
A systematic review on the biochemical threshold of mitochondrial genetic variants
- Karan K. Smith1,
- Jesse D. Moreira1,2,
- Callum R. Wilson1,
- June O. Padera1,
- Ashlee N. Lamason1,
- Liying Xue1,
- Deepa M. Gopal1,
- David B. Flynn3 and
- Jessica L. Fetterman1
- 1Evans Department of Medicine and Whitaker Cardiovascular Institute, Boston University Chobanian and Avedisian School of Medicine, Boston, Massachusetts 02118, USA;
- 2Programs in Human Physiology, Department of Health Sciences, Boston University Sargent College, Boston, Massachusetts 02215, USA;
- 3Medical Sciences and Education, Boston University Chobanian and Avedisian School of Medicine, Boston, Massachusetts 02118, USA
Abstract
Mitochondrial DNA (mtDNA) variants cause a range of diseases from severe pediatric syndromes to aging-related conditions. The percentage of mtDNA copies carrying a pathogenic variant, variant allele frequency (VAF), must reach a threshold before a biochemical defect occurs, termed the biochemical threshold. Whether the often-cited biochemical threshold of >60% VAF is similar across mtDNA variants and cell types is unclear. In our systematic review, we sought to identify the biochemical threshold of mtDNA variants in relation to VAF by human tissue/cell type. We used controlled vocabulary terms to identify articles measuring oxidative phosphorylation (OXPHOS) complex activities in relation to VAF. We identified 76 eligible publications, describing 69, 12, 16, and 49 cases for complexes I, III, IV, and V, respectively. Few studies evaluated OXPHOS activities in diverse tissue types, likely reflective of clinical access. A number of cases with similar VAFs for the same pathogenic variant had varying degrees of residual activity of the affected complex, alluding to the presence of modifying variants. Tissues and cells with VAFs <60% associated with low complex activities were described, suggesting the possibility of a biochemical threshold of <60%. Using Kendall rank correlation tests, the VAF of the m.8993T > G variant correlated with complex V activity in skeletal muscle (τ = −0.58, P = 0.01, n = 13); however, no correlation was observed in fibroblasts (P = 0.7, n = 9). Our systematic review highlights the need to investigate the biochemical threshold over a wider range of VAFs in disease-relevant cell types to better define the biochemical threshold for specific mtDNA variants.
Mitochondria produce ∼90% of all cellular energy, primarily through oxidative phosphorylation (OXPHOS) (Wallace 2013). Genetic variants that impair the OXPHOS complexes result in mitochondrial diseases. Mitochondrial diseases occur in one in 4300 individuals and can present at any age from the neonatal period through late adulthood (Gorman et al. 2015). Uniquely, the OXPHOS complexes are encoded by both the mitochondrial genome (mtDNA) and the nuclear genome with genetic aberrations in either genome resulting in mitochondrial disease.
The mtDNA encodes 13 OXPHOS subunits and its own translational machinery (22 tRNAs, two rRNAs). Complex II is the only complex entirely encoded by the nuclear genome. Unlike the nuclear genome that contains only two alleles for every gene, a single cell contains multiple mitochondria, with each mitochondrion containing five to 10 copies of mtDNA (Wallace 2005); hence, each cell contains ∼103 to 104 mtDNAs (Larsson and Clayton 1995). The mtDNAs in a cell or tissue may all be identical in sequence (homoplasmy), or some mtDNA copies may contain different alleles at the same position, a condition called heteroplasmy. Emerging studies indicate that mitochondrial heteroplasmy is more prevalent than previously appreciated, and it is likely that most of the population carries heteroplasmic mtDNA variants (Chinnery et al. 2002; Payne et al. 2013; Liu et al. 2021; Laricchia et al. 2022). Further, a higher variant allele frequency (VAF) of heteroplasmic variants is associated with advancing age in the general population and mitochondrial disease onset (Shoffner et al. 1990; Chinnery et al. 1997, 2002; Liu et al. 2021; Stewart and Chinnery 2021).
The mere presence of a pathogenic mtDNA variant is not sufficient to alter mitochondrial function and result in disease. Defects in protein synthesis, OXPHOS complex and supercomplex assembly or stability, and enzymatic function depend on the location and type of mtDNA variant and the number of mtDNA copies carrying the pathogenic variant (VAF) (Fig. 1).
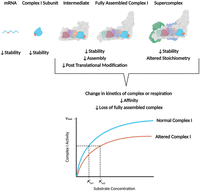
Effects of mtDNA variants on OXPHOS complex stability, assembly, and activity. A mtDNA variant can affect OXPHOS complex function at each stage of OXPHOS complex translation and assembly (illustrated using complex I but applicable for the other OXPHOS complexes). mtDNA variants can reduce the stability of mRNA, complex subunits, fully assembled complexes, or supercomplexes. mtDNA variants can hinder assembly of the OXPHOS complex or supercomplexes or prevent post-translational modifications. Additionally, mtDNA variants can alter the ratio of OXPHOS complexes forming a supercomplex. Ultimately, mtDNA variants can result in a loss of one or more OXPHOS complexes or induce a change in the kinetic activity of the affected complex, shown as a change in Michaelis–Menten kinetics of the affected enzyme. A higher Km for the affected complex I (Km2) compared with the normal complex I (Km1) indicates a decrease in affinity for a substrate.
Further complicating the matter, the VAF associated with disease presentation is tissue specific and thought to be reflective of random segregation during development (Liu et al. 1998; Chinnery et al. 1999; Shanske et al. 2004). Highly energetic tissues are more sensitive to impairments in OXPHOS (Rossignol et al. 1999, 2000) and, hence, may have a lower VAF threshold for a pathogenic mtDNA variant. In a given tissue, the proportion of mtDNAs carrying a pathogenic variant must reach a “threshold” to result in disease, referred to as the “phenotypic threshold effect” (Rossignol et al. 2003). Similarly, the level of OXPHOS impairment required to have an effect on mitochondrial function is known as the “biochemical threshold effect” (Rossignol et al. 2003).
Prior studies have suggested that the phenotypic and biochemical thresholds of mitochondrial disease–causing variants are at VAFs of ≥60% (Tuppen et al. 2010). However, whether the often-cited biochemical threshold of ≥60%–80% VAF is similar across all variants, genes, complexes, and tissues is unclear. We performed a systematic review of the literature to identify the biochemical threshold of specific pathogenic mtDNA variants based on the protein-encoding gene affected, tissue type, and degree of heteroplasmy (VAF). Our findings suggest that few studies have evaluated mitochondrial function across a wide range of VAFs in order to sufficiently identify the biochemical threshold for pathogenic mtDNA variants in protein-encoding genes.
Methods
A subset of relevant articles identified before the literature search was used to obtain controlled vocabulary terms (Majander et al. 1991; Makino et al. 2000; Vilarinho et al. 2000; Legros et al. 2001; Lamantea et al. 2002; Lebon et al. 2003; Simon et al. 2003; Acı´n-Pérez et al. 2004; Bugiani et al. 2004; Kirby et al. 2004; Leshinsky-Silver et al. 2005; Moslemi et al. 2005; Blok et al. 2007; Castagna et al. 2007; Srivastava et al. 2007; López-Gallardo et al. 2014; Gorman et al. 2015; Zhang et al. 2018; Shimura et al. 2022). The controlled vocabulary terms for the OXPHOS enzymes and mitochondrial diseases from the subset of relevant articles were chosen to refine the results (Table 1). Searches in PubMed and Embase were performed with the controlled vocabulary terms to identify relevant publications. The Boolean operator “AND” was also used to refine the results.
Search terms
Articles were identified using keywords in the title or abstract. Keywords included mtDNA-encoded OXPHOS subunits in the context of quantitative assessment of function or biochemical analysis. Included articles contained human participants, a variant in a respiratory chain complex, VAF, and quantitative assessment of function of at least the affected complex. Few papers evaluated mitochondrial respirometry, leading us to focus on complex activity measures. Reviews, commentary, editorials, and articles in non-English language were excluded from the results (Table 2). We did not confine our results by publication year.
Exclusion criteria
We identified a total of 76 publications that met the inclusion criteria spanning the years 1991–2023 (Fig. 2). For the publications after 1999, 23 explicitly state that the revised Cambridge reference sequence (rCRS) was used as the reference sequence. Of the 10 papers published before the rCRS in 1999, we were able to confirm the use of the CRS (published in 1981) for five of the manuscripts and checked that the reported positions were not later corrected and changed in the rCRS. Because the variants in the publications identified have a pathogenic status on MSeqDR and MITOMAP and because the CRS and rCRS have long been considered the standard mitochondrial genetic reference by most in the field (Anderson et al. 1981; Andrews et al. 1999; Bandelt et al. 2014), it is possible, but unlikely, that the position coordinates for the publications identified in our search differ from the rCRS. Each individual in a publication that met our criteria were termed “cases.” We include in Supplemental Table 1 the pathogenic classification of the variants by MSeqDR myTool and MITOMAP (Lott et al. 2013; Shen et al. 2018), as well as the population allele frequency, if available, from gnomAD V3.1.2 (Chen et al. 2024) and MITOMAP.
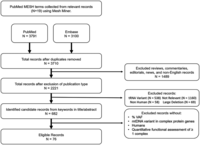
Flowchart of article selection.
Calculation of residual OXPHOS activity
The majority of studies reported OXPHOS complex activity normalized to the same metric of mitochondrial mass, citrate synthase activity. Further, most studies present the OXPHOS complex activities as a percentage of residual activity compared with cells/tissue from referents. For studies that did not express the OXPHOS complex activity as a percentage of residual activity, we calculated the percentage of residual activity by dividing the complex activity measured in the cells/tissue by the mean complex activity of the referent group, and then multiplied by 100 to obtain the percentage. If the OXPHOS activity data was not normalized to citrate synthase but citrate synthase activity was provided, we divided the OXPHOS complex activity by citrate synthase before calculating the residual activity.
Statistical analysis of the relation of VAF with OXPHOS complex functional assessment
Kendall rank correlation tests were conducted to evaluate the association between the VAF and residual complex activity for the following: Complex I activity in muscle biopsies from patients carrying the m.13513G > A variant in MT-ND5, and ATP synthase activity in muscle biopsies and dermal fibroblasts from patients carrying the m.8993T > G variant in MT-ATP6. VAFs reported as >95%, >99%, or homoplasmic were interpreted as 100% for statistical analysis. Analyses were performed using R version 4.0.5 (R Core Team 2022).
Results
In the 76 publications that met our inclusion criteria, individuals carrying 68 unique variants in protein-encoding mitochondrial genes were described (Fig. 3).
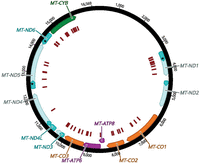
mtDNA structure and location of variants. The mtDNA protein-encoding genes are indicated by blocks of color with complex I subunits in teal, the complex III subunit in green, complex IV subunits in orange, and complex V subunits in magenta. The variants characterized in the publications that met our inclusion criteria are indicated in the innermost track by red bars. The numbers on the outermost track are the nucleotide positions.
Complex I variants
Complex I (NADH:ubiquinone oxidoreductase) is the largest complex of the mitochondrial respiratory chain, consisting of 45 subunits (Vinothkumar et al. 2014). The nuclear genome encodes 38 of the 45 complex I subunits, whereas the mtDNA encodes seven subunits (Mimaki et al. 2012). Of the 45 subunits that make up complex I, 14 subunits are considered “core” subunits, meaning they are evolutionarily conserved (Formosa et al. 2018; Hock et al. 2020). All seven mtDNA-encoded subunits are core subunits (Hock et al. 2020).
Complex I is L-shaped with a mtDNA-encoded arm embedded in the inner mitochondrial membrane and a nuclear DNA–encoded arm facing the matrix, termed the peripheral arm (Formosa et al. 2018). At the top of the peripheral arm, NADH is oxidized by flavin mononucleotide. The electrons from NADH funnel through the iron–sulfur clusters of the peripheral arm to the ubiquinone reduction site (Hunte et al. 2010; Giachin et al. 2016; Kampjut and Sazanov 2020). In the membrane arm, the mtDNA-encoded subunits facilitate the translocation of protons into the intermembrane space coupled to the reduction of ubiquinone (Giachin et al. 2016). The mechanism of proton translocation is theorized to be through conformational changes resulting from ubiquinone reduction (Gutiérrez-Fernández et al. 2020; Kampjut and Sazanov 2020).
Pathogenic variants have been identified in all seven complex I mtDNA genes (Rodenburg 2016). Complex I variants are the most prevalent, likely owing to the greater number of subunits encoded by the mtDNA (Kleist-Retzow et al. 1998; Swalwell et al. 2011). Pathogenic complex I mtDNA variants are associated with several mitochondrial diseases, including mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), Leigh syndrome, and Leber hereditary optic neuropathy (LHON) (Yu-Wai-Man and Chinnery 2011; Rodenburg 2016; Danhelovska et al. 2020). Variants in mtDNA-encoded subunits often impede complex I assembly or decrease complex I stability, resulting in complex I deficiency (Bai and Attardi 1998; Jiang et al. 2016).
Given that complex I deficiency is one of the most prevalent respiratory chain deficiencies (Kleist-Retzow et al. 1998; Swalwell et al. 2011), it is not surprisingly that majority of the records identified report variants in the mtDNA-encoded complex I genes. A total of 69 cases, carrying 29 unique variants in complex I genes were identified (Table 3). Publications reporting complex I activity in tissues possessing a variant in MT-ND2 and MT-ND4L were not found; however, pathogenic variants in MT-ND2 and MT-ND4L have been reported in the literature (Mackey and Howell 1992; Ugalde et al. 2007; Behbehani et al. 2014; Destiarani et al. 2020; Dahadhah et al. 2021). Only a single variant (m.11778G > A) in MT-ND4 was identified with a VAF of 100% in lymphoblastoid cells generated from an individual with LHON (Majander et al. 1991). The lymphoblastoid cells with the m.11778G > A had normal complex I activity, suggesting that the pathogenic variant may have cell type–specific effects not captured through the use of lymphoblastoid cell lines (Majander et al. 1991).
Complex I variants
MT-NDI
We identified 18 cases carrying a variant in MT-ND1 with assessment of complex I activity. The most frequently reported variants in MT-ND1 were m.3460G > A (N = 4) and m.3697G > A (N = 3) (Majander et al. 1991; Cock et al. 1999; Kirby et al. 2004; Spruijt et al. 2007; Iommarini et al. 2014). The m.3460G > A variant replaces an alanine with a threonine in a hydrophilic domain of MT-ND1 (Leman et al. 2015), which may interfere with ubiquinone binding or reduction as the ubiquinone binding site is located at the intersection of MT-ND1 and several nuclear DNA–encoded subunits (Leman et al. 2015; Yi et al. 2023). Complex I activity was only measured in cells carrying the m.3460G > A variant at VAFs ≥95% (Majander et al. 1991; Cock et al. 1999; Iommarini et al. 2014). At homoplasmic levels, the m.3460G > A variant was associated with 43%–50% residual complex I activity in cybrid cell lines and fibroblasts (Cock et al. 1999; Iommarini et al. 2014). In contrast, lymphoblastoid cell lines homoplasmic for the m.3460G > A variant had lower residual complex I activities ranging between 20% and 33% (Majander et al. 1991). Additionally, some studies evaluated complex I-linked ATP synthesis in cells harboring the m.3460G > A variant. ATP synthesis with complex I substrates (pyruvate/malate, glutamate/malate) was unaffected by the variant in fibroblasts, whereas 60% lower ATP synthesis was observed in cybrids, indicating cell type–specific differences in the effect of the m.3460G > A variant (Cock et al. 1999; Iommarini et al. 2014).
The m.3697G > A variant in MT-ND1 changes glycine 131 to a serine. Like the m.3460G > A variant, the m.3697G > A variant is also located in the hydrophilic domain of MT-ND1. In skeletal muscle from two siblings, one sibling carried the m.3697G > A variant at a VAF of >97% and the other sibling had a VAF of <1% (Spruijt et al. 2007). In the skeletal muscle from these two siblings, only 8% and 16% residual complex I activities were detected, respectively (Spruijt et al. 2007). Why the individual with nearly undetectable levels of the m.3697G > A pathogenic variant in skeletal muscle showed low residual complex I activity is unclear. Functional validation of the m.3697G > A variant using dermal fibroblasts or cybrids was not performed in this study (Spruijt et al. 2007). Skeletal muscle from an unrelated patient carrying the m.3697G > A variant at a VAF of 80% had 60% residual complex I activity, further indicating a wide range of residual activity in relation to VAF for this variant (Kirby et al. 2004). Cultured fibroblasts from this patient with a VAF of 79%, similar to that in skeletal muscle, had only 35% residual complex I activity, indicative of cell/tissue-type differences in the effects of the m.3697G > A variant (Kirby et al. 2004).
Cybrids carrying the m.3634A > G missense variant at homoplasmic levels had 36% residual complex I activity. Cybrids carrying the m.3634A > G variant had similar levels of assembled complex I as referent cells, indicating that the m.3634A > G variant may alter activity without interfering with assembly or stability of complex I (Carreño-Gago et al. 2017). The m.3380G > A variant was reported in a single individual at a VAF of 50% in skeletal muscle with 77% residual complex I activity (Horváth et al. 2008). Two cases carrying a frameshift variant, m.3571insC, were also identified (Iommarini et al. 2014; Lou et al. 2023). Cybrids generated from one of the individuals homoplasmic for the m.3571insC variant had no residual complex I activity owing to a loss of complex I (Iommarini et al. 2014). Many of the remaining variants in MT-ND1 (m.3365T > C, m.3376G > A, m.3481G > A, m.3796A > G, m.3946G > A, m.3949T > C, m.4175G > A) were present in a single individual at VAFs ≥60% in skeletal muscle with residual complex I activities ranging from <1% to 36% (Simon et al. 2003; Kirby et al. 2004; Blakely et al. 2005a; Malfatti et al. 2007; Gorman et al. 2015).
MT-ND3
Complex I activity was evaluated in tissues and cells from 18 cases carrying an MT-ND3 pathogenic variant. A single case carrying the m.10134C > A variant at nearly homoplasmic levels in skeletal muscle had only 17% residual complex I activity, which was likely caused by a loss of complex I protein (Miller et al. 2014). The most frequently reported MT-ND3 variants were the m.10158T > C (N = 4), m.10191T > C (N = 5), and m.10197G > A (N = 8) (Taylor et al. 2001; Lebon et al. 2003; Crimi et al. 2004; Leshinsky-Silver et al. 2005; Chae et al. 2007; Malfatti et al. 2007; Sarzi et al. 2007; Bakare et al. 2021). All patients carrying the m.10158T > C variant had a VAF of 83%–85% skeletal muscle, which was associated with similar residual complex I activities of 15%–27% (Lebon et al. 2003; Crimi et al. 2004; Bakare et al. 2021). Fibroblasts from one patient carrying the m.10158T > C variant had a VAF of 30% and a residual complex I activity of 98% (Bakare et al. 2021).
The m.10191T > C variant changes the hydrophilic serine at amino acid 45 to a hydrophobic proline, which could interfere with the confirmation of MT-ND3 (Taylor et al. 2001). The m.10191T > C variant was measured in skeletal muscle for all cases (N = 5) with VAFs of 77%–100% and a range of residual complex I activities of 17%–40% reported (Taylor et al. 2001; Lebon et al. 2003; Leshinsky-Silver et al. 2005; Malfatti et al. 2007). In contrast, despite the narrow range of VAFs (80%–100%) for the m.10197T > C variant in skeletal muscle across seven affected individuals, a wide range of residual complex I activities were observed (<1%–55%) (Taylor et al. 2001; Lebon et al. 2003; Leshinsky-Silver et al. 2005; Malfatti et al. 2007). The wide range of residual complex I activity associated with the m.10197T > C in muscle between different individuals may be owing to additional genetic factors that modify the penetrance of the variant.
MT-ND5
Complex I activity was reported for tissues from 21 individuals carrying seven unique variants in MT-ND5. The m.13513G > A variant was the most frequently reported variant (N = 12), which results in the replacement of an aspartic acid at amino acid position 393 with an asparagine (Fig. 4; Santorelli et al. 1997; Chol et al. 2003; Kirby et al. 2003; Blok et al. 2007; Ruiter et al. 2007; Van Karnebeek et al. 2011; Brecht et al. 2015). In a Kendall rank correlation test, the VAF of the m.13513G > A variant did not correlate with residual complex I activity (τ = 0.21, P = 0.4, n = 10) in skeletal muscle with VAFs ranging from 14% to 90% and residual complex I activity ranging from 8% to 54% (Fig. 5; Santorelli et al. 1997; Chol et al. 2003; Kirby et al. 2003; Blok et al. 2007; Ruiter et al. 2007; Van Karnebeek et al. 2011; Brecht et al. 2015). In a single study, skeletal muscle biopsies with VAFs of >90% for the m.13513G > A across all three individuals had varying residual complex I activities ranging from 12% to 47%, arguing against the wide range of activities being the result of measurement differences between different groups (Chol et al. 2003). In one case, 8% residual complex I activity in skeletal muscle was associated with a VAF as low as 26% for the m.13513G > A variant, indicating that low VAFs may significantly affect complex I activity (Kirby et al. 2003). The cases with relatively high complex I residual activities in association with a VAF of >90% indicate that the pathogenicity of the m.13513G > A variant may be modified by other genetic variants.
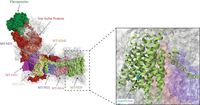
Complex I structure. The complex I structure with the mtDNA-encoded subunits (MT-ND1-4, MT-ND4L, MT-ND5-6) highlighted. An enlarged image indicates of the location of the m.13513G > A variant that results in an Asp393Asn within an alpha helix (PBD ID: 5XTD) (Guo et al. 2017).
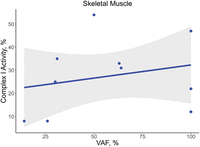
No correlation of m.13513G > A VAF with complex I activity. VAF for the m.13513G > A variant in MT-ND5 was not correlated with residual complex I activity in skeletal muscle (τ = 0.21, P = 0.4, n = 10).
Additional genetic variants reported in MT-ND5 include m.12338T > C (N = 1), m.12706T > C (N = 3), m.13042G > A (N = 1), m.13045A > G (N = 2), m.13063G > A (N = 1), and m.13376T > C (N = 1) (Taylor et al. 2002; Lebon et al. 2003; Naini et al. 2005; Malfatti et al. 2007; Zhang et al. 2018; Panadés-de Oliveira et al. 2020; Sasaki et al. 2020; Bakare et al. 2021). Normal complex I activity was observed in skeletal muscle from one individual carrying the m.13045A > G at a VAF of 50%, whereas another individual carrying the same variant at a higher VAF of 84% had only 36% residual activity (Panadés-de Oliveira et al. 2020). Surprisingly, skeletal muscle from an individual carrying the m.13376T > C at a VAF of ∼100% had normal complex I activity (Sasaki et al. 2020). The association of homoplasmic m.13376T > C with normal complex I activity suggests that the variant may not affect electron transfer through the complex but, instead, may alter complex I proton pumping, which is not measured in the spectrophotometric assays used to evaluate complex I activity. The m.12706T > C variant present at a VAF of 60% in muscle was associated with only 10% residual activity (Lebon et al. 2003). The m.13063G > A variant present at a VAF of 80% in skeletal muscle was associated with lower complex I and IV activities, suggesting that the m.13063G > A variant may interfere with the interactions of complex I with complex IV (Malfatti et al. 2007).
MT-ND6
Complex I activity was assessed in 11 cases carrying five unique variants in MT-ND6. The MT-ND6 variants m.14453G > A, m.14577T > C, and m.14600G > A were only reported in single patients at high VAFs (>82%) and residual activities of 28%–36% in skeletal muscle or cybrids (Tawata et al. 2000; Ravn et al. 2001; Malfatti et al. 2007). The m.14459G > A variant was reported in four cases, all with a VAF > 60% and residual activities ranging from 36% to 72% in skeletal muscle (Ronchi et al. 2011; Kurt et al. 2016; Yu et al. 2021). One of the most commonly reported MT-ND6 variants was m.14487T > C (N = 4 cases), which results in the replacement of a methionine with a valine in a transmembrane helix of MT-ND6 (Lebon et al. 2003; Malfatti et al. 2007; Spyropoulos et al. 2013). In cases harboring the m.14487T > C variant in skeletal muscle at VAFs of 50%, 80%, 84%, and 95%, residual complex I activities were 28%, 60%, 21%, and 15%, respectively (Lebon et al. 2003; Malfatti et al. 2007; Spyropoulos et al. 2013). The higher complex I activity in the case possessing a VAF of 80% compared with the case having a VAF of 50% may indicate that other genetic variants modify the effect of the m.14487T > C variant.
Complex III variants
Complex III (ubiquinol:cytochrome c–oxidoreductase, cytochrome bc1 complex) is a homodimer that couples the transfer of electrons from ubiquinol to cytochrome c, concomitant with the pumping of protons across the inner mitochondrial membrane (Vercellino and Sazanov 2022). Each monomer consists of 11 subunits, with three of the 11 subunits serving as catalytic cores: cytochrome b, cytochrome c1, and a Rieske iron–sulfur protein (Fig. 6) (Signes and Fernandez-Vizarra 2018). Cytochrome b (MT-CYB) is the only complex III subunit encoded by the mtDNA. Cytochrome b contains two heme groups (heme bL and heme bH) of differing redox potentials that transfer electrons from ubiquinol to the Rieske iron–sulfur protein within the complex (Guo et al. 2017; Vercellino and Sazanov 2022). The Rieske iron–sulfur protein transfers the electrons to cytochrome c1, which then transfers the electrons to the electron carrier cytochrome c (Vercellino and Sazanov 2022). In addition to serving as a catalytic core to complex III, cytochrome b is also the first subunit inserted into the inner mitochondrial membrane during complex III assembly (Tang et al. 2020). Like complex I, variants in MT-CYB can interfere with complex III and supercomplex assembly and stability (Legros et al. 2001; Blakely et al. 2005b). They may also induce conformational changes that block the proton exit pathway of complex III, interfere with ubiquinol binding, or decrease the electron transfer rate (Blakely et al. 2005b; Lloyd et al. 2015).
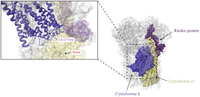
Complex III structure. The mtDNA-encoded subunit (cytochrome b) is indicated in blue with the cytochrome c1 and a Rieske protein indicated in yellow and purple, respectively. The m.15498G > A variant that results in a Gly251Asp is indicated in the enlarged image, along with a heme group in cytochrome c1 that it is located nearby (PBD ID: 5XTE) (Guo et al. 2017).
MT-CYB
Compared with the other respiratory complexes, complex III deficiency is rare, comprising 2%–15% of respiratory enzyme deficiencies described (Rustin et al. 1994; Mourmans et al. 1997; Kleist-Retzow et al. 1998). In our review, we identified 13 cases carrying 10 unique pathogenic variants in MT-CYB (Table 4). Assessment of complex III function in relation to specific variants was mainly limited to skeletal muscle, circulating lymphocytes, and fibroblasts.
Complex III variants
Most of the functional assessments of complex III variants were performed in tissues with a VAF of ≥80%, except for three cases carrying the m.14864T > C, m.15150G > A, and m.15800C > T variants. The m.14864T > C variant present at a VAF of 39% in muscle was associated with 72% residual complex III activity (Emmanuele et al. 2013). The m.14864T > C variant changes an amino acid in the ubiquinone binding site of complex III, which likely interferes with ubiquinone binding and electron transfer (Emmanuele et al. 2013). At a VAF of 45% in muscle, the m.15800C > T variant was associated with 27% residual complex III activity (Lamantea et al. 2002). The m.15800C > T variant introduces a premature stop codon, forming a truncated cytochrome b polypeptide. The low residual complex III activity observed in the m.14864T > C and m.15800C > T cases suggests that disruption of cytochrome b could destabilize complex III; however, the levels of complex III protein, or supercomplex formation and composition, were not evaluated in these studies. Consistent with this idea, a single case describing the m.15150G > A variant present at a VAF of 60% in muscle showed decreased complex III protein levels and only 7% complex III residual activity (Legros et al. 2001).
Among the complex III cases, several studies evaluated residual complex III activity alongside the activity of complex I. In one case, the m.15170G > A variant present in muscle at a VAF of 99% was associated with 10% residual complex III activity (Bruno et al. 2003). Like the m.15800C > T variant, the m.15170G > A variant results in a premature stop codon, a truncated cytochrome b polypeptide, and the loss of complex III (Bruno et al. 2003). Additionally, only 30% residual complex I activity was observed, suggesting disruption of an interaction between complexes III and I when cytochrome b is lost; however, no studies evaluating supercomplex assembly were conducted (Bruno et al. 2003). Furthermore, a skeletal muscle biopsy from an individual carrying the m.15699G > C missense variant at a VAF of 88% revealed low protein levels of both complexes I and III, suggestive of impaired supercomplex formation (Blakely et al. 2005b). Complex I and III activities were evaluated in a single case carrying the m.15579A > G variant (Wibrand et al. 2001). Complex I and III activities were decreased to 89% and 6% residual activities, respectively, in skeletal muscle with a VAF of 88% (Wibrand et al. 2001). The m.15579A > G variant is located in a helix of the ubiquinone binding site and, hence, may interfere with ubiquinone binding and electron transfer. How this missense variant interferes with the function of complex I is unclear as complex I and III protein levels were not measured nor was supercomplex assembly evaluated in the biopsied tissue or cybrids.
A single case was reported carrying the m.15197T > C variant (Legros et al. 2001). The m.15197T > C variant results in the substitution of a proline for a serine at position 151. The m.15197T > C variant was present at a VAF of 80% in muscle and was associated with 17% residual complex III activity, likely owing to low complex III protein levels (Legros et al. 2001). Notably, the substitution of serine 151 with a proline that occurs in the cd2 α-helix may disrupt the association of the cd2 α-helix with the catalytic center of cytochrome b (Esposti et al. 1993; Iwata et al. 1998; Legros et al. 2001).
Three cases carrying the m.15498G > A variant, a variant located near the ubiquinone binding site of complex III (Fig. 6), have been reported (Haut et al. 2004). Two individuals carrying m.15498G > A variant at homoplasmic levels in lymphocytes had 43% and 57% complex III residual activities (Haut et al. 2004). Although the m.15498G > A variant was present at near homoplasmic levels in lymphocytes and fibroblasts with similar complex III deficiencies, one individual was phenotypically normal, whereas the other showed severe growth restriction (Haut et al. 2004). The phenotypically normal individual may have higher levels of the pathogenic variant in blood cells but not in other tissues as a result of uneven segregation during development (Stewart and Chinnery 2015). It is also possible that the biochemical and phenotypic thresholds may not yet have been reached in tissues that would result in a clinical phenotype in the asymptomatic individual, or that other environmental or genetic factors affect the penetrance of this variant.
A single individual carrying an m.15635T > C variant at homoplasmic levels in skeletal muscle, fibroblasts, leukocytes, and liver has been reported (Fragaki et al. 2009). The residual activities of complex III in skeletal muscle, fibroblasts, and liver were 83%, 7%, and 66%, respectively (Fragaki et al. 2009). Contrarily, the activities of the other OXPHOS complexes were within the reference range, indicative of isolated complex III deficiency (Fragaki et al. 2009). The m.15635T > C variant replaces a highly conserved serine with a bulky proline at position 297, located within a transmembrane hydrophobic helix near an iron–sulfur binding site. Fully assembled complex III was absent in cybrids generated from the patient, suggesting that the m.15635T > C variant destabilizes complex III or interferes with its assembly (Fragaki et al. 2009).
The m.15243G > A variant was identified in one case (Valnot et al. 1999). In this individual, presenting with hypertrophic cardiomyopathy, a 90% VAF was associated with 22% residual complex III activity in cardiac tissue (Valnot et al. 1999). The m.15243G > A variant replaces a highly conserved glycine at position 166 with a glutamic acid within the transmembrane helices of cytochrome b, rather than the ubiquinone binding site. Evaluation of complex III activity in the patient's cardiac tissue compared with referent tissue revealed a fourfold lower Vmax; however, the Vmax/Km ratio, reflective of ubiquinone binding activity, was similar between tissue from the patient compared with the referent (Valnot et al. 1999). Hence, the m.15243A > G variant likely alters the conformation of complex III. Complex III protein levels were not evaluated and could also explain the differential complex III activity in cardiac tissue from the patient carrying the m.15243A > G compared with the referent tissue.
Complex IV variants
Complex IV (cytochrome c oxidase) catalyzes the transfer of electrons from reduced cytochrome c to molecular oxygen, coupled to the pumping of protons into the intermembrane space (Timón-Gómez et al. 2018). Complex IV consists of 14 subunits, including subunit NDUFA4, which was originally thought to belong to complex I (Balsa et al. 2012; Zong et al. 2018). Of the 14 complex IV subunits, three are encoded by the mtDNA: MT-CO1, MT-CO2, and MT-CO3 (Fig. 7). The mtDNA subunits form the catalytic core of complex IV with the remaining subunits encoded by the nuclear genome playing structural and regulatory roles. In the inner mitochondrial membrane, cytochrome c is oxidized by the CuA center of MT-CO2. The electrons are transferred to the heme within a catalytic center of MT-CO1 and then transferred to the heme a3–CuB binuclear center, in which O2 is reduced to water (Kadenbach and Hüttemann 2015; Timón-Gómez et al. 2018). MT-CO3 is essential for stabilizing the complex for proton pumping (Kadenbach and Hüttemann 2015).
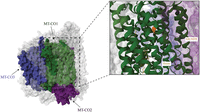
Complex IV structure. Complex IV with the three mtDNA-encoded subunits highlighted in green (MT-CO1), purple (MT-CO2), and blue (MT-CO3; PDB ID: 5Z62). The m.6930G > A variant that introduces a premature stop codon at position 343 is indicated in orange in the enlarged image (Zong et al. 2018).
MT-CO1
Pathogenic variants have been identified in all three mtDNA-encoded complex IV subunits (Manfredi et al. 1995; Hanna et al. 1998; Bruno et al. 1999; Clark et al. 1999; Rahman et al. 1999; Aguilera et al. 2001; Campos et al. 2001; Varlamov et al. 2002; Horváth et al. 2005; Kollberg et al. 2005; Herrero-Martín et al. 2008; Marotta et al. 2011; Wang et al. 2021). A total of 16 cases, each carrying a unique mtDNA variant, with VAF and complex IV activity assessment were identified (Table 5). Two nonsense variants were identified in MT-CO1: m.6930G > A and m.6708G < A (Bruno et al. 1999; Kollberg et al. 2005). The m.6930G > A variant results in a loss of the last 170 amino acids of MT-CO1 (Fig. 7). In muscle with the m.6930G > A variant at a VAF of 75%, only 10% of residual complex IV activity remained (Bruno et al. 1999). The m.6708G > A variant was found to result in a loss of the last 245 amino acids of MT-CO1. Functional assessment of complex IV activity in muscle harboring the m.6708G > A variant at a VAF of 81%–89% revealed 21% residual complex IV activity (Kollberg et al. 2005). Immunoblot results showed that the m.6930G > A variant resulted in lower levels of MT-CO1, MT-CO2, and COX4I1. In contrast, the m.6708G > A was associated with lower levels of only MT-CO1 and MT-CO2 (Bruno et al. 1999; Kollberg et al. 2005). Hence, both the m.6930G > A and m.6708G > A variants likely alter the assembly or stability of complex IV. The higher residual activity observed in the m.6708G > A case, despite the loss of more amino acids, may indicate some form of compensation for the partial loss of complex IV.
Complex IV variants
In addition to the two nonsense variants, three missense variants have been described in MT-CO1: m.6489C > A, m.6955G > A, and m.7444G > A (Aguilera et al. 2001; Varlamov et al. 2002; Herrero-Martín et al. 2008). Functional assessment of complex IV activity in muscle harboring the m.6489C > A variant in one case and the m.6955G > A variant in another case, both with at a VAF of ≥95%, revealed 74% and 15% residual complex IV activities, respectively (Varlamov et al. 2002; Herrero-Martín et al. 2008). The m.6489C > A variant was not found to alter hydrophobicity of the subunit, which may contribute to the less severe loss of complex IV activity despite the high VAF. In contrast, the m.6955G > A variant replaced glycine with aspartate, which interferes with the positions of histidine residues in the catalytic domain of complex IV and may account for the greater loss of complex IV activity compared with the m.6489C > A variant when present at a similar VAF (Herrero-Martín et al. 2008). Interestingly, the mother and brother of the patient carrying the m.6489C > A variant at a VAF of 90% in blood were phenotypically normal (Varlamov et al. 2002). This finding suggests that the VAF in blood is likely not reflective of other tissues, which is consistent with prior studies showing no correlation of the VAF of variants in blood and other tissues from individuals with mitochondrial diseases, likely owing to purifying selection (Rajasimha et al. 2008; Parikh et al. 2015; Stewart and Chinnery 2021).
The m.7444G > C variant in MT-CO1 present in muscle at homoplasmic levels was associated with 35% residual complex IV activity (Aguilera et al. 2001). The m.7444G > C variant replaces a stop codon with a lysine, resulting in the addition of three amino acids until the next stop codon (Brown et al. 1992). Despite being present at homoplasmic levels, the m.7444G > C variant was associated with a more similar residual complex IV activity to that of the m.6489C > A variant but with higher residual activity than the m.6955G > A variant. The m.7444G > C case may have higher complex IV activity than the m.6955G > A case because it is not located near a binding site of complex IV, unlike the m.6955G > A variant (Zong et al. 2018; UniProt Consortium 2023). Hence, the position of a missense variant within the complex may, in part, explain the differences in residual activity.
MT-CO2
Two missense variants (m.7671T > A, m.7587T > C) and two nonsense variants (m.7896G > A, m.7970G > T) were identified in MT-CO2 (Clark et al. 1999; Rahman et al. 1999). The m.7671T > A variant replaces methionine with a lysine in the membrane domain of MT-CO2 (Rahman et al. 1999). The m.7587T > C variant is located in the stop codon in MT-CO2 (Clark et al. 1999). MT-CO2 and MT-CO3 levels were lower in tissue from individuals carrying the m.7671T > A and m.7587T > C variants, suggesting that both variants destabilize or impair the assembly of complex IV (Clark et al. 1999; Rahman et al. 1999). It is also possible that the m.7671T > A variant may have resulted in aberrant MT-CO2 translation by removing the stop codon. The m.7896G > A nonsense variant was detected in skeletal muscle at a VAF of 76% and associated with 13% residual complex IV activity (Campos et al. 2001). Similarly, the m.7970G > T nonsense variant present at a VAF of 90% in skeletal muscle was associated with 3% residual complex IV activity (Horváth et al. 2005). Both the m.7896G > A and m.7970G > T nonsense variants resulted in lower levels of complex IV (Campos et al. 2001; Horváth et al. 2005). Collectively, the variants in MT-CO2 likely destabilize or interfere with assembly of complex IV, resulting in complex IV deficiency.
MT-CO3
One deletion, three missense variants, and three nonsense variants were identified in MT-CO3 (Hanna et al. 1998; Horváth et al. 2005; Marotta et al. 2011; Wang et al. 2021; Xu et al. 2021). Muscle was used for all measurements of complex IV function for all cases describing variants in MT-CO3. The m.9957T > C missense variant replaces a phenylalanine with a leucine in a conserved region of MT-CO3 (Wang et al. 2021). In skeletal muscle with a VAF of 81% for the m.9957T > C variant, residual complex IV activity was 79% with complex IV protein levels intact (Wang et al. 2021). The m.9789T > C missense variant replaces a conserved hydrophilic amino acid, serine, with a proline, a hydrophobic amino acid. The residual complex IV activity for the m.9789T > C variant (VAF of 50%) was 10% in skeletal muscle with lower levels of complex IV subunits detected by immunohistochemistry compared with the referent tissue (Horváth et al. 2005). Skeletal muscle from a case carrying the m.9396G > A variant at a VAF of 94% had 78% residual complex IV activity, suggesting that even at high VAFs, some variants may have only modest effects on enzymatic activity (Xu et al. 2021).
Three cases carried nonsense variants (m.9952G > A, m.9553G > A, 9379G > A) in MT-CO3 (Hanna et al. 1998; Horvath et al. 2002; Wang et al. 2021). Skeletal muscle from the case carrying the m.9952G > A variant had a VAF of 57% and 17% residual complex IV activity, owing to a loss of MT-CO1 and MT-CO2 (Wang et al. 2021). Similarly, the m.9553G > A variant at a VAF of 89% in skeletal muscle was associated with 59% residual complex IV activity owing to lower complex IV levels (Wang et al. 2021). An individual harboring the m.9379G > A variant at a VAF of 93% in muscle had 1% complex IV residual activity when normalized to citrate synthase (Horvath et al. 2002). Immunoblotting of tissue from the individual with the m.9379G > A variant indicated lower in MT-CO3 and MT-CO2 levels compared with tissue from referents, with only a nuclear encoded subunit of complex IV showing immunoreactivity. A m.9559delC variant that caused a frameshift was detected at a VAF of 58% in skeletal muscle and associated with 38% residual complex IV activity, likely the result of a loss of complex IV (Marotta et al. 2011). The association of lower levels of MT-CO1 and MT-CO2 in tissues harboring MT-CO3 variants shows the importance of MT-CO3 for stabilizing the catalytic core of complex IV.
Complex V variants
The final step of OXPHOS is catalyzed by complex V (ATP synthase, F0–F1 ATPase). Complex V uses the proton motive force generated by the electron transport chain to phosphorylate ADP to ATP. Complex V is composed of two components: the F0 domain and the F1 domain. Embedded in the inner mitochondrial membrane, the F0 domain contains a multisubunit c ring that is responsible for proton translocation. The matrix facing F1 domain contains the catalytic site for ADP phosphorylation. The two domains are held together by a peripheral and central stalk that prevents unnecessary rotation (Fig. 8). Protons flow down the electrochemical gradient through the F0 domain, powering the rotation of the c ring. The rotation of the c ring subsequently confers conformational changes in the F1 domain, which results in ADP phosphorylation (Xu et al. 2015; He et al. 2018; Signes and Fernandez-Vizarra 2018; Tang et al. 2020).
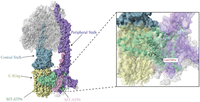
Complex V structure. The complex V structure is depicted with the mtDNA-encoded subunits indicated in pink (MT-ATP8) and seafoam green (MT-ATP6), and key structural elements are in blue-gray (central stalk), purple (peripheral stalk), and yellow (c-ring; PDB ID: 8H9S). The m.8993T > C variant changes a leucine to a proline at position 156 within MT-ATP6, which is located at the intersections of the c-ring and central and peripheral stalks, as indicated in the enlarged image (Lai et al. 2023).
Complex V consists of 29 subunits, with MT-ATP6 and MT-ATP8 encoded by the mtDNA (He et al. 2018). A total of 49 cases carrying seven unique mtDNA variants in MT-ATP6 or MT-ATP8 were identified (Table 6). Most of the studies identified measured ATP synthase activity in skeletal muscle, fibroblasts, lymphocytes, and cybrids. Variants in the mtDNA-encoded complex V subunits can affect the assembly or stability of the enzyme, uncouple respiration from ATP synthesis, and decrease the rate of ATP synthesis.
Complex V variants
MT-ATP6
The most frequently reported variant in MT-ATP6 was the m.8993T > G missense variant (N = 29 cases) (Tatuch et al. 1992; Tulinius et al. 1995; Uziel et al. 1997; García et al. 2000; Carelli et al. 2002; Carrozzo et al. 2004b; Morava et al. 2006; Bakare et al. 2021). The m.8993T > G variant replaces a leucine with an arginine that changes the confirmation of a helix in MT-ATP6, resulting in possible interference with proton translocation or the rotation of subunit c. A higher VAF of the m.8993T > G variant was associated with lower complex V activity in skeletal muscle (τ = −0.58, P = 0.01, n = 13) but not in fibroblasts (τ = −0.11, P = 0.7, n = 9) (Fig. 9).The lower complex V activities associated with the m.8993T > G variant likely result from a loss of fully assembled complex V (Morava et al. 2006; Verny et al. 2011).
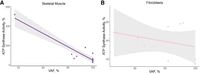
Association of m.8993T > G VAF with complex V activity. The VAF for the m.8993T > G variant in MT-ATP6 correlated with the percent residual complex V activity in skeletal muscle (A; τ = −0.58, P = 0.01, n = 13) but not in fibroblasts (B; τ = −0.11, P = 0.7, n = 9). However, it should be noted that the correlation in skeletal muscle is likely driven by a single case.
The range of complex V activities identified in our study suggests the possibility that other genetic variants influence the penetrance of the m.8993T > G variant. Complex V activity ranged from 27% to 63% in skeletal muscle across multiple patients (n = 6), despite similar VAFs of ≥95% (Uziel et al. 1997; Morava et al. 2006). Similarly, lymphoblastoid cell lines with the m.8993T > G variant at VAFs >95% had residual complex V activities ranging from 34% to 54% (Tatuch et al. 1992) Intriguingly, skeletal muscle from some patients had higher complex V activities compared with tissue from referents, suggestive of a compensatory mechanism (Tatuch et al. 1992; Tulinius et al. 1995). Lower complex V activity (77% residual activity) was also observed at a VAF as low 10% in platelets (Carelli et al. 2002), suggesting that the biochemical threshold can exist well below a VAF of 60%. However, it should be noted that many of the individuals carrying the m.8993T > G variant were diagnosed with NARP/Leigh syndrome or other neurologic conditions, and the VAF of platelets may not be reflective of the affected tissues such as those in the brain.
Another variant frequently observed in the literature was the m.8993T > C missense variant in MT-ATP6 (N = 9 cases) (Fig. 8; Santorelli et al. 1996; Morava et al. 2006). Like the m.8993T > G variant, the residual complex V activity in skeletal muscle harboring the m.8993T > C variant at a VAF of >95% exists at a range from 25% to 52% (Morava et al. 2006). Several studies found that skeletal muscle from individuals carrying the m.8993T > G or m.8993T > C variant had lower levels of complex V compared with skeletal muscle from the referents, suggesting that the m.8993T > G/C variants may destabilize the complex or impair assembly of the holoenzyme (Morava et al. 2006; Verny et al. 2011).
The missense variants m.9176T > C (N = 6) and m.9185T > C (N = 1) in MT-ATP6 change a leucine to proline. Although both leucine and proline are hydrophobic amino acids, the large alkyl side-chain on proline likely interferes with protein folding and assembly, which is consistent with the lower levels of fully assembled complex V observed in fibroblasts from several cases carrying the m.9176T > C (Verny et al. 2011). Complex V activity ranged from 74% to >100% in patient-derived fibroblasts with VAFs >95% for the m.9176T > C variant (Dionisi-Vici 1998; Verny et al. 2011). The modest effect of the m.9176T > C variant on complex V activity, despite being present at high VAFs, suggests a possible compensatory mechanism that may be cell type specific. Complex V activity, however, was not evaluated in any other cell or tissue type with the m.9176T > C variant. Two cases carrying a different allele at position m.9176 (m.9176T > G), resulting in the replacement of leucine with an arginine, were also described (Carrozzo et al. 2004a,b). Cybrids and fibroblasts from the patients were homoplasmic for the m.9176T > G variant, which was associated with residual complex V activities of 40% and 20%, respectively (Carrozzo et al. 2004a,b). A single case carrying the m.9127-9128delAT frameshift variant was reported with a VAF of 50% in fibroblasts associated with 40% residual complex V activity (Mordel et al. 2017).
MT-ATP8
Only one case was identified that assessed the effect of a variant in MT-ATP8, a m.8529G > A nonsense variant, with complex V activity (Jonckheere et al. 2008). The m.8529G > A variant in skeletal muscle at a VAF of 90% was associated with 17% complex V residual activity (Jonckheere et al. 2008). Immunoblot results from muscle homogenate showed higher levels of assembly intermediates of ATP synthase compared with the referent tissue, indicating that the variant disrupts complex V assembly or stability (Jonckheere et al. 2008).
Discussion
In our systematic review, we identified reports describing cases of mitochondrial diseases in which the level of heteroplasmy for a pathogenic mtDNA variant was evaluated in the same tissue/cells used to measure the activity of the affected complex. Most studies evaluated the VAF of the pathogenic mtDNA variant and OXPHOS complex activity in skeletal muscle, circulating white blood cells, dermal fibroblasts, lymphoblastoid cell lines, or cybrids. Although mitochondrial mass was accounted for by normalizing to citrate synthase, a number of studies did not account for possible differences in protein levels of the affected complex. Most studies did not evaluate supercomplex levels or composition or evaluate mitochondrial respiration. With a few exceptions, the majority of the pathogenic mtDNA variants described were identified in only several individuals and were present at VAFs >60% in the tissues and cell types evaluated; hence, our ability to draw conclusions regarding the effect of a particular variant across a range of VAFs on OXPHOS complex activity is limited. However, a number of pathogenic, missense variants present at lower VAFs (<50%) were associated with low residual activity of the affected complex. Therefore, the often cited >60%–80% VAF threshold may not be applicable for all variants in the mtDNA protein-encoding genes. Lastly, a number of the pathogenic variants were present at similar VAFs in the same tissue/cell type across individuals with a wide range of residual OXPHOS complex activity detected, even when the measures were performed by the same group. The heterogeneity of residual OXPHOS complex activity across individuals carrying the same pathogenic variant at similar VAFs suggests that other genetic factors likely alter the biochemical threshold.
Several studies have sought to identify the biochemical threshold for each OXPHOS complex by treating isolated mitochondria or cells in vitro with pharmacologic inhibitors of the complexes across a range of doses (Rossignol et al. 1999, 2000). The biochemical thresholds identified through pharmacologic inhibition of each OXPHOS complex in isolated mitochondria from multiple different tissues from rats revealed tissue-specific differences (Barrientos and Moraes 1999; Rossignol et al. 1999, 2000). Mitochondria isolated from highly energetic tissues (heart, brain) were more sensitive to pharmacologic inhibition of complexes I and IV and had lower biochemical thresholds compared with mitochondria from other tissues (Rossignol et al. 1999, 2000). In an osteosarcoma cell line treated with increasing doses of rotenone, a decline in mitochondrial respiration was observed once complex I was inhibited by ∼30%–40% (Barrientos and Moraes 1999). Pharmacologic inhibition of an OXPHOS complex is likely reflective of mtDNA variants that result in a loss of the holoenzyme. However, the relevance of pharmacologic inhibition of OXPHOS complexes to mtDNA variants that likely have a more modest effect on complex function and that do not result in the loss of the fully or partly assembled complex is less clear.
In human tissues and cell types, mechanistic studies have primarily used cybrids generated from patients carrying a pathogenic mtDNA variant, mostly those in the tRNA-encoding genes (Shoffner et al. 1990; Boulet et al. 1992; Yoneda et al. 1994). Variants in the mtDNA-encoded tRNAs appear to require higher VAFs before a biochemical threshold is reached, and such studies are the most frequently cited when referring to the biochemical threshold being at VAFs ≥60%–80% (Shoffner et al. 1990; Boulet et al. 1992; Morgan-Hughes et al. 1995). The high VAF required for pathogenic tRNA variants to exert an effect on mitochondrial function is likely because tRNAs are present in excess, and the tRNAs carrying the referent allele may be more stable than tRNAs containing the pathogenic variant (Yoneda et al. 1994; Rossignol et al. 2003). Although cybrids provide a model for validating pathogenic mtDNA variants, it is important to consider that cybrids are generated using cancer cell lines that have metabolic disturbances at baseline. Such metabolic abnormalities confound the ability to deeply interrogate the multifaceted effects of mtDNA variants on mitochondrial metabolism and cellular phenotype.
Our systematic review has several limitations. Our approach consisted of identifying studies based on MESH terms describing complex deficiency, genetic variants in the mtDNA, and specific mitochondrial diseases. It is possible that publications describing individuals carrying a pathogenic mtDNA variant were not captured using our MESH terms and, consequently, were not included in our review. It is possible that differences in referent groups used by research or clinical site contribute to the variability in OXPHOS complex activity measurements. Only a few studies indicated the use of a referent group consisting of individuals belonging to the same mitochondrial haplogroup as the patient, whereas other studies used a reference range, presumably determined across multiple genetic ancestries, but such details are often not provided. Future studies should include details such as whether the referents are related to the patient or belong to the same macrohaplogroup as the patient. The age of the cases at the time of biosample collection for OXPHOS activity assessment was often not provided, limiting our ability to account for age in our models evaluating VAF with residual complex activity.
The assays used to evaluate OXPHOS activities do not comprehensively assess all aspects of the functions of each OXPHOS enzyme or the efficiency of ATP production. For example, the complex I activity assay indirectly measures the electron transfer rate through the enzyme but does not evaluate proton pumping efficiency of the complex. Ideally, mitochondrial respiration studies in cells and tissues from the patients and well-matched (age, sex, mitochondrial haplogroup) referent should be performed. Additionally, whether a clear cutoff can be used in clinical diagnosis is controversial as the range of OXPHOS complex activities is wide and likely exists on a continuum (Rustin et al. 1994; Thorburn et al. 2004). However, our objective was not to define a cutoff for clinical diagnosis but rather to evaluate the relation of the pathogenic variant VAF with residual OXPHOS complex activity. Pathogenic variants in the OXPHOS complexes may also interfere with other mitochondrial functions such as fatty acid oxidation, amino acid biogenesis, or apoptosis (Shaham et al. 2010; Nsiah-Sefaa and McKenzie 2016; Spinelli and Haigis 2018; Sharma et al. 2021; Li and Hoppe 2023), which would not be captured by evaluating the OXPHOS complex activities. Future studies of cells carrying a range of VAFs for a pathogenic variant should evaluate multiple different mitochondrial functions, including mitochondrial dynamics, metabolism through metabolomics and metabolic flux analysis, and effects on epigenetic and post-translational modifications, among others (Kopinski et al. 2019; Monzel et al. 2023). Because of tissue mosaicism, it is also possible that the site biopsied contains cells with low VAFs for the pathogenic variant or cells lacking the pathogenic variant, which can result in a false-negative finding upon OXPHOS activity measures. Such limitations can be overcome using newer approaches such as the use of induced pluripotent stem cells generated from patients with mitochondrial disease and emerging tools for editing the mtDNA to more comprehensively and systematically define the biochemical threshold for specific variants in disease-relevant cell types.
The importance of studying the biochemical threshold of mtDNA variants across different cell types relevant to human disease will be critical for gaining mechanistic insights into mitochondrial diseases. Buffering of pathogenic mtDNA variants can occur through increased mtDNA replication to sustain a threshold of mtDNA's with the referent allele (Durham et al. 2007), as well as through the rerouting of metabolites through alternative pathways to sustain ATP generation (Rossignol et al. 2003; Mick et al. 2020). Tissue- and cell type–specific differences exist in both the sensitivity to OXPHOS complex impairments and their ability to buffer OXPHOS impairment through alternative metabolic pathways (Rossignol et al. 1999, 2000; Mick et al. 2020). Additionally, the OXPHOS complexes are often organized into supercomplexes, particularly in highly energetic tissues, with tissue-specific stoichiometries (Vercellino and Sazanov 2021, 2022). Further, tissue-specific isoforms of some OXPHOS complex subunits have been identified (Kadenbach et al. 1990; Arnaudo et al. 1992; Wolz et al. 1997). However, studies of human mtDNA genetic variation to date have focused on a limited number of tissues and cell types that are likely not reflective of the tissues affected in mitochondrial and aging-related diseases. Future studies should evaluate pathogenic mtDNA variants across a range of VAFs and cell types using respirometry to identify the variant- and cell type–specific biochemical thresholds.
Our systematic review raises the question of whether a universal biochemical threshold exists for each OXPHOS complex or variant. The heterogeneity in OXPHOS complex residual activity measured in the same tissue or cell type with similar VAFs for the same pathogenic mtDNA variant across individuals suggests other genetic modifiers are at play that may limit our ability to define a universal biochemical threshold. It is also possible that the biochemical threshold exists across a wider range of VAFs as a result of such heterogeneity in OXPHOS activity. A greater understanding of the biochemical threshold of mtDNA variants in cell types relevant to disease will provide insights into the different phenotypes and ages of onset observed across individuals carrying the same pathogenic variant. Defining the biochemical threshold of mtDNA variants will also aid in determining whether heteroplasmic variants at particular VAFs are expected to induce aging-related decline in mitochondria respiration, in a tissue-dependent manner.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This research was supported by the National Heart, Lung, and Blood Institute, T32 HL007224-45 (J.D.M.) and K01 HL143142 (J.L.F.); National Institute of Aging, R03 AG081053 (J.L.F.); and an American Heart Association grant, 17FTF33670369 (D.M.G.).
Author contributions: K.K.S. performed the literature search, coded the manuscripts, created the conceptual figure, and drafted the manuscript with guidance from J.L.F. J.D.M., C.R.W., J.O.P., and A.N.L. also coded the manuscripts identified by the literature search. L.X. performed the statistical analyses and generated the plots of variant VAFs and residual activities. D.B.F. provided guidance on the terms selected for the literature search and methodology for the systematic literature search. D.M.G. reviewed and provided critical feedback on the manuscript. J.L.F. conceived of the review and oversaw all aspects of the project.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278200.123.
-
Freely available online through the Genome Research Open Access option.
- Received June 19, 2023.
- Accepted March 15, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵








