PD-1 mAb for multiple myeloma is also awaited (Clara and Childs 2022). The multifaceted functions of KIRs stem from the development of NK and T cells (Bashirova et al. 2006; Yawata et al. 2006; Fauriat et al. 2010) and are influenced by factors such as binding affinity, specificity, ligation angle, signal transduction capability, and intracellular trafficking (Gardiner et al. 2001; Saunders et al. 2016; Wu et al. 2021).
KIR regions are characterized by the gene compositions as A and B haplotypes, both containing four framework genes: KIR3DL3, KIR3DP1, KIR2DL4, and KIR3DL2. Haplotype A is the majority worldwide, with homozygous AA predominant (de Brito Vargas et al. 2021). However, many unknown haplotypic structures and gene contents may exist, challenging current methods. One large-scale sequencing-based KIR study presumes a diploid genome with two copies of each framework gene (Sakaue et al. 2022), potentially overlooking CNVs of framework genes. Duplications and deletions of KIR3DP1, KIR2DL4, and KIR3DL1/S1 appear to form a highly conserved triad with frequent unequal crossover events (Martin et al. 2003; Williams et al. 2003; Gómez-Lozano et al. 2005; Amorim et al. 2021). Many short-read-based copy number determination algorithms (Norman et al. 2016; Marin et al. 2021; Song et al. 2023) rely on aligned depth within a data set, potentially leading to limited results. Traditional methods require manual crafting of haplotypes by incorporating gene content and copy number information, followed by estimation based on the linkage disequilibrium derived from known haplotypes (Amorim et al. 2021), for which the correct phasing information is absent. These challenges are compounded by ongoing expansion and evolution of KIR genes in all populations (Alicata et al. 2020; Kevin-Tey et al. 2022; NurWaliyuddin et al. 2022).
The Kass assembly method efficiently interprets KIR diploid haplotypes in contigs assembled from probe-captured Pacific Biosciences (PacBio) CCS long-read sequencing data (Roe et al. 2020). Assembly quality, including completeness and contiguity, largely depends on the sensitivity and specificity of the in vitro and in silico probes and the assembly algorithm employed. Therein lies the potential scope for refinement to further enhance output quality. The Telomere-to-Telomere (T2T) consortium sequenced the complete haploid human genome, T2T-CHM13, encompassing each autosome and X Chromosome (Nurk et al. 2022). Later, the Human Pangenome Reference Consortium (HPRC) released 47 high-quality, haplotype-resolved genome assemblies (HPRC Year-1 release) (Wang et al. 2022; Liao et al. 2023), using Hifiasm (Cheng et al. 2021) and family trio samples from The 1000 Genomes Project (1KGP) (The 1000 Genomes Project Consortium 2015). The assembly of HG002, one of the 47 genomes, was characterized by more accurate variant calls than other methods (Jarvis et al. 2022). The paternal and maternal assemblies of HPRC autosomes cover ∼92.8% and 94.1% of T2T-CHM13, respectively. The reliability score of the HPRC assemblies was estimated at 99.12%, whereas T2T-CHM13 was 99.91% (Liao et al. 2023). This high level of accuracy, near-complete coverage, and almost telomere-to-telomere continuity underscores the value of the 47 HPRC diverse diploid genome assemblies.
Kass annotates its own assembly with high-resolution allelic genotyping information for KIR genes (Roe et al. 2020). However, some erroneous annotations were generated when tested with the same HPRC-released HG002 data (Supplemental Table S1), suggesting a need for improved methods. T1K (Song et al. 2023) also utilized 26 of the 47 HPRC genome assemblies to benchmark its KIR genotyping algorithm but did not account for potential novel structural variations in the 52 haplotypes analyzed.
In this study, we developed an improved structural KIR annoTator (SKIRT) method for high-resolution allelic and CNV identification of KIR genes. This method accurately annotates complete haplotypes, gene copy numbers, and structural variations. Our study on long-read assembled genome data confirmed the exact flanking positions, enabling the accurate, complete haplotypes of the KIR complex.
Results
Method overview
The 47 HPRC phased diploid genome assemblies, produced using Hifiasm with PacBio HiFi reads, were processed using our proprietary SKIRT pipeline (Fig. 1A). This pipeline was engineered to yield the highest possible resolution of all official KIR alleles assigned by the KIR nomenclature committee (Robinson et al. 2010; Maccari et al. 2020; https://www.ebi.ac.uk/ipd/kir/about/nomenclature/allele/). Similar to HLA allele nomenclature, the KIR nomenclature uses the gene name followed by an asterisk as a separator and an up-to-seven-digits numerical allele designation. The first three digits are designed for distinct encoded protein sequences. The subsequent two digits are used to determine alleles based on the synonymous differences within the coding sequence (CDS). The last two digits distinguish alleles based on the sequence differences in the noncoding regions. Initially, SKIRT identified the coding regions of all KIR genes in the assembly contigs and distinguished five-digit alleles by mapping IPD-KIR CDS-only sequences against the contigs. Upon perfect alignment with five-digit alleles, seven-digit alleles were further identified using IPD-KIR genomic sequences. However, when a CDS-only full allele (five-digit) aligned with variations, a further investigation was conducted to ascertain if the observed variations led to amino acid changes. Variations that altered amino acids resulted in nonsynonymous novel transcripts, whereas those that did not were considered synonymous (three-digit resolution) (Fig. 1B).
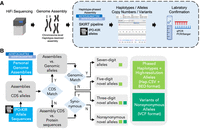
KIR allele annotation of the diploid genome assemblies. (A) Overview of the high-resolution allelic KIR identification workflow. The segment shaded blue denotes the primary focus of this study. We developed a specialized pipeline, the structural KIR annoTator (SKIRT), to facilitate annotation of high-resolution KIR alleles and structural variations in human genome assemblies. A series of laboratory experiments were conducted to validate the computational findings and ensure their accuracy. (B) The operational flow of the SKIRT pipeline. This pipeline merges a long sequence alignment tool, minimap2 (Li 2018), with in-house Python and Shell scripts. These scripts map the KIR Repository of Immuno Polymorphism Database (IPD-KIR) allele sequences against genome assemblies, such as the HPRC Year-1 release data, to identify existing KIR genes on each contig with an accuracy of up to seven-digit high-resolution alleles.
For nonsynonymous alleles, we conducted further verification using Illumina whole-genome short-read sequences (WGS) and, when necessary, PacBio HiFi reads to authenticate the presence of gene sequences from 1KGP and HPRC. When the variant percentage surpassed 1%, we manually examined the likelihood of novel structural variations such as gene fusion and segmental deletion. Any structural variations identified in our study, including CNVs, deletions, and gene fusion, were further confirmed through a series of laboratory experiments.
Overview of the 94 HPRC KIR haplotypes
We fully characterized the high-resolution alleles of KIR genes for all known KIR alleles in 47 HPRC human genome assemblies (Fig. 2A–D; Supplemental Table S2). According to the KIR haplotypes definition on the IPD-KIR website (https://www.ebi.ac.uk/ipd/kir/about/#haplotypes), the KIR A haplotype includes nine genes arranged as follows: KIR3DL3, KIR2DL3, KIR2DP1, KIR2DL1, KIR3DP1, KIR2DL4, KIR3DL1, KIR2DS4, and KIR3DL2. Any haplotype differing from this typical gene content arrangement is categorized as a KIR B haplotype. We further classified all HPRC haplotypes into four groups based on the types of centromeric and telomeric regions. For example, the centromeric A (cA) group includes five genes: KIR3DL3, KIR2DL3, KIR2DP1, KIR2DL1, and KIR3DP1, whereas the telomeric A (tA) group includes KIR2DL4, KIR3DL1, KIR2DS4, and KIR3DL2 genes. Haplotypes differing from the typical KIR A haplotypes are classified as centromeric B and telomeric B. Our results indicated that 57 out of 94 KIR A haplotypes (57/94, 60.64%) fell into the cA and tA, as shown in Figure 2A. Besides, six haplotypes were considered variations of KIR A haplotypes, resulting in the total to 63 haplotypes (63/94, 67.02%) in the cA-tA group. Similarly, seven (7.45%) were cA and telomeric B (Fig. 2B), 14 (14.89%) were centromeric B and tA (Fig. 2C), and 10 (10.64%) were centromeric B and telomeric B (Fig. 2D). Among the 94 haplotypes of the 47 genome assemblies, eight (8.51%) displayed at least one KIR gene duplication, six (6.38%) exhibited deletions of both KIR3DP1/KIR2DL4 framework genes (Fig. 2B,D), and one haplotype showed KIR3DL2 framework gene deletion (Fig. 2A). In summary, 15.96% of HPRC haplotypes (15/94, 15.96%) revealed structural variations in KIR genes that had not been previously investigated at the full-genomic sequence base resolution. This emphasizes the significance of SKIRT analyses for gaining a comprehensive understanding of genetic variation in KIR genes.

KIR haplotypes and novel alleles in the 47 HPRC phased assemblies characterized by SKIRT. Diverse KIR haplotypes and structural variations in the HPRC cohort. The KIR haplotypes were categorized as follows: (A) cA-tA haplotypes (67.02%, 63/94), (B) cA-tB haplotypes (7.45%, 7/94), (C) cB-tA haplotypes (14.89%, 14/94), and (D) cB-tB haplotypes (10.64%, 10/94).
In this study, we used the notation SampleID-P to denote the paternal haplotype of the sample and SampleID-M to denote the maternal haplotype, with SampleID representing the sample identity. For example, HG002-P and HG002-M correspond to the paternal and maternal haplotypes of HG002.
Within the HPRC assemblies, we identified a recurrent duplication event in the KIR3DP1-KIR2DL4-KIR3DL1/S1 triad in six samples. We also detected a known fusion gene, KIR2DL3/2DP1, in the paternal haplotype of sample HG02723, denoted as HG02723-P, and another recurrent novel fusion gene, KIR2DS4/3DL1, in HG02630-M and NA19240-P (Fig. 2A, third row). Moreover, we identified 26 KIR alleles with variants in the CDS and 428 KIR alleles with non-CDS variants. Of the 26 alleles with CDS variants, seven were detected as synonymous with existing IPD-KIR alleles, 15 were detected as nonsynonymous, and three were detected as fusion genes composed of segments of known KIR alleles (Fig. 3A; Supplemental Table S3). Five haplotypes had 10 novel alleles, 75 harbored at least five novel alleles, and all 94 haplotypes contained at least two novel alleles (Fig. 3B; Supplemental Table S2). By annotating IPD-KIR alleles to 94 HPRC haplotypes, 115 CDS-only alleles were complemented with the full genomic sequences derived from HPRC assemblies.
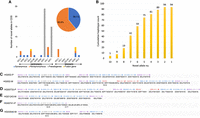
Representation of typical and atypical haplotype compositions within the HPRC cohort. (A) The identified novel alleles with variances in the CDS of KIR genes. Blue indicates alleles with synonymous variants, and orange indicates alleles with nonsynonymous variants. Counts of 2DS3 and 2DS5 were observed in the centromeric region. The pie chart only counts novel alleles of functional KIR genes, excluding fusion genes. (B) Haplotype counts with novel alleles identified. The number in each bar indicates the cumulative number of haplotype counts containing at least the number of novel alleles specified on the x-axis. There were five haplotypes with 10 novel alleles, and all 94 haplotypes contained at least two novel alleles. (C–G) Representative KIR haplotypes compositions within the HPRC cohort. (C) HG002 paternal (HG002-P) and maternal (HG002-M) haplotypes. The paternal haplotype of HG002, a typical cA01-tB01 haplotype, is presented from the centromeric to the telomeric motifs without deletions, duplications, ambiguities, and other structural variations. (D) The paternal haplotype of HG00733 features a duplication of KIR3DP1-KIR2DL4-KIR3DL1/S1, integrated into a cB01-tA01 haplotype. (E) The maternal haplotype of HG01243 is distinguished by the absence of both the KIR3DP1 and KIR2DL4 framework genes. (F) The paternal haplotype of HG00741 is characterized by the deletion of both the KIR3DP1 and KIR2DL4 framework genes as well as a truncated KIR3DL2 gene containing only exons 1–3. (G) The maternal haplotype of HG03540 is marked by a deletion of KIR3DL2 framework gene with an African high-frequency KIR3DL1 allele resulting from a deletion between KIR3DL1 and KIR3DL2. Purple represents framework genes; red, duplicated genes; blue, nonframework genes; and gray, pseudogenes. (#) An allele having nonsynonymous variant(s) in CDS, segmental deletion, or fusion with another gene; ($) an allele having variant(s) in non-CDS; and (+) a genomic allele matching an IPD-KIR CDS-only allele. The percentage (%) represents the proportion of identical base pairs in the CDS of the KIR gene loci to the assigned allele in a nonsynonymous allele or pseudogene case.
Duplications and deletions of KIR3DP1 and KIR2DL4 framework genes
We identified duplications of KIR3DP1 and KIR2DL4 in six (6.38%) haplotypes: HG002-M, HG00733-P, HG01123-P, HG02622-P, HG02630-P, and HG02717-M. These haplotypes contained two sequential copies of the KIR3DP1-KIR2DL4-KIR3DL1/S1 triad with an intergenic length of ∼14 kb between KIR3DP1 and KIR2DL4 (Figs. 2, 3C,D). Two consecutive triads, previously reported as tA01-ins3/4 (Pyo et al. 2013), were formed by special recombination events and typically included a specific downstream KIR3DP1*004 allele (Pyo et al. 2013), carried by HG002-M, HG00733-P, and HG01123-P. In an earlier study, the KIR3DP1*004 allele was confirmed to be transcribed in peripheral blood mononuclear cells because of the nature of the recombinant KIR3DP1 and KIR2L5A (Gómez-Lozano et al. 2005).
In contrast, deletions in KIR3DP1 and KIR2DL4 framework genes were observed in six (6.38%) haplotypes: HG00741-P, HG01243-P, HG01243-M, HG01358-M, HG03098-P, and NA19240-M. HG00741-P resulted from a deletion event involving KIR2DL1-KIR3DP1-KIR2DL4-KIR3DS1-KIR2DL5A-2DS3S5 in the cA01-tB01 haplotype, which generated the cA01-tB01-del7 haplotype (Figs. 2B, 3F; Pyo et al. 2013). The remaining five (5.32%) haplotypes, HG01243-P, HG01243-M, HG01358-M, HG03098-P, and NA19240-M, were formed by the deletion of KIR2DP1-KIR2DL1-KIR3DP1-KIR2DL4-KIR3DS1-KIR2DL5A-2DS3S5 in the cB01-tB01 haplotype, creating the cB01-tB01-del7 haplotype (Figs. 2D, 3E; Pyo et al. 2013).
PING (Norman et al. 2016) also analyzed KIR3DL1/S1 and KIR3DL2 alleles and their copy numbers in more than 2000 1KGP samples, including HG00733 and NA19240, both of which are part of the HPRC. In our study, the phased haplotypes of HG00733 were identified as one typical haplotype A (Fig. 2A), and the other featured both KIR3DL1 and KIR3DS1 together (Fig. 3D). The KIR3DL1, KIR3DS1, and KIR3DL2 alleles in HG00733, as well as the KIR3DL2 alleles in NA19240, were consistent with PING's results. However, PING identified an additional KIR3DL1*022 allele in NA19240 (Supplemental Table S16), a discrepancy potentially attributable to the KIR2DS4/3DL1 fusion we identified in NA19240-P.
Full and partial deletions of KIR3DL2
Previous studies have reported haplotypes lacking KIR3DL2 or exhibiting a partial deletion of KIR3DL2 in which sequences from intron 3 to exon 9 are missing (Jiang et al. 2012; Pyo et al. 2013). In our analysis, HG00741-P exhibited a partially deleted KIR3DL2 with a sequence matching only exons 1–3 of KIR3DL2*00601 (equivalent to exons 1–3 of KIR3DL2*10002 in our computational assignment) (Fig. 3F). This sequence was identical to the coding regions of exons 1–3 of KIR3DL2*007, as reported previously (Norman et al. 2009; Traherne et al. 2010). We also identified HG03540-M, the daughter of a Gambian trio that lacks the KIR3DL2 framework gene and carries KIR3DL1*05901. This allele type is frequently observed in Africa and has been reported as a fusion gene of KIR3DL1 and KIR3DL2 resulting from a deletion event in KIR3DL1 exons 6–9, KIR2DS4, and KIR3DL2 exons 1–5 within the tA01 haplotype, forming the tA01-del9-hybd1 haplotype (Fig. 3G; Norman et al. 2009; Pyo et al. 2013).
Real-time quantitative polymerase chain reaction (qPCR) was used to further verify CNVs in the KIR framework genes in 11 HPRC samples (HG002, HG00733, HG00741, HG01243, HG01358, HG02622, HG02630, HG02717, HG03098, HG03540, and NA19240) (Supplemental Table S4; Jiang et al. 2016). First, we confirmed that all 11 samples had two copies of the KIR3DL3 framework gene compared with the internal control gene STAT6. Five samples (HG002, HG00733, HG02622, HG02630, and HG02717) carried three copies of KIR3DP1 and KIR2DL4; four samples (HG00741, HG01358, HG03098, and HG03540) had only one copy of the pair; and one sample (HG01243) lacked both genes. Regarding KIR3DL2, a CNV with one copy was detected in two samples (HG00741 and HG03540) of the 11 studied. Because the HG00741 paternal haplotype contained only a part of KIR3DL2 (exons 1–3), it was not detectable by qPCR. The determination of CNV in the KIR framework genes was consistent with the results of the SKIRT pipeline.
Identification of a novel fusion gene, KIR2DS4/3DL1
We identified two copies of KIR2DS4 in HG02630-M and NA19240-P, both of which are cA01-tA01 haplotypes. One of the two KIR2DS4 copies in each haplotype was a fusion of KIR2DS4 and KIR3DL1 genes, comprising exons 1–7 of KIR2DS4*00101 and exons 8–9 of KIR3DL1*03501, which are also identical to many other KIR3DL1 alleles. Both haplotypes contained complete versions of KIR2DS4*00101 and KIR3DL1*03501 (Fig. 4A). We verified the presence of the KIR2DS4/3DL1 fusion gene using PacBio HiFi reads obtained from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) for both HG02630 and NA19240. We found that multiple PacBio HiFi reads perfectly matched at least exon 7 to exon 9 sequences of the novel fusion gene in both samples (Supplemental Table S5). Because KIR2DS4 was absent in the other haplotypes of HG02630 and NA19240 (i.e., HG02630-P, the second row in Fig. 2B, and NA19240-M, the first row in Fig. 2D), these PacBio HiFi reads supported the existence of the KIR2DS4/3DL1 fusion gene. We also explored the potential evolutionary relationship between the KIR2DS4, KIR3DL1, and KIR2DS4/3DL1 fusion genes. Several regions within intron 7 of KIR2DS4 and KIR3DL1 exhibited high sequence similarity, as indicated by the alignment tool and represented the crossover region of the fusion gene (Fig. 4B).

Identification of a novel fusion gene, KIR2DS4/3DL1. (A) A schematic diagram illustrating the formation of a novel KIR2DS4/3DL1 fusion gene composed of exons 1–7 from KIR2DS4 and exons 8–9 from KIR3DL1. (B) Sequence alignments comparing the intron 7 region of the fusion gene with corresponding regions in KIR2DS4 and KIR3DL1 for HG02630-M and NA19240-P. The highlighted yellow regions, signifying the sequences common to all three genes, are potential sites of the crossover event leading to the formation of the fusion gene. (C) An electrophoresis image displaying the results from the two trios of HG02630, NA19240, and their respective parents. Five out of the six samples contain the KIR2DS4/3DL1 fusion gene (indicated by FG), whereas all six samples carry the KIR3DL1 control gene as an internal control (indicated by IC). (D) The translated protein sequences of the KIR2DS4*00101, KIR3DL1*03501, and the KIR2DS4/3DL1 fusion genes were aligned to assess potential functional alterations in the fusion gene. Variations in the transmembrane domain are highlighted in bold. The transmembrane domain is encoded by the DNA sequence of exon 7, so the KIR2DS4/3DL1 fusion gene shares identical transmembrane domain residues with KIR2DS4*00101. The cytoplasmic domain, encoded by exons 7, 8, and 9, incorporates contributions from KIR3DL1*03501, including two inhibitory tyrosine-kinase motifs (ITIMs; I/VxYxxL/V), which are underscored and in bold in the alignment (Carr et al. 2005; Campbell and Purdy 2011; Oszmiana et al. 2016).
We further confirmed the KIR2DS4/3DL1 fusion gene by PCR amplification with sequence-specific primers (PCR-SSP) (Gómez-Lozano and Vilches 2002), followed by Sanger sequencing on the two trios of HG02630, NA19240, and their parents. Both HG02630 and her mother HG02629 carried the KIR2DS4/3DL1 fusion gene, whereas her father HG02628 did not. Similarly, NA19240 and her father, NA19239, carried the fusion gene, whereas her mother, NA19238, also carried the fusion gene unexpectedly. All six samples from the two trios were positive for KIR3DL1, which was used as an internal control (Fig. 4C). We subsequently performed Sanger sequencing of five samples positive for the fusion gene (HG02630, HG02629, NA19240, NA19239, and NA19238) using the PCR-SSP amplicons. All fusion gene sequences in the five samples were identical to the sequences from the HPRC assemblies, confirming the presence of the KIR2DS4/3DL1 fusion gene. Sequencing results for the KIR3DL1 control gene showed that all five samples had identical KIR3DL1 sequences in the amplicon region, providing evidence for the coexistence of KIR3DL1 and KIR2DS4/3DL1 in these samples.
The fusion gene's translation from exons 7 to 9 was in frame, resulting in intact transmembrane and cytoplasmic domains (Fig. 4D). Because the transmembrane domain is encoded by the DNA sequence of exon 7, the KIR2DS4/3DL1 fusion gene shares identical transmembrane domain residues with KIR2DS4*00101. However, the cytoplasmic domain, encoded by exons 7, 8, and 9, incorporates contributions from KIR3DL1*03501, including two inhibitory tyrosine-kinase motifs (ITIMs; I/VxYxxL/V). A positively charged lysine at position 9 within the transmembrane domain enhances interaction with DAP12, which is associated with activating KIRs. Additionally, the presence of valine at the first position of the transmembrane domain may alter the conformation of the extracellular domain, potentially impacting its inhibitory function (Carr et al. 2005; Campbell and Purdy 2011; Oszmiana et al. 2016).
Identification of the KIR2DL3/2DP1 fusion gene in HG02723-P
We identified the KIR2DL3/2DP1 fusion gene in the paternal haplotype of HG02723 (HG02723-P), consistent with previous reports (Traherne et al. 2010; Hou et al. 2012; Pyo et al. 2013). In these earlier studies, the same fusion gene was detected and verified using the sequences obtained under the NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) accession numbers CU041340 and CU633846. Our analysis of HG02723-P revealed that the fusion gene, composed of exons 1–5 of KIR2DL3*00101 and exons 6–9 of KIR2DP1*00201, matched the sequences in GenBank accessions CU041340 and CU633846.
The identification of the KIR2DL3/2DP1 fusion gene in the KIR gene complex confirms its presence and highlights the need to investigate the potential functional implications and roles of such hybrid genes in immune responses and disease susceptibility.
Difficulties and incompleteness in the HG002 assembly
The paternal haplotype of HG002 contained KIR3DL3*00101-KIR2DL3*00101-KIR2DP1*00201-KIR2DL1*0030218-KIR3DP1*0030202-KIR2DL4*0050101-KIR3DS1*0130101-KIR2DL5A*0010103-KIR2DS5*0020111-KIR2DS1*00201-KIR3DL2*00701 from 5′-end to 3′-end in one single contig. However, the HG002 maternal haplotype assembled into four different contigs containing both KIR2DL4 and KIR3DL1/S1, indicating four possible copies of the KIR genes. The four contigs of the maternal haplotype were KIR3DL3*00301-KIR2DS2*005-KIR2DP1*00102-KIR2DL1*0040105-KIR3DP1*0030101-KIR2DL4*0050101-KIR3DS1*01301 in contig 1, KIR2DL4*0050101-KIR3DS1*01301 in contig 2, KIR2DL4*0050101-KIR3DS1*0130118-KIR2DL5A*00101-KIR2DS5*00201-KIR2DS1*00201-KIR3DL2*00701 in contig 3, and KIR3DS1*0130118-KIR3DP1*004-KIR2DL4*0050101 in contig 4, respectively (Fig. 5; Supplemental Table S1). Based on the annotation, we observed that there were two complete KIR3DS1*0130118 and KIR2DL4*0050101 alleles in both contigs 3 and 4 but displayed in reverse order, whereas only partial KIR3DL1/S1 alleles in contigs 1 and 2 were of the same order. According to the HPRC data, we could only assume that there may be more than one copy of the triad of KIR3DP1-KIR2DL4-KIR3DS1 genes.

The discontinuous KIR contigs of HPRC HG002 maternal assembly. The HG002 maternal assembly is segmented into four contigs matching with IPD-KIR alleles, representing an unusual case (one of 94 haplotypes) in the HPRC Year-1 release. No single contig encompasses the entire KIR complex. One contig (4), KIR3DS1-KIR3DP1-KIR2DL4, aligns exclusively between the centromeric and telomeric motifs, suggesting potential structural variations and gene CNVs around the recombinant region.
We assessed the qualities of the KIR region in the maternal haplotype of HG002 using assemblies generated from different algorithms (Supplemental Table S6). To address the discontinuity of the HG002 maternal haplotype, we incorporated assembly data from an extended study that enhanced Hifiasm to produce more complete versions of the haplotype-resolved HG002 contig. The data, which were derived from a combination of PacBio HiFi reads and Hi-C data (Cheng et al. 2022), affirmed our primary assumption of duplication of the KIR3DP1-KIR2DL4-KIR3DS1 triad (Figs. 2D, 3C; Supplemental Table S15). Furthermore, we compared the KIR alleles of high-quality HG002 assemblies released in a recent study that utilized a combination of Oxford Nanopore Technologies long-read sequences, Hi-C, trio data, and/or HiFi read sequences (Rautiainen et al. 2023). We observed maximum concordance among the various data sets with hifiasm.hic.0.16.1.rep4.hap1 as a representative example (Figs. 2D, 3C; Supplemental Table S15). The only discordance was the allele of KIR2DS5 of HG002-M, which appeared in all results of samples named as starting with downsampled_verkko. The telomeric motifs of the paternal and maternal haplotypes of HG002 were identical, particularly in the three copies of KIR2DL4 and KIR3DS1. This extreme similarity makes the phasing of the two haplotypes particularly challenging. Despite the difficulties and incompleteness of HG002 assembly, the SKIRT pipeline comprehensively identified all KIR alleles of HG002 using HPRC assembly, including KIR2DP1, KIR3DP1, and all KIR framework genes in various copy numbers (zero/one/two copies), with no mix-ups among KIR2D genes.
Comparisons with Kass in HG002 genome
The Kass assembly and annotation workflow did not identify the duplication of the KIR3DP1-KIR2DL4-KIR3DS1 triad as disclosed in the study. As for HPRC HG002 assembly, Kass annotation could not generate annotations for KIR2DP1 and KIR3DP1 even if it perfectly matches some alleles, whereas SKIRT identified a perfectly matched allele (i.e., paternal KIR2DP1) and other partially matched ones. Furthermore, Kass annotation seemed to operate under the presumption of a single copy for all KIR framework genes on one haplotype and exhibited confusion regarding some KIR2D genes (Supplemental Table S1). However, SKIRT identified all the KIR alleles and CNVs of the KIR framework genes in the HPRC HG002 assembly.
Comparisons with T1K in HPRC genomes
T1K (Song et al. 2023) offered an innovative methodology for KIR genotyping using Illumina short-read WGS, termed hereafter as T1K-genotype. The study also independently annotated KIR alleles for 26 out of the 47 HPRC phased assemblies to validate their T1K-genotype results. We termed their annotation results hereafter as T1K-annotate. The data released by T1K-annotate (https://github.com/mourisl/T1K_manuscript_evaluation/tree/master/HPRC_process/Genome) and T1K-genotype (https://github.com/mourisl/T1K_manuscript_evaluation/tree/master/HPRC_process/Illumina) were compared with SKIRT results. The T1K-annotate results for 26 HPRC genomes were assumed to be the benchmark for validating T1K-genotyping results. However, the results for the T1K-genotype and T1K-annotate were not identical, implying the potential ambiguity. For example, SKIRT and T1K-annotate both identified KIR2DL1 as two copies, one in each phased haplotype, in HG00438, but T1K-genotype identified it as only one copy in the genome. When we used the SKIRT copy number results as the ground truth (N = 499 for all KIR genes in 26 HPRC individuals), T1K-genotype showed an overall 85% (426/499) recall rate, also known as sensitivity. There were 13 out of 17 genes with a <90% gene level recall rate. T1K-annotate showed a 99.6% (497/499) overall recall rate (Fig. 6A). Furthermore, the precision, also known as the positive predictive value, for copy numbers of individual genes were largely inconsistent between T1K-genotype and T1K-annotate. For example, T1K-annotate failed to identify the KIR2DS4/3DL1 fusion gene in the HG02630-P and the truncated KIR3DL2 in HG00741-P, the latter of which led to incorrect identification of copy number variation of KIR3DP1 in HG00741-P, impacting the sensitivity of KIR2DS4 and KIR3DL2 and the precision of KIR3DP1. Both of the correct copy number determinations have been demonstrated by independent molecular validations in this study.
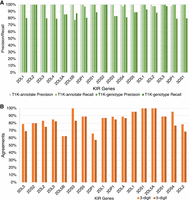
The comparisons of KIR gene copy number results of T1K and SKIRT. (A) The precision and recall rate of copy numbers determined by T1K-annotate and T1K-genotype as compared with SKIRT in 26 HPRC genomes. The gene copy numbers detected by T1K-annotate and T1K-genotype are compared separately with the results of SKIRT as the ground truth. T1K-annotate exhibits perfect precision and recall of 100% in almost all KIR genes, except for a 98% recall rate for both KIR2DS4 and KIR3DL2 and a 98% precision rate for KIR3DP1. Nevertheless, T1K-genotype shows recall rates of <90% for 13 out of 17 genes. (B) The agreement between T1K-annotate and SKIRT for the allele annotation of 26 HPRC genomes. Each allele annotation of T1K is compared with the results of SKIRT at two resolution levels: three digit and five digit. If a candidate allele annotated by T1K matches that of SKIRT in the same haplotype, it is marked as one in agreement. If T1K gives one or multiple candidate alleles with differences only in the sixth and seventh digits compared with the one assigned by SKIRT, the agreement was assigned on the five-digit level. If the first three digits were the same but the fourth and fifth digits were different, the agreement was assigned on three-digit level.
Although T1K-annotate demonstrated proficiency in identifying the copy numbers of KIR genes, its performance in allele identification showed an overall agreement of 85% and 79% with the three-digit and five-digit resolution results from SKIRT, respectively (Fig. 6B; Supplemental Table S7). We further validated the identified alleles from both T1K and SKIRT by visually inspecting the alignments with IPD-KIR allele CDSs, which confirmed the accuracy of the annotations made by SKIRT (Supplemental Fig. S1A,B). Given that T1K-annotate selects alleles based solely on the alignment of exonic sequences to HPRC assemblies using BWA-MEM (Song et al. 2023), we confined our comparison of allele resolutions to the CDS, representing the first five digits. In contrast, SKIRT provides a more comprehensive genomic resolution, extending to all seven digits. The low consistency between SKIRT and T1K-annotate can be partly attributed to T1K's early versions, which ignored alleles without intronic sequences in IPD-KIR. T1K-annotate excluded such alleles from its reference file, thereby not reporting them in annotation results. For instance, KIR3DL3*01403, labeled as “partial” in IPD-KIR and lacking an intronic sequence, was not considered by T1K-annotate (Supplemental Fig. S1A).
KIR alleles of human reference genomes
We compared the SKIRT KIR allele annotations for GRCh37 (hg19) (Church et al. 2011), GRCh38 (hg38) (Schneider et al. 2017), and T2T CHM13v2.0 (hs1) (Nurk et al. 2022) with the latest GENCODE annotations available on the UCSC Genome Browser (Kent et al. 2002; Raney et al. 2014; Nassar et al. 2023) for the three reference genomes (Supplemental Fig. S2A–C; Supplemental Tables S8–S10; Fiddes et al. 2018; Frankish et al. 2021; Shumate and Salzberg 2021). SKIRT annotations were added as custom tracks on the UCSC Genome Browser for comparison with the GENCODE annotations in the KIR region. SKIRT provided IPD-KIR allele annotations for the genomes and only displayed coding regions, excluding untranslated regions (UTRs), as no UTR information was available for the IPD-KIR allele sequences. The annotations for KIR2DP1 and KIR3DP1 pseudogenes were more comprehensive: Exon 3 of KIR2DP1, identified as a pseudoexon, was not displayed in our annotations but was shown in the GENCODE annotations, whereas exon 3 of KIR3DP1, which is not a pseudoexon, was displayed in our annotations but missed in the GENCODE annotations. GENCODE sometimes provides multiple alternative splicing annotations for a single KIR gene; however, our annotations identified the exact correct coding regions for each KIR gene. The KIR gene content identified by SKIRT and GENCODE was consistent with the GRCh37 reference sequence and regions (Chr 19 of hg19) identified in an earlier study (Sakaue et al. 2022).
Incomplete alleles in the IPD-KIR database and our genomic identification
In our study, we uncovered that several alleles listed as “partial” in the IPD-KIR database, regardless of their categorization as CDS-only or genomic, had either portions (KIR2DS4*00104 and KIR2DS5*009) or the entirety (KIR3DL2*024, KIR3DL2*01101, and KIR3DL3*030) of exons 1 and 2 missing from their IPD-KIR listings. Our comprehensive investigation led to the complete genomic identification of these alleles. Furthermore, we identified the exon 2 presence of two additional allele sequences, KIR3DP1*00601 and KIR3DP1*01001, which were confirmed as “full genomic” but without exon 2 and its flanking region in the IPD-KIR database. The KIR3DP1*00601 allele from HG01123-M diverged from the KIR3DP1*0060101 allele in the IPD-KIR database, primarily by the inclusion of an additional exon-2 segment and its flanking region, with the others of the CDS remaining identical. Similarly, the KIR3DP1*01001 allele from NA21309-P mainly differed from the KIR3DP1*0100102 allele in the IPD-KIR database in the presence of an additional exon-2 segment and its flanking region, whereas the others of the CDS were completely identical. Previous studies have indicated that certain KIR3DP1 alleles show variations in the presence or absence of exon 2 (Gómez-Lozano et al. 2005; Bono et al. 2018). There are currently no deterministic rules for detecting the synonymity and completeness of KIR2DP1 and KIR3DP1 alleles. We could not compare the putative transcripts between the novel and known alleles nor define an allele with an extra exon compared with a known “full” allele in IPD-KIR. Because of insufficient information, further discussion with the IPD-KIR committee is needed before submitting these pseudogene sequences to GenBank and IPD-KIR databases (Supplemental Data).
Discussion
This study presented a comprehensive investigation of the KIR complex in HPRC Year-1 assemblies, efficiently describing the high-resolution allele-level haplotypic structure of each chromosome pair with all existing KIR gene duplications, deletions, and CNVs. The SKIRT pipeline is a practical tool for analyzing KIR genes in human genome assembly data, enabling accurate identification and annotation of KIR alleles and their structural variations. We demonstrate that the copy numbers of the three KIR framework genes except for KIR3DL3, presumably one for each haplotype in some previous studies using NGS, were mostly one but sporadically two or none for a single haplotype of the HPRC samples studied. The identification of the KIR2DS4/3DL1 fusion gene in HG02630 and NA19240 individuals and subsequent PCR verification highlighted the importance of using a robust analytical pipeline to uncover novel KIR gene structural variations. This is particularly relevant considering the potential presence of undiscovered KIR receptors resulting from continuously occurring unequal crossover and rearrangement of gene segments in the KIR complex. The duplication and deletion of KIR genes may alter their expression and thus modulate the reciprocal immune response, such as KIR3DL1/S1 influences some infectious diseases (Boudreau et al. 2016; Boelen et al. 2018; Li et al. 2022).
Utilizing the highest-resolution and accurate annotation of KIR alleles, our study effectively identified all known KIR alleles listed in the IPD-KIR database. Moreover, we provided full genomic sequences for known CDS-only alleles, as well as novel synonymous and nonsynonymous alleles. In total, we generated 570 full-genomic sequence alleles, of which 26 alleles exhibited CDS variants, 428 alleles displayed non-CDS variants, and an additional 116 alleles showed no variants but complemented their corresponding CDS-only alleles in the IPD-KIR database. This achievement paves the way for the submission of these sequences to the database, thereby catalyzing further KIR studies. Correct submission to the IPD-KIR database is critical for the advancement of KIR research. For instance, our study first identified the KIR2DL5*0020102 allele for HG01952-P, KIR2DL5*0020106 for HG02145-M, and KIR2DL5*0020104 for HG02257-P. Currently, these three allele sequences are available only for CDS, instead of full genomic sequences, in the IPD-KIR database. The well-organized KIR nomenclature synergizes with the IPD-KIR database (Marsh et al. 2003), curating hundreds of nucleotide and peptide sequences and facilitating the digital identification of KIR allele sequences, ideally to benefit our study.
The HPRC Year-1 release provides data from multiple sequencing technologies, including genome assemblies, their PacBio HiFi long-read sequences, and their parental Illumina short-read sequences. These raw data, sequenced from publicly available cell line samples from the National Human Genome Research Institute Sample Repository for Human Genetics Research at the Coriell Institute, were selected from 1KGP, which offers diverse human genetic variations (The 1000 Genomes Project Consortium 2015). These cell line samples serve as invaluable resources for further experiments and assays, and the variety of sequencing data facilitates the development of novel analysis methods. Although there have been many studies focusing on 1KGP, Kass, T1K, and SKIRT were the only three generating KIR alleles using 1KGP genome assemblies, and SKIRT outperformed the other two (Roe et al. 2020; Song et al. 2023). Expanding the analysis of KIR gene diversity to include a broader range of populations could help elucidate population-specific patterns of KIR gene variations and their potential implications for disease risk and transplantation outcomes.
Our results revealed several specific patterns of KIR gene duplication within the haplotypes. The most noticeable was duplication of the KIR3DP1-KIR2DL4-KIR3DS1 triad. Long-range deletion and duplication of sequence segments in the KIR region create a great variety of copy numbers and polymorphic alleles. Identifying accurate and high-resolution alleles for all presence of KIR genes in the HPRC assemblies paves the way for future exploration and development of tools and functional assays aimed at the novel KIR alleles and genes uncovered. Lin et al. (2023) utilized SKIRT's results as a reference ground truth to evaluate the performance of PING's KIR genotyping methods on short-read data (Marin et al. 2021). Their preliminary findings show that PING (Marin et al. 2021) achieved high accuracy levels, scoring 0.99 with 100 simulated short-read WGS data sets and 0.92 with a subset of HPRC short-read WGS data sets (44 out of 47 samples). The accuracy in identifying KIR alleles may be highly affected by the quality of the contigs around the KIR complex region. This underscores the potential of the SKIRT pipeline as an effective and valuable tool for evaluating the quality of assembled contigs.
Methods
HPRC assembly data collection and IPD-KIR database
The SKIRT pipeline annotates publicly available HRPC genome assemblies with all official KIR alleles assigned by the KIR nomenclature committee (Robinson et al. 2010; Maccari et al. 2020; https://www.ebi.ac.uk/ipd/kir/about/nomenclature/allele/). We used version v2.12.0 of the IPD-KIR hosted by the European Molecular Biology Laboratory's European Bioinformatics Institute (EMBL-EBI) (https://www.ebi.ac.uk/ipd/kir/). We obtained 47 HPRC genome assemblies from Year-1 sequencing data (https://github.com/human-pangenomics/HPP_Year1_Assemblies). The 47 high-quality haplotype-resolved genome assemblies were produced as diploid contigs using parental Illumina short-read sequence data to sort filial haplotypes of PacBio HiFi long-read sequence data and de novo assembly using the graph trio binning algorithm of Hifiasm (Cheng et al. 2021; Wang et al. 2022), providing the highest resolution for haplotype-resolved contigs. With a long sequencing length (10–20 kb) and highly accurate consensus quality (Q30) of HiFi-reads, the reads could cover each KIR gene (4–16 kb) or intergenic region (2.4–14 kb). Consequently, phased diploid genome assemblies and algorithms offer better ways to identify each parental KIR haplotype.
The PacBio HiFi data used in this study were obtained from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA701308 for HG00411, HG00423, HG00438, HG00480, HG00621, HG00673, HG00730, HG00735, HG00741, HG01071, HG01106, HG01123, HG01138, HG01175, HG01258, HG01358, HG01361, HG01888, HG01891, HG01928, HG01952, HG01978, HG01998, HG02027, HG02083, HG02148, HG02257, HG02486, HG02523, HG02559, HG02572, HG02622, HG02630, HG02717, HG02886, HG03453, HG03471, HG03516, HG03540, and HG03579; and PRJNA731524 for HG002, HG005, HG00733, HG01109, HG01243, HG01442, HG02055, HG02080, HG02109, HG02145, HG02723, HG02818, HG02970, HG03098, HG03486, HG03492, NA18906, NA19030, NA19240, NA20129, NA20300, and NA21309. The NCBI BioProject accession number PRJNA730823 is an umbrella project for all HPRC-related projects, and PRJNA730822 is an umbrella project for all 47 HPRC assembly projects. The other two assembly data sets for HG002 are available publicly at Zenodo (Cheng et al. 2022; Rautiainen et al. 2023; https://doi.org/10.5281/zenodo.5948487; https://doi.org/10.5281/zenodo.7400747).
SKIRT: algorithms of the structural KIR annoTator pipeline
The pipeline was designed to identify new variants, novel alleles (both synonymous and nonsynonymous), and structural variations, including gene duplications and deletions. (Fig. 1A) To discern multiple polymorphic alleles at each potential locus, we employed minimap2 (Li 2018), a versatile pairwise long-read sequence aligner, to map all KIR alleles (including CDS-only and genomic) as query sequences against human genome assemblies as target sequences. A separate mapping process was conducted to detect the short (8 bp) exon 9 of KIR3DS1 in the CDS-only queries, employing the “‐‐end-seed-pen 5” setting to overcome the minimap2 limitation for “tiny exons”. To detect long intragenic insertions, we activated the splice mode and limited the insertion size with the “-G16k” setting. This approach facilitated the recognition of splicing sites and intragenic distances of CDS-only alleles and prevented the potential overextension issue of a single CDS-only allele mapping across multiple KIR loci owing to high polymorphism among certain alleles of different genes.
Candidate aligned alleles were assessed based on several criteria for each KIR gene, including a reasonable flanking region with an intergenic length of 2.4 kb among most KIR genes and 14 kb between KIR3DP1 and KIR2DL4, full genomic length of each gene ranging from 4 to 16 kb, starting and ending coordinates, and numbers of mismatches and gaps. We compared all mapped candidates to select the best-matching KIR alleles, achieving up to seven digits of KIR allele nomenclature indicative of genomic resolution (Fig. 1B). The mappings of genomic alleles were examined only when the same CDS-only allele perfectly matched the locus. Allele resolution was presented as seven-digit when the genomic allele matched perfectly. In the case of any variance in the mapping of the query CDS-only allele, we identified the CDS of the KIR locus in the target assemblies using the cs tag of each alignment generated by minimap2. The target CDS was mapped against the IPD-KIR protein sequences using TBLASTN (Camacho et al. 2009) and Biopython (Cock et al. 2009) to confirm whether the genetic variances were synonymous with an existing allele (Fig. 3A; Supplemental Table S3).
The SKIRT outputs the haplotype data into a comma-separated value (CSV) file named “<sample name>.hap.csv,” providing a comprehensive overview of the entire haplotype with all high-resolution KIR alleles arranged according to their chromosomal order. In this CSV table, each haplotype is presented as a row, with the presence or absence of each KIR allele denoted in its dedicated columns. To aid in the identification of gene duplications within the haplotype, particularly when gene orders do not follow typical patterns, a “central region” column is included between columns for common centromeric and telomeric motifs. Moreover, it generates variant call format (VCF) and browser extensible data (BED) format files, capturing allele annotations and their variants compared with the most similar alleles identified during the process. The assembly FASTA, VCF, and BED files were loaded into the Integrative Genomics Viewer (IGV) (Robinson et al. 2011) for a graphical view of the KIR allele annotations. This visualization facilitated the understanding of relative gene locations, coordinates, and structural variations, such as CNVs, deletions, gene fusions, and variants specified in the BED files (Supplemental Fig. S3; Supplemental Methods).
Identification of the fusion genes KIR2DS4/3DL1 and KIR2DL3/2DP1
Initially, we only identified a part of KIR2DS4*00101 as the KIR2DS4/3DL1 fusion gene in both the maternal haplotype of HG02630 and the paternal haplotype of NA19240 because the allele mapped 96.1% of the gene segment. The predominant part, encompassing only exons 1 to 7 of KIR2DS4*00101, exhibited the absence of the subsequent exons 8 and 9. This is primarily because of the mapping preference of minimap2, which tends to identify the more extensive mapping when one locus maps to two allele sequences. In such cases, minimap2 does not present the mapping of the minor part, that is, the exons 8 and 9 in KIR3DL1 of the two haplotypes, showing only those mapped to the majority of the loci. Further manual investigation of the downstream flanking sequences of this partial KIR2DS4 revealed the presence of two other exon sequences completely identical to exons 8 and 9 of KIR3DL1*03501 in the HG02630-M and NA19240-P contigs. The sequence of these two exons is also identical to many other KIR3DL1 alleles.
We initially observed an unusually 3.2 kb long intragenic region, spanning much longer than the usual length (2.4 kb), between the two identified KIR2DS4 alleles, with the upstream one lacking exons 8 and 9. Subsequent detailed mapping of exons 8 and 9 only sequences of other KIR alleles, excluding KIR2DS4, led to the identification of the fusion gene. Without long and continuous genomic sequences exceeding 16 kb in such regions, it would not have been possible to identify the fusion genes and determine the accurate copy numbers of the related genes in the haplotype, that is, KIR2DS4 and KIR3DL1. To assure the existence of the fusion gene and eliminate the possibility of assembly errors, we cross-verified the fusion gene sequences using both Illumina WGS short reads and PacBio HiFi long reads, confirming our results.
We identified the KIR2DL3/2DP1 fusion gene in the paternal haplotype of HG02723 (HG02723-P), consistent with previous reports (Traherne et al. 2010; Hou et al. 2012; Pyo et al. 2013). In these earlier studies, the same fusion gene was detected, verified, and submitted to GenBank (accession numbers CU041340 and CU633846). Our analysis of HG02723-P revealed that the fusion gene, composed of exons 1–5 of KIR2DL3*00101 and exons 6–9 of KIR2DP1*00201, matched the CDSs of GenBank accessions CU041340 and CU633846. This fusion gene was not a novel finding. However, the allele sequence was not submitted to IPD-KIR. We used Illumina WGS short reads and PacBio HiFi long reads to certify the sequence of this fusion gene of HG02723-P and confirmed the existence of the fusion gene at the locus. The KIR2DL3/2DP1 fusion gene was first discovered in a sample of European ancestry (Traherne et al. 2010) and was reported again in a sample of Asian ancestry (Hou et al. 2012), both as cA and telomeric B haplotypes. However, HG02723-P is a Gambian individual with the cA and tA haplotypes.
TaqMan multiplex real-time PCR for CNV verification
For KIR copy number determination, we conducted TaqMan multiplex real-time PCR on an Applied Biosystems (ABI) QuantStudio 5 real time detection system (Thermo Fisher Scientific) using the qKAT protocol (Jiang et al. 2016). For the KIR framework genes KIR3DL3, KIR3DP1, KIR2DL4, and KIR3DL2 and the internal control gene STAT6, a reaction volume of 20 μL contained a final concentration of 1× qPCRBIO probe mix Lo-ROX (PCR BIOSYSTEMS), optimized concentrations of primers and probes (Supplemental Tables S11, S12), and 8 ng of genomic DNA (Supplemental Table S13; Supplemental Methods).
PCR-SSP primer design and confirmation of the KIR2DS4/3DL1 fusion gene
To amplify the correct product of the KIR2DS4/3DL1 fusion gene, primers were designed for PCR-SSP (Gómez-Lozano and Vilches 2002), specifically for HG02630 and NA19240. Our design ensured that the primer sequences perfectly matched the specific target alleles, whereas others had certain nucleotide mismatches, particularly at the 3′ end of the primers. We examined the exon 7 sequence of KIR2DS4 to determine the forward primer, KIR2DS4_fusion, and the exon 8 sequence of KIR3DL1 to obtain the reverse primer, KIR3DL1_reverse. These primers were designed to produce PCR amplicons 596 bp in length. We also designed another forward primer targeting on the exon 7 of KIR3DL1, KIR3DL1_control, to ensure the effectiveness of the primer sets. Together with the KIR3DL1_reverse primer, this primer set produced 634-bp amplicons when the KIR3DL1 gene was present in the samples (Supplemental Table S17; Supplemental Methods).
Data access
All HPRC cell lines were obtained from Coriell, with the exception of HG01123, HG02486, and HG02559. All data generated or analyzed during this study are included in this published article, its Supplemental Files, and Zenodo (https://doi.org/10.5281/zenodo.8094803). SKIRT source code is publicly available on GitHub (https://github.com/calvinckhung/skirt) and as Supplemental Code. All genomic nucleotide sequence data of novel KIR alleles, except pseudogenes KIR2DP1 and KIR3DP1, reported in this study have been submitted to the NCBI Genbank database (https://www.ncbi.nlm.nih.gov/genbank/) and are available in the Third Party Annotation (TPA) section of the DDBJ/ENA/GenBank databases under accession numbers BK064711–BK064753 and BK067265–BK067627 (Supplemental Table S14).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This study was funded by the Ministry of Science and Technology, Taiwan (MOST-109-2622-B-002-004-CC2). We thank the National Center for High-performance Computing (NCHC) of the National Applied Research Laboratories (NARLabs) in Taiwan for providing computational and storage resources. The English editing of this article was sponsored by National Taiwan University with the support of the Higher Education Sprout Project from the Ministry of Education, Taiwan.
Author contributions: P-L.C., J.S.H., Y-C.Y., and Y-C.L. proposed the initial idea of annotating KIR alleles with high-quality assemblies, initiated the project, and assisted with discussions. T-K.H. conceived the study, designed the analysis pipeline, acquired the HPRC data, performed the analysis, interpreted the analysis results, and produced the related figures and tables with the guidance of P-L.C., Y-C.Y., J.S.H., and C-L.H. W-C.L. and Y-C.Y. designed all laboratory experiments, and W-C.L. performed the experiments and produced related figures and tables under the guidance of Y-C.Y. T-K.H. and W-C.L. managed the writing and organization of the manuscript with suggestions and proofreading by Y-C.Y. and J.S.H. H-W.C. and H-Y.L. assisted with the WGS data access and analysis under the guidance of J.S.H. and C-Y.C. P-L.C. managed the project. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278358.123.
-
Freely available online through the Genome Research Open Access option.
- Received November 12, 2023.
- Accepted August 14, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








