
Large-scale genomic analysis of the domestic dog informs biological discovery
Abstract
Recent advances in genomics, coupled with a unique population structure and remarkable levels of variation, have propelled the domestic dog to new levels as a system for understanding fundamental principles in mammalian biology. Central to this advance are more than 350 recognized breeds, each a closed population that has undergone selection for unique features. Genetic variation in the domestic dog is particularly well characterized compared with other domestic mammals, with almost 3000 high-coverage genomes publicly available. Importantly, as the number of sequenced genomes increases, new avenues for analysis are becoming available. Herein, we discuss recent discoveries in canine genomics regarding behavior, morphology, and disease susceptibility. We explore the limitations of current data sets for variant interpretation, tradeoffs between sequencing strategies, and the burgeoning role of long-read genomes for capturing structural variants. In addition, we consider how large-scale collections of whole-genome sequence data drive rare variant discovery and assess the geographic distribution of canine diversity, which identifies Asia as a major source of missing variation. Finally, we review recent comparative genomic analyses that will facilitate annotation of the noncoding genome in dogs.
The domestic dog has increasingly been recognized as a uniquely informative model system. Dogs are divided into more than 350 breeds recognized worldwide by organizations such as the Fédération Cynologique Internationale (FCI; https://www.fci.be/en/) and American Kennel Club (AKC) (Rogers and Brace 1995; Fogle 2000; https://www.akc.org). There also exists an untold number of nonbreed, geographically defined populations, often referred to as village dogs, that are not under human selection but that make up most of the world's dog populations (Boyko et al. 2009; Shannon et al. 2015). Studies of both breed and village dogs have generated exciting opportunities to identify genetic mechanisms underlying both morphological and behavioral traits, as well as genetic variants associated with disease, aging, and domestication.
Dogs were domesticated from wolves over a very short evolutionary time, with recent estimates suggesting 15,000–40,000 years before the present (Skoglund et al. 2015; Perri et al. 2021; Bergström et al. 2022). Although the timing, location, and number of primary domestication events remain topics of debate (Savolainen et al. 2002; Larson et al. 2012; Frantz et al. 2016, 2020; Sinding et al. 2020; Bergström et al. 2022), most recent analyses are consistent with a dual origin of domestication from east and west Eurasia that continues to shape the population structure of modern dogs today (Bergström et al. 2022).
Breed-associated differences in morphology and behavior have garnered particular interest and are often included in “breed standards,” which are the ideal set of characteristics that define each breed. The stringent criteria and closed breeding practices set for each breed suggests that breed standard traits are highly heritable. Such traits are often synonymous with a breed, like the spotted coat of the Dalmatian, but can also be more nuanced, like the requirement that every Norwegian Lundehund have a minimum of six toes. Much harder to study are well-recognized stereotypic dog behaviors (e.g., herding, pointing) and personality traits (e.g., protective, affable, stubborn, etc.) (MacLean et al. 2019; Dutrow et al. 2022; Morrill et al. 2022; Salonen et al. 2023). This is largely because of variability in presentation, a lack of quantifiable metrics, complex underlying genetics, and the degree to which such phenotypes are truly breed traits (Morrill et al. 2022). Herein, we discuss recent advances in canine genomics, strategies for optimizing canine genetic studies, and future perspectives.
Behavior and morphology in dogs
Interest in canine behaviors led to studies of stereotypic breed behaviors from the earliest stage of the canine genome project (McCaig 1996). Although such studies were generally underpowered, more recent analyses have readdressed some of the same questions, yielding interesting results (Dutrow et al. 2022; Morrill et al. 2022). Dutrow et al. (2022) collated genetic data from more than 4000 canids, identifying 10 major genetic lineages. They showed that the lineage membership of breeds was associated with behavioral trait data collected from 46,000 dogs. Further, lineage-specific variation was associated with genes in neurodevelopmental coexpression networks, suggesting that the accumulation of many small effect variants drove behavioral diversification between breeds (Dutrow et al. 2022). Interestingly, the investigators found that sheepdog-associated variation was enriched among genes with roles in axon guidance. Eight of the 14 identified axon-guidance genes were important in midline patterning, suggesting a relationship between binocular vision and motor behavior in sheepdogs. In comparison, Morrill et al. (2022) paired survey and genotype data from large numbers of pure and mixed breed dogs to measure the relationship between breed and behavior. Although they found that many behavioral traits had high heritability, breed composition itself had only a modest value for predicting the behavior of any one dog. They conclude that modern breeds are defined by morphological and not behavioral traits.
Most modern breeds were developed during Victorian times and were formed on the basis of aesthetics, leading to striking differences in breed morphology (Fogle 2000; Worboys et al. 2018). Considerable effort has gone into identifying genes for body size and shape, leg length, coat texture and color, skull shape, ear position, and others (see the following and references therein, Plassais et al. 2019; Bannasch et al. 2020; Brancalion et al. 2022). These results indicate a recurring theme in canine genetics: A small number of loci account for a large fraction of phenotypic variance. For instance, fewer than 30 genes are responsible for >80% of the variance in body size across breeds, supporting an earlier study in which variation in just six genes accounted for ∼50% of body size variation (Fig. 1A; Rimbault et al. 2013). Conversely, for complex morphologic traits in humans, such as height, hundreds of genetic variants often contribute to only a small fraction of phenotypic variance (Conery and Grant 2023). This difference speaks to the strong artificial selection for morphological traits that dogs have undergone, forcing single alleles to disperse rapidly throughout a breed. It also means that genes for complex traits, such as body shape and size, are much easier to identify in dogs than in humans owing to reduced genetic complexity (Boyko et al. 2010; Hayward et al. 2016). As more dogs with individual-level phenotype data are included in such studies, additional loci contributing to more nuanced parts of a phenotype will be found.
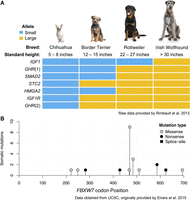
Genomic analyses help resolve the genetics of shared human–canine traits. (A) Approximately 50% of variance in dog breed body size is explained by association at seven genetic markers. The figure shows several combinations of small and large body-size alleles contributing to the heights of different breeds. Raw data provided by Rimbault et al. (2013). (B) Recurrent mutations in FBXW7 are a feature of canine lymphoma and human cancer. The figure shows a single-amino-acid site, R470, which accounts for 41% of all FBXW7 somatic mutations in a cohort of whole-exome-sequenced lymphoma samples. Data obtained from UCSC, originally provided by Elvers et al. (2015).
Disease gene studies in modern dogs
Strong selection for morphological and behavioral traits is often accompanied by changes in disease susceptibility among one or a small number of breeds sharing recent common ancestry. This makes the study of breed-enriched disorders a major focus of comparative genomics communities. This area of investigation offers the opportunity to solve the problem of locus heterogeneity, which has proven intractable in many human disorders (Ostrander et al. 2019a; Leeb et al. 2023). It should also be noted that research into canine genetic diseases is a high priority not just for humans, but for the 49 million households in the United States alone that, in aggregate, own 70 million dogs (U.S. Census Bureau and U.S. Department of Housing and Urban Development 2021).
The importance of genetic analyses in dogs is reflected in the growing market of direct-to-consumer tests available to dog owners. To date, nearly 450 spontaneous diseases with suspected genetic components have been described in dogs, with strong support for at least 350 variants (Nicholas 2003). Although some companies offer tests to predict morphology, behavior, and disease susceptibility, others focus on just the latter, with at least one company offering simultaneous testing for 270 diseases. Identification of component ancestry for mixed breed dogs is also big business. The widespread use of these tests is helping researchers to characterize the broad population-based allele frequencies associated with many diseases and to also help map new disease risk variants (Donner et al. 2018, 2023; Kawakami et al. 2022).
Many heritable diseases in dogs have a similar clinical presentation as the comparable human disorder (Kaur et al. 2023). For instance, progressive retinal atrophy (PRA) is an umbrella term referring to a group of heritable degenerative eye diseases in dogs known to affect retinal photoreceptor cells, ultimately resulting in blindness. Different presentations of the disease may be limited to one or a small number of related breeds, inferring a strong genetic component (Mellersh 2014; Hitti et al. 2019). Many forms of canine PRA mimic or present identically to retinitis pigmentosa (RP) disorders in humans, which are often caused by variants in the same genes (Bunel et al. 2019). However, in humans, variants in more than 270 genes are associated with retinal dysfunction and deterioration and more than one-third of cases remain unexplained (Martin-Merida et al. 2018), highlighting the ongoing need for genetic studies of vision disorders in dogs. One additional advantage of the dog genetic system is that some diseases can also be found in small pedigrees, providing systems for the study of rare human disorders, such as Stickler syndrome, a hereditary cataract disorder found in the Old English sheepdog (Stanbury et al. 2023). Also, progress in gene therapy suggests the dog has a considerable amount to offer in the development of therapeutics (Tuohy and Megaw 2021).
Studies of canine cancer are a particular focus as the clinical presentation, histopathology, molecular features, and treatment responses often mimic human cancers, and breed predispositions suggest strong genetic components for risk (Knapp et al. 2015; Megquier et al. 2019b; London et al. 2023). For instance, osteosarcoma is observed at increased frequency in long-limbed breeds such as the Scottish deerhound (OR 118.4, 95% CI 41.12–340.95) and Great Dane (OR 34.24%–95% CI 17.8165.83) (O'Neill et al. 2023). Importantly, recent studies have identified dozens of contributing loci, highlighting the utility of the dog as a genetic model (Simpson et al. 2020; Sarver et al. 2023). Additional examples include Scottish terriers, which are at a 22-fold increased risk for bladder cancer (Knapp et al. 2014), and histiocytic sarcoma (HS), a lethal disease that affects 25% of Bernese mountain dogs and 20% of flat-coated retrievers (Abadie et al. 2009; Dobson et al. 2009). Encouragingly, studies of HS have recently been undertaken using multiomic approaches, revealing that regulatory variants for PIK3R6 and TNFAIP6 explain 35% of disease risk (Evans et al. 2021).
The study of tumor DNA facilitates canine precision medicine and improves prognostic biomarkers (Chon et al. 2023). For example, B cell lymphoma is the most common hematological malignancy in dogs, accounting for ∼50%–60% of cancer cases (Avery 2020). Sequencing of lymphoma tumor DNA reveals recurrent somatic mutation profiles in tumor-suppressor genes, such as FBXW7. Somatic mutations in this gene are also associated with shorter survival times in affected dogs (Fig. 1B; Elvers et al. 2015; White et al. 2020). In humans, FBXW7 mutations are associated with tumor promotion in multiple types of cancers. Moreover, proteomic profiling of FBXW7 mutant tumors aids the identification of potential downstream therapeutic targets, suggesting this gene could provide a viable therapeutic target in dogs (Urick and Bell 2020; Kawaguchi et al. 2021; Urick et al. 2021).
Sequencing strategies for large-scale genomic analyses
In the past 10 years, there has been a steady decrease in the cost of short-read whole-genome sequencing (WGS) (Cullen and Friedenberg 2023), facilitating an explosion in sequencing data for domestic species (Fig. 2A; Supplemental Table S1). As a result, the domestic dog now has almost 3000 high-coverage genomes available to the public, powering large-scale genome analyses and capturing a significant amount of canine genomic diversity (Meadows et al. 2023). However, for most domestic species, low-pass WGS (coverage < 5×) is the preferred strategy (Fig. 2B), as lower sequencing costs allow for additional resources to be directed toward bolstering sample sizes. This widely employed strategy is effective for population genetic analysis, demography studies, and genome-wide association studies (GWAS). When paired with imputation, the resolution of low-pass sequencing is vastly improved (Rubinacci et al. 2021). In dogs, low-pass sequencing and imputation can achieve >95% genotyping accuracy for variants at 1% allele frequency in reference populations (Meadows et al. 2023). However, an important limitation relates to diversity of the reference panel, as samples whose ancestry is poorly represented achieve lower accuracy rates (Buckley et al. 2022).
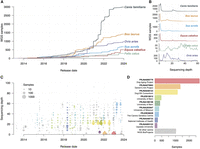
Illumina whole-genome sequences (WGSs) publicly released by animal genomics communities. (A) Number of samples per species sequenced at depths between 20× and 40×. (B) Distribution of sequencing depths for six different domestic mammalian species. (C) Public release of canine WGS Illumina data from 2013 to 2024 on the NCBI Sequence Read Archive (SRA). Size of the data points represents the number of samples released within a yearly quarter at a similar sequencing depth. Points are colored according to their listed NCBI BioProject in D. (D) The total number of samples released for the nine largest canine WGS BioProjects.
Recently, the improved accessibility and quality of genome sequencing have profoundly affected the adoption of various sequencing strategies (Fig. 2C). Initially, genome sequencing was only performed for a small number of cases and controls to identify potential functional variants within a candidate locus, usually defined using array-based technologies. Now, larger data sets exist that have powered mapping studies for breed standard traits and led to the creation of WGS reference panels. Despite these improvements, harmonization of genomic data from a broad array of sources remains challenging, and as a result, the community has pivoted toward large-scale sequencing efforts (Fig. 2D).
Sequencing strategies will continue to update as new technologies become more accessible. Advances in long-read sequencing make it possible to capture the complexities of individual-level genome structure across human populations (Beyter et al. 2021). This technology will improve detection of structural variants (SVs) and help to identify new genotype–phenotype relationships in dogs (Chaisson et al. 2019; Mastrorosa et al. 2023), as SVs are already known to be a major component of canine genomic variation (Halo et al. 2021; Wang et al. 2021; Meadows et al. 2023). Dog genomes also have elevated levels of retrogene activity and associations between SVs and morphological variation have already been characterized, such as an FGF4 retrogene associated with chondrodysplasia (Parker et al. 2009; Serres-Armero et al. 2021; Bannasch et al. 2022; Batcher et al. 2022). Alignment between multiple long-read dog genome assemblies has led to the identification of additional SVs of consequence (Edwards et al. 2021; Wang et al. 2021; Nguyen et al. 2023). For example, a heterozygous duplication of CYP1A2, a gene involved in xenobiotic metabolism, was found in the UU_CFam_GSD_1.0 reference assembly of a donor German shepherd dog, Mischka (Wang et al. 2021). Although this duplication was not associated with any change in transcript abundance, potential phenotypes may only be observable after a drug challenge, owing to the inducible nature of the gene (Graham et al. 2002). Other long-read assemblies, each with the potential to reveal new genetic variants, include an updated sequence of the boxer Tasha (Jagannathan et al. 2021), two new basenji assemblies (Edwards et al. 2021), an additional German shepherd dog (Field et al. 2020), a Labrador retriever (Player et al. 2021), and a Great Dane (Halo et al. 2021). Eventually, long-read sequencing will become common practice in animal genomics, permanently altering sequencing strategies and analysis techniques while capturing the full spectrum of variation within a single genome.
Large-scale sequencing efforts
The International Dog10K consortium was founded to create a single high-quality data set representative of global canine breed diversity (Ostrander et al. 2019b). Although samples were contributed by multiple consortium members, all sequencing and bioinformatic processing was performed using a uniform approach. Altogether, the project encompasses 1987 genomes and is composed of 1611 breed dogs from 321 distinct breeds (Meadows et al. 2023). The overarching strategy was aimed at sampling as many breeds and village dog populations as possible, as well as previously uncharacterized populations (Ostrander et al. 2019b). The project also included 67 wild canids, most of which were wolves. The final data set represents the largest single public release of high-coverage canine WGS data to date, consisting of more than 48 million single-nucleotide, indel, and structural variants.
Other large-scale canine sequencing efforts include the Darwin's Ark Project and the Dog Aging Project, both of which use low-pass WGS and imputation for genotyping, allowing these projects to genotype many more samples at lower cost. The Darwin's Ark Project is focused on determining the genetic basis of complex traits, including behavior. Thus far, the project reports sequencing 2155 dogs and has collected survey data for more than 18,000 individuals (Morrill et al. 2022). The Dog Aging Project is a longitudinal study aimed at collecting environmental, clinical, and biochemical data on several thousand dogs and has plans to sequence 10,000 dogs in total. The goal of the project is to investigate how genetic and environmental factors contribute to aging (Creevy et al. 2022).
Although Dog10K, Darwin's Ark, and the Dog Aging Project each consist of thousands of samples, they vary significantly in terms of sequencing coverage, associated metadata, and sample diversity. The Dog10K Project is designed with sufficient WGS coverage levels to identify new variants, focusing sampling efforts on previously uncharacterized breeds and populations. One tradeoff of this approach is that sample metadata are primarily restricted to breed standard metrics or geographical data, limiting the scope of traits that can be mapped in this cohort. Conversely, the Darwin's Ark Project and Dog Aging Project limit genotyping to known variants that are captured through imputation of low-pass sequence data. These projects also focus their sampling efforts on pet dogs, allowing owners to fill out detailed surveys on individual dogs, thereby facilitating investigation of complex traits and gene-by-environment interactions.
A role for rare variation in canine genomics
To date, canine genomic research has prioritized analysis of common over rare variations in studies of trait mapping, as breed-specific population structure and selection history make it difficult to assess the impact of rare mutations. However, given the well-established role of rare variants in human disease, the field of canine genetics is beginning to focus on rare variant discovery and characterization (Halvorsen et al. 2021; Momozawa and Mizukami 2021).
Rare variants are usually defined as having a minor allele frequency (MAF) < 1%, whereas larger cohorts tend to define rare variants at MAF < 0.1%. One reason for classifying rare variants as distinct from common is that their analysis requires alternative statistical approaches. For example, rare variants are typically removed from GWAS, as allele counts for those sites are often underpowered. A major challenge in canine genomics research is determining whether variant allele frequency is predictive of variant age. Most rare variants in human populations are derived from recent mutations, reflecting low levels of inbreeding in recent human history (Albers and McVean 2020; Ceballos et al. 2021). In contrast, canine variant frequencies are less likely to be predictive of variant age and are instead likely to be sensitive to population sampling effects, reflecting the outcomes of recurrent population bottlenecks, multiple admixture events, the use of popular sires, and small founding populations (Karlsson et al. 2007; Freedman et al. 2014, 2016). Consider, for example, a dog from an underrepresented breed within a large multibreed cohort. This dog's genome will contain many rare variants, as defined by low MAF within the larger cohort. Without sufficient representation of this dog's ancestry within the cohort, it is impossible to determine which low MAF variants represent recent mutations versus those that are common to the breed and only seem rare because the dog's ancestry is underrepresented. This need for precise classification of rare variants has important outcomes for assessing selection effects acting upon genes. If rare variants are recent, their distribution across coding sequences is expected to be the result of gene mutability and evolutionary constraint, which together cause genes with essential roles to be depleted of functional mutations (Samocha et al. 2014). However, if variants only appear rare owing to sample acquisition bias, their presence in the genome may have persisted owing to population dynamics like drift or artificial selection rather than their impact on fitness. In these cases, the accumulation of functional variation within a gene may be a poor indicator of the gene's importance.
To better characterize variant allele frequencies within dog populations, we measured each Dog10K individual's site frequency spectrum (ISFS) and then placed each dog into one of four groups according to the skew of the resulting curve (Fig. 3A; Supplemental Table S2). Groupings approximately represented the ratio of rare to common variants, in which dogs from group 1 carried the lowest number of rare variants, and dogs from group 4 carried the highest. The types of dogs represented in each group also varied (Fig. 3B). Most breed dogs belonged to group 1, indicating that most of their variants were shared across multiple breeds. Dogs from group 2 and group 3, which had more rare variants than group 1 dogs, made up ∼10% of breed dogs and ∼80% of village dogs, which is indicative of the comparatively higher levels of genetic diversity found in village dog populations (Shannon et al. 2015; Meadows et al. 2023). Finally, dogs belonging to group 4 were either wolves or wolf–dog hybrids and contained the highest level of individual variation.
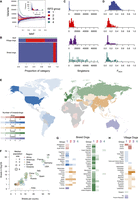
(See following page for legend.) Genetic diversity of dog breeds represented within Dog10K. (A) Individual site frequency spectrum (ISFS) from the Dog10K data set (Meadows et al. 2023). Each curve represents the number of variants that each dog carries and the MAF of those variants across the Dog10K cohort. Individual dogs were grouped according to the skewness of their ISFS curve. Most of the genetic variation for dogs in ISFS group 1 is common across dog populations, whereas dogs in ISFS group 4 carry a high number of variants that are rare within Dog10K. (B) ISFS group proportions for each Dog10K sample category (wolf, village dogs, or breed dogs). Height of the horizontal bars is proportional to the number of Dog10K dogs belonging to each category. (C) Distribution of singleton variants per individual, plotted according to ISFS group. (D) Degree of inbreeding for each dog depicted as the fraction of the genome within runs of homozygosity, plotted according to ISFS group. (E) Global distribution of dog breeds. Colors represent global regions, and depth of shading represents the number of breeds originating in each country. (F) Sample collection within the Dog10K Project is representative of global breed diversity. The number of breeds collected was proportional to the number of breeds that originated in each country. The size of the data point indicates the median breed population size collected within Dog10K. Data points are colored according to the global region of breed origin. (G) Number of Dog10K breed dogs per country that belong to each ISFS group depicted in A. Depth of shading in G and H represents the numbers of dogs in each category according to the key shown in E. (H) Number of Dog10K village dogs per country that belong to each ISFS group depicted in A.
Another metric for evaluating canine variation is the number of singleton variants per dog. Dogs with well-represented ancestry will only carry a small number of singletons, which mostly consist of recently occurring mutations. Dogs with poorly represented ancestry will instead have many singletons, consisting of both recently acquired mutations and ancestral variants that are shared with dogs not included in the data set. For the purposes of identifying rare variants that represent recent mutations, only dogs with a small number of singletons should be considered, such as those in group 1 (Fig. 3C).
Individual inbreeding coefficients, calculated as the fraction of the genome within runs of homozygosity, can also indicate the diversity of the population from which a dog was sampled. For inbred populations, the landscape of common variation can be captured with fewer samples than in outbred populations. Importantly, dogs in groups 2, 3, and 4 all have lower mean inbreeding coefficients compared with that of group 1 (Fig. 3D). However, some individuals from these groups appear relatively inbred, suggesting that additional sampling of their source populations will lead to vastly improved breed representation for these dogs. Together, these results indicate that assumptions made in the analysis of human genetic data do not necessarily hold for dogs.
Geographical distribution of missing canine genetic variation
To identify potential sources of missing genetic variation by country of origin, we cataloged data for more than 500 dog breeds and varieties (Fig. 3E; Supplemental Table S3). We also used breed origins from Dog10K samples to capture genetic variation of breeds and populations from each country (Fig. 3F). We observed that Dog10K lacks proportional representation of breeds from regions such as Southeast Asia, Eastern Europe, and South America. In addition, India was a particularly large outlier as it has 18 distinct breeds, none of which are represented in Dog10K. Moreover, peer-reviewed analyses regarding the genetics of Indian dog breeds are rare, indicating that India could provide a large source of missing genetic variation once samples can be exported from the country (Shannon et al. 2015). Other sources of missing variation may be found in dogs that do not belong to recognized breeds. For example, different varieties of dogs are found throughout Africa, but only nine breeds in our data set have an African country listed as their origin. Most importantly, African village dogs carry ancestry components distinct from other canine populations (Boyko et al. 2009; Wang et al. 2016; Liu et al. 2018), indicating that further sample collection within this region will greatly increase genomic diversity within canine variant catalogs.
Although the absence of particular breeds from large WGS cohorts can be indicative of missing variation, genetic variants from unsampled breeds can often be found within closely related breeds that share recent common ancestry reflecting shared identity-by-descent (IBD) segments (Parker et al. 2017). This is evidenced by high imputation accuracy for dogs whose breeds are absent from reference panels (Buckley et al. 2022; Meadows et al. 2023), reflecting breeding strategies aimed at transferring favorable traits during breed formation or reflecting the predominance of a source population that shares and distributes favorable traits to multiple breeds. In the latter case, breed dog genetics may be reflective of local village dog populations (Shannon et al. 2015; Dutrow et al. 2022). Regardless, capturing ISFS is useful for determining how much unique variation an individual dog is contributing to a cohort.
Most breed dogs from Europe, the Americas, Oceania, and Africa are from ISFS group 1 (Fig. 3G). Europe is the most well represented in the data set; thus, many breeds of European origin can be organized into distinct lineages with extensive haplotype sharing (Parker et al. 2017; Dutrow et al. 2022). The degree to which this reflects small source populations versus simply the effort to collect breeds of European origin is not clear. However, breeds from the Americas, Oceania, and Africa are also likely to be reflective of European colonization (Shannon et al. 2015). The genetic structure of Asian dog breeds is highly distinct, and most Asian dogs are from ISFS group 2 or 3, indicating that Asian breeds could also be a source of missing variation. This high level of diversity has been used to argue for an Asian origin of dog domestication (Savolainen et al. 2002; Vonholdt et al. 2010; Wang et al. 2016).
Village dogs tend to carry more diversity than breed dogs, likely owing to the absence of selected breeding or closed breeding populations (Fig. 3H). However, in the Americas and Oceania, village dogs harbor a significant level of Western European–associated variation. Because pre-European-contact dog populations are largely extinct or never existed in these countries, populations in the Americas and Oceania may have therefore arrived with the later introduction of European breeds (Shannon et al. 2015). Conversely, Asian village dogs and breed dogs are most enriched for rare variation. Ultimately, greater sampling of these populations, along with adequate phenotyping, will likely lead to new genotype–phenotype discoveries (Surbakti et al. 2020).
Interpretation of variant effects
In trait mapping, the single nucleotide polymorphisms (SNPs) with the strongest phenotype association are often within the noncoding portion of the genome. Determining how these SNPs exert their functional consequences often requires annotation of gene regulatory elements. In human genomics, considerable effort has been directed toward this goal, leading to the creation of a wide array of gene regulatory annotation data sets, such as ENCODE (The ENCODE Project Consortium et al. 2020), the Roadmap Epigenomics Program (Satterlee et al. 2019), and GTEx (The GTEx Consortium 2020). Conversely, in canine genomics, similar functional data are often lacking. The need for genome regulatory annotation in dogs is further emphasized by additional challenges specific to the canine system. For instance, although pedigrees can be extensive in dogs (Jónasdóttir et al. 2000), it is unusual to have samples from three generations to establish robust linkage. In addition, few relevant canine-derived cell lines exist for testing variant function in the appropriate contexts. Also, although possible, performing CRISPR-Cas9 in dogs is expensive and inefficient, and animal use issues are a major concern (Zou et al. 2015; Tian et al. 2023). Variant prioritization is also made more challenging in dogs owing to extensive linkage disequilibrium (LD) caused by breeding practices that favor specific traits and reduce overall haplotype diversity (Sutter et al. 2004; Lindblad-Toh et al. 2005; Halo et al. 2021; Wang et al. 2021; Meadows et al. 2023).
The scarcity of regulatory annotation within the dog genome can be addressed by leveraging evolutionary conservation between dogs and humans. Approximately 1.4 Gb of DNA sequence, or ∼40% of the human genome, is shared between humans and dogs, which is typical of the evolutionary distance between humans and other non-primate mammalian species (Lindblad-Toh et al. 2005; Armstrong et al. 2020). Tools like liftOver use human–dog alignments to map human noncoding regulatory elements within the canine genome, enhancing prediction of variant impacts in dogs (Hinrichs et al. 2006). However, the use of species pairwise alignment-based approaches for regulatory annotation assumes that conserved noncoding sequences have conserved epigenomic activity, which is often not the case (Zemke et al. 2023). Also, dogs lack a functional copy of PRDM9; hence, the recombination landscape is biased toward CpG islands and promoters, causing the gene regulatory landscape of canids to diverge from other mammals (Auton et al. 2013). Finally, robust regulatory element annotation requires capturing epigenomic activity in the appropriate cellular contexts. To overcome these limitations, the Zoonomia Consortium has created whole-genome alignments of 240 species to detect evolutionarily constrained genomic elements of potential functional importance (Zoonomia Consortium 2020). Almost half of all constrained bases had no functional annotations in any ENCODE cell types (Christmas et al. 2023), highlighting the difficulty of identifying functional DNA elements through epigenomic profiling alone. Importantly, human GWAS SNP heritability and canine disease mutations were both enriched within constrained sites (Meadows et al. 2023; Sullivan et al. 2023), indicating that genomic constraint will aid in prioritizing noncoding variation for trait mapping within the dog's characteristic large LD blocks (Tengvall et al. 2022; Lingaas et al. 2023).
One limitation of using evolutionary constraint to predict functional importance is that species-specific functional elements will be overlooked. In these cases, conserved sequence can be contrasted against rapidly evolving lineage-specific sequence to identify species-specific accelerated regions (Pollard et al. 2006). In humans, these accelerated regions (HARs) often act as enhancer elements during neural development and are important in neurodivergent conditions (Girskis et al. 2021; Whalen et al. 2023). For example, excess rare biallelic point mutations in HARs are at a significant excess in individuals with autism spectrum disorder risk, often affecting active enhancers for genes implicated in neural function (Doan et al. 2016). Similar approaches could be applied to dogs to identify genomic regions related to canine-specific traits, as has been done in other species (Ferris et al. 2018).
Species alignments have also advanced variant interpretation within coding sequences after being combined with machine learning approaches to distinguish pathogenic from benign missense changes. The programs PrimateAI and PrimateAI-3D take advantage of naturally occurring amino acid substitutions across primates to learn the tolerable landscape of missense mutations in humans (Sundaram et al. 2018; Gao et al. 2023). Because most protein sequences from closely related species have conserved structural/functional roles, amino acid substitutions likely have benign functional consequences. Coding variation across primates is therefore sufficient to characterize the pathogenicity of human missense mutations. A similar approach may also be suitable for the dog genome. Amino acid substitutions between carnivores would provide suitable input data, obviating the need to sequence thousands of individual dogs.
Machine learning approaches combined with multispecies alignment can also be used to annotate gene regulatory elements within noncoding regions (Chen et al. 2018; Kelley 2020; Minnoye et al. 2020; Kaplow et al. 2022). The Tissue-Aware Conservation Inference Toolkit (TACIT) (Kaplow et al. 2023) uses chromatin accessibility data from several species to learn sequence features predictive of tissue-specific and cell type–specific enhancer activity. The tool can then use this information to predict enhancer activity across a phylogeny. One application of TACIT demonstrates how predicted enhancers can be associated with brain size, a highly complex phenotype. The development of such approaches makes it possible to predict the regulatory landscape for trait-relevant cell types in dogs using data previously produced in other mammals.
Although the sophisticated use of species alignment and machine learning can greatly expand our knowledge of the dog genome, there is still an important role for experimentally derived annotations. Two initiatives characterizing the epigenome of the dog include BarkBase and Epigenome Catalog of the Dog (EpiC Dog). BarkBase contains bulk ATAC-seq data from five dogs, with five to 10 tissues available per individual (Megquier et al. 2019a). EpiC Dog contains data from three dogs across 11 tissues for five different histone marks and DNA methylation, allowing for the identification of 13 different chromatin states, each reflecting different types of regulatory activity (Son et al. 2023). However, analysis of epigenomic activity in bulk tissue is always subject to effects of cellular heterogeneity within the sample. Single-cell profiling of open chromatin in trait-relevant tissues and developmental time points will help prioritize variants with phenotypic impacts as well as disease and trait-relevant cell types (Corces et al. 2020; Son et al. 2023).
The future of canine genomics
Comparative genomics relies on the observation that species divergence has led to a high degree of biological innovation, without disruption of essential biological processes. Analyses therefore depend either on using shared sequences between divergent species to identify conserved genomic elements or on using species-specific genetic associations with shared traits to identify genes in conserved biological pathways. Canine genomics primarily employ the latter approach, utilizing genotype–phenotype associations to define new roles for genes and refine existing paradigms. The future of canine genomics will therefore be shaped by improvements to the collection of samples and associated metadata, capturing the full repertoire of genomic variation, linking genome variation to gene function, and validating gene–trait associations.
Existing sample collection efforts focus on ascertainment of rare breeds and pet dogs, with metadata often provided by owners. Future efforts should prioritize mixed breed dogs of known ancestry with detailed health data, as canine genetic studies are often confounded by breed ancestry. A cohort of mixed breed dogs will also retain causal variants for breed-related traits and have low levels of population stratification. Multiple prospective cohorts need to be initiated using much the same structure as the Golden Retriever Lifetime Study (Labadie et al. 2022). Such studies should include frequent sampling, owner questionnaire data, and access to detailed health care data and related samples. In addition, the use and accessibility of electronic medical records, the backbone of human disease genetic studies, should become a part of standard veterinary practice.
Without the full repertoire of canine genomic variation, particularly SVs, variant impact is difficult to define. Long-read sequencing technologies and new assembly algorithms have resulted in telomere-to-telomere sequencing for humans (Rautiainen et al. 2023). As scalability improves, this technology should be applied to dogs, thus facilitating the association of complex, largely uncharacterized SVs and their associated phenotypes (Miga and Eichler 2023).
Studies that link genetic variation to gene function and expression should also be prioritized, as canines offer unique opportunities to identify new functions for recognized DNA sequence motifs. To accomplish this, the development of expression quantitative trait loci data sets and massively parallel reporter assays linking variants to nearby gene activity are needed.
Finally, affordable strategies for validation of gene and trait associations in dogs are needed. The strongest evidence for a gene's role in a particular trait is to show that purposeful modulation of the gene's activity causes changes in the corresponding phenotype. In addition to exploiting existing approaches, large repertoires of canine cell lines need to be developed and characterized, and an economical system for distribution established. For example, reference cell lines, such as those used by The ENCODE Project Consortium, would greatly enhance canine genetic studies (The ENCODE Project Consortium 2011). Together, the advances summarized here will increase the utility of the dog as a genetic system uniquely powered to inform studies of human conditions. In addition, the same advances will improve canine health, revealing ever more about these important members of our families and positioning the dog in its rightful place, as it has always been, by our side.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank members of the Ostrander laboratory for careful reading of this manuscript and critical comments. We especially thank Aitor Serres-Amero and Tatiana Feuerborn for their contributions and Dayna Dreger for sharing her compiled list of dog breed countries of origin. E.A.O. and R.M.B. are funded by the Intramural Program of the National Human Genome Research Institute at the National Institutes of Health (HG200377).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278569.123.
-
Freely available online through the Genome Research Open Access option.
This is a work of the US Government.








