
Full-resolution HLA and KIR gene annotations for human genome assemblies
Abstract
The human leukocyte antigen (HLA) genes and the killer cell immunoglobulin-like receptor (KIR) genes are critical to immune responses and are associated with many immune-related diseases. Located in highly polymorphic regions, it is difficult to study them with traditional short-read alignment-based methods. Although modern long-read assemblers can often assemble these genes, using existing tools to annotate HLA and KIR genes in these assemblies remains a nontrivial task. Here, we describe Immuannot, a new computation tool to annotate the gene structures of HLA and KIR genes and to type the allele of each gene. Applying Immuannot to 56 regional and 212 whole-genome assemblies from previous studies, we annotate 9931 HLA and KIR genes and found that almost half of these genes, 4068, have novel sequences compared with the current Immuno Polymorphism Database (IPD). These novel gene sequences are represented by 2664 distinct alleles, some of which contained nonsynonymous variations, resulting in 92 novel protein sequences. We demonstrate the complex haplotype structures at the two loci and report the linkage between HLA/KIR haplotypes and gene alleles. We anticipate that Immuannot will speed up the discovery of new HLA/KIR alleles and enable the association of HLA/KIR haplotype structures with clinical outcomes in the future.
The human leukocyte antigen (HLA) genes and killer cell immunoglobulin-like receptor (KIR) genes play a central role in immune responses. They are clustered in two genomic regions, respectively. The HLA genes are located in the major histocompatibility complex (MHC) region on the short arm of Chromosome 6. The MHC region spans ∼5 Mb. Coding genes in MHC, such as HLA-A, -B, -C, -DR, -DP, and -DQ are highly polymorphic among human populations, which is maintained by balancing selection (DeGiorgio et al. 2014; Meyer et al. 2018). Many HLA alleles (Marsh et al. 2010), or subgroups distinguished by gene sequences, were reported to be associated with hundreds of immune-related diseases (Gough and Simmonds 2007; Medhasi and Chantratita 2022), and HLA allele matching (HLA-A, -B, -C, -DRB1, and -DQB1) has been widely used in pairing unrelated donors and recipients to lower the failure risk of graft transplantation (Buck et al. 2016; Tiercy 2016; Dehn et al. 2019).
The KIR gene cluster is a smaller region located on Chromosome 19, ranging from 150 kb to 200 kb in different individuals. It contains 17 genes, including 15 coding genes and two pseudogenes, regulating the development, tolerance, and activation of natural killer (NK) cells. Different from HLA genes’ high polymorphism, KIR genes share similar gene structures and have diverse copy numbers for each haploid genome (Carrington and Norman 2003). The most common KIR haplotype is called the “A-haplotype” with nine genes, and all other KIR haplotypes are defined as the “B-haplotype” (Cisneros et al. 2020). The A-haplotype is also called the inhibitory haplotype because most of its KIR genes inhibit the activating of NK cells, whereas the B-haplotype tends to cover more activating genes. Similar to the HLA genes, KIR genes also have a nomenclature system to functionally distinguish each KIR allele sequence (Robinson et al. 2018).
High-throughput sequencing with short reads has become a common approach to typing HLA/KIR alleles, which enables the discovery of the connection between special gene groups and clinical outcomes in large cohorts (Sakaue et al. 2023). By searching the best combinations of alleles that are explained by the read data, many computational methods can achieve high sensitivity and accuracy when the gene has been well-documented (Szolek et al. 2014; Norman et al. 2016; Kawaguchi et al. 2017; Lee and Kingsford 2018; Dilthey et al. 2019; Kim et al. 2019; Sverchkova et al. 2019; Gao et al. 2022; Song et al. 2023). For example, the classic HLA genes (A, B, C, DRB1, DQB1, DPB1) have hundreds to thousands of allele sequences in the IPD-IMGT/HLA Database (Barker et al. 2023). However, for underrepresented nonclassical HLA genes, these tools would have reduced power owing to difficulties in accurately recovering novel allele sequences with only short reads, and most of them would not even report those alleles. Genotyping with long reads may overcome this limitation as each gene sequence can be fully assembled before comparing with the Immuno Polymorphism Database (IPD), which would be less affected by incomplete reference gene sequences. Thus, it is more straightforward to annotate and call alleles on an assembly rather than from raw read data (Dilthey et al. 2019). Recently, more and more long-read full-genome assemblies have been released. For example, the Human Pangenome Reference Consortium (HPRC) released 94 haploid assemblies in phase I (Liao et al. 2023); the number will go up to several hundred in phase III by mid-2024. The Chinese Pangenome Consortium (CPC) released 114 haploid assemblies in phase I (Gao et al. 2023). There is an urgent need to study HLA/KIR regions on those assemblies.
To this end, we developed a new computation tool, Immuannot, to annotate HLA and KIR gene structures on human assemblies and output the allele calls at the same time. Taking IPD-IMGT/HLA and IPD-KIR as references, Immuannot employs full-gene sequences to resolve gene structures and uses all available coding sequences (CDSs), including the ones with partial CDSs, to type alleles. We applied our pipeline to 212 full-genome assemblies, 50 KIR reference contigs, and six HLA reference genomes. Immuannot reported thousands of novel HLA and KIR allele sequences that were missing in the IPD database, particularly for those nonclassical coding genes and pseudogenes. Through a series of genetic analyses, we validated Immuannot and identified numerous new genetic patterns. As long-read data become more and more popular, our tool will improve the genetic research in HLA and KIR regions.
Results
Method overview
Immuannot types 36 HLA genes in the IPD-IMGT/HLA Database v3.52.0 and 17 KIR genes in the IPD-KIR Database (v2.12) (Robinson et al. 2013; Barker et al. 2023). It first aligns gene sequences, including introns, to contigs of interest (Fig. 1A). A gene is initially detected on a contig if 90% of the gene sequence is mapped. When multiple reference allele sequences match the same location of the target contig, the one with the largest gap-compressed identity and longest matching length is chosen as the template allele, which is further used for determining gene structure, such as exon boundaries, and allele type. If the template allele has a perfect match to the contig, the contig is reported to carry that allele. Otherwise, CDS is extracted for allele calling with extra steps. We use alleles with full-gene-length sequences for template selection and use all alleles with full or partial CDS for allele typing.
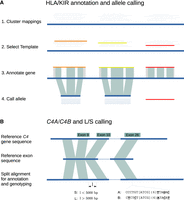
Overview of the Immuannot algorithm. (A) Annotate HLA/KIR genes using known alleles from the IPD-IMGT/HLA or the IPD-KIR databases. Gene sequences (thin line) from the IPD databases are mapped to the target contig sequence (thick line) at step 1. At step 2, the best-matching allele with the smallest number of mismatches and the longest alignment length is chosen as the template allele (in different colors) for each mapping cluster. Project the gene structure of template allele to the contig to annotate exons, introns, UTRs, CDS, and start and stop codons (step 3). At step 4, determine the allele type based on the CDS similarity. (B) Annotate the C4 genes. Align the C4 transcript sequence from GRCh38 to the contig sequence. Infer C4A versus C4B based on the sequence on exon 26 and infer the long (L) versus short (S) form based on the intron size between exons 9 and 10.
To annotate complement component 4 (C4), an MHC class III gene that is not covered by the IPD-IMGT/HLA database, we apply a different strategy (Fig. 1B). In this case, exons are extracted from one reference gene sequence and aligned to the target contig with splicing. We can annotate the gene structure and identify the length of the ninth intron and the key peptides in the 26th exon to infer the long versus short form and C4A versus C4B types (Yu et al. 1986; Blanchong et al. 2001; Wang and Liu 2021).
Immuannot can distinguish multiple copies of a particular gene because each reference allele sequence is allowed to be mapped to multiple regions/contigs. It outputs in the GTF format, including the gene structure annotations and the allele type defined by the nomenclature system (Marsh et al. 2010; Robinson et al. 2018). When a novel allele is detected, we will provide a consensus call with a “new” tag in the corresponding naming field to summarize the effects of new nucleotide variants. More details about our pipeline can be found in the Methods section.
Notably, IPD-IMGT/HLA and IPD-KIR include both protein-coding genes and pseudogenes. In their master annotation file, they provide “CDS” coordinates for pseudogenes but without translated proteins owing to frameshifts or in-frame stop codons in the CDS. We could not find out how CDSs for pseudogenes were determined. Either way, following the IPD-IMGT/HLA and IPD-KIR annotations, Immuannot reports CDSs for all genes as well, including both protein-coding genes and pseudogenes.
Evaluation
To evaluate our pipeline, we first applied Immuannot to eight MHC haplotypes (Houwaart et al. 2023). This is the only published data set we know with complete assembly. It provided computational annotation of nine protein-coding HLA genes (A, B, C, DRB3/4/5, DRB1, DQA1, DQB1, DPA1, and DPB1) and C4. We compared our results to the published annotation. Among 72 full-resolution comparisons of HLA protein-coding genes across eight haplotypes (Supplemental Table S1), we only observed one inconsistency: In sample OK649235, we called 03:01:01:02 for the HLA-DRB1 gene, whereas Houwaart et al. (2023) called 03:01:01:01, different in the fourth field. Further investigation shows that allele DRB1*03:01:01:02 is perfectly aligned to the assembly OK649235, whereas allele DRB1*03:01:01:01 has 11 mismatches (a 10 bp deletion and a substitution) in introns. Our C4 typing is identical to the results by Houwaart et al. (2023) as well (Supplemental Table S2).
NA12878 is a sample commonly used for evaluating HLA genotyping. We acquired NA12878 HiFi reads (AC: SRP194450), assembled the data with hifiasm (version 0.19.8) (Cheng et al. 2021), and genotyped HLA genes with Immuannot. Our typing agrees with previous reports.
We also tested HiFiHLA (https://github.com/PacificBiosciences/hifihla), an unpublished proprietary tool, to genotype HLA genes. The current version (0.2.3) of HiFiHLA did not type the HLA-U, HLA-N, HLA-S, and TAP2 genes. It agreed with Immuannot on all remaining HLA genes across all HPRC samples. Because it needs to align target assembly to a reference genome, the typing pipeline was more than 10 times slower than Immuannot when using a full-genome assembly as input (Supplemental Table S3). Because HiFiHLA could not type KIR genes, we focus the comparison among HLA genes.
We also compared Immuannot with the recently released software SKIRT (Hung et al. 2024) for KIR allele annotation on assembled genomes. We tested both software on HPRC assemblies. On average, Immuannot took ∼4 min to annotate KIR alleles on one full-genome haploid assembly with three threads, whereas SKIRT spent ∼24 min with three threads (Supplemental Table S3). SKIRT was slower because it heavily relied on minimap2 v2.17 (Li 2018) with split-alignment mode, which was more time-consuming than the nonsplit mode. Immuannot annotated 901 genes that were all annotated by SKIRT, and 99.7% (898/901) of them shared the same location and allele name. For the three mismatches, SKIRT called them as gene fusions, whereas Immuannot only annotated these genes without specific allele groups. SKIRT annotated five more genes not reported by Immuannot. Three of them have partial deletions with the remaining regions covering <90% of the reference sequence, and the other two include large intronic indels that fragmented the alignment and failed our template selection step (Supplemental Notes).
We further applied Immuannot and SKIRT to 50 KIR contigs that were part of the GRC38.p14 release for a three-way comparison (Supplemental Table S4). Immuannot annotated 427 genes, in which 425 of them perfectly matched the SKIRT annotation. Of the two differences, the first, at NW_003571057.2:827290-839484, was annotated by SKIRT as a fusion of genes KIR2DL3 and KIR2DP1, but Immuannot reported KIR2DP1 without allele group information. As to the second difference, Immuannot found gene KIR2DL1 at NW_016107303.1:71161-85920, which was missed by SKIRT but was confirmed by the GRC annotation (Supplemental Table S5). SKIRT identified 18 additional genes that were not found by Immuannot. We manually checked six cases and found all of them to be partial genes. Among these 18 genes, 12 were reported by GRC, and 11 of them had identical annotations. GRC annotated two genes not reported by either SKIRT or Immuannot. Both genes were annotated as KIR3DX1, which was absent from the IPD-KIR Database (Supplemental Table S6).
Novel alleles
We applied Immuannot to 212 full-genome haploid assemblies, including HPRC (n = 94) (Liao et al. 2023), CPC (n = 114) (Gao et al. 2023), CHM13.0 (Nurk et al. 2022), GRCh38.p14 (Schneider et al. 2017), and CN-T2T (n = 2) (He et al. 2023); 50 KIR reference contigs; and six HLA reference assemblies (Houwaart et al. 2023). This resulted in 7519 HLA genes, represented by 2673 unique alleles as some unique alleles were present in multiple samples. Furthermore, 2696 of these genes (1531 unique alleles) were not seen in the IPD-IMGT/HLA Database. As to KIR, Immuannot annotated 2412 genes, represented by 1503 unique alleles, and 1372 of these genes (1133 unique alleles) were novel. In total, 41.0% [(2696 + 1372) / (7519 + 2412)] of HLA/KIR genes had novel alleles absent from the IPD databases (Fig. 2; Supplemental Fig. S1).
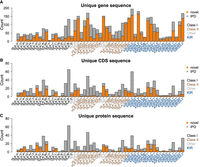
Summary of unique allele sequences. Alleles with identical sequences are merged. Known allele sequences in the IPD databases are shown in gray, and novel allele sequences are shown in orange. (A) Gene sequences with intron included. (B) CDS sequences. (C) Protein sequences.
Across the 41 protein-coding genes in the IPD-IMGT/HLA and IPD-KIR databases, the rate of novel CDS mutations not seen in IPD is 5.1 × 10−5 per base pair, or 42.9 in the Phred scale. This rate is more than 10 times higher than the estimated base error rate of HPRC contigs (Liao et al. 2023). Furthermore, 94% of the novel CDS mutations are single-nucleotide substitutions, whereas HiFi reads are enriched with insertion/deletion errors (Wenger et al. 2019). These observations suggest most novel CDS alleles identified by Immuannot are not caused by base errors in contigs.
In more detail, among the six classical HLA protein-coding genes (HLA-A, -B, -C, -DRB1, -DPB1, and -DQB1), 22.1% (289/1310) genes annotated by Immuannot were novel, but only 2.4% (7/289) carried mutations that led to amino acid changes (Fig. 2C; Supplemental Table S7). All seven novel proteins had distinct sequences. Among other HLA protein-coding genes (i.e., not pseudogenes), 31.3% (1080/3454) were novel, and 10.2% (110/1080) had amino acid changes. These 110 protein-coding genes were represented by 43 unique alleles. At the KIR locus, 57.8% (1116/1931) of the KIR protein-coding genes were novel, and 4.2% (47/1116) had amino acid changes, represented by 42 novel unique protein sequences (Supplemental Table S7). Among pseudogenes in the IPD databases, 48.9% (1583/3236) were novel.
HLA gene diversity
We investigated the CDS diversity of HLA genes (Fig. 3A,B; Supplemental Fig. S2). HLA-DQB1 had the highest diversity in our data set with the average pairwise sequence divergence approaching 5% (Fig. 3B). Several other well-studied HLA genes, such as HLA-A, -B, -C, and -DRB1, were also highly diverse, and their average pairwise sequence divergence is >2.5% among either Africans in HPRC or Asians in CPC.
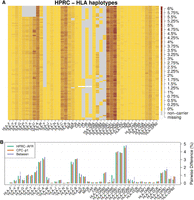
The diversity of HLA coding sequences. (A) HLA haplotypes among HPRC samples. Each row represents one HLA haplotype. For each haplotype, heat from light yellow to the dark brown indicates the average pairwise CDS divergence between the haplotype and all the other haplotypes. A gray box denotes the deletion of a gene in the middle of a contig, and a white box denotes an unassembled gene in assembly gaps. A version with detailed HPRC haplotype names is included as Supplemental Figure S10. (B) HLA within-population diversity and between-population divergence. Each group of bars shows average pairwise CDS difference within HPRC African samples (AFR; n_hap = 24), within CPC Asian samples (n_hap = 114), and between the two populations. Permutation tests were performed to test whether the two populations are distinguishable in terms of CDS divergence. P-values are adjusted with the Bonferroni method. Number of stars indicates different significance level: (*) P-value < 0.05, (**) P-value < 0.01, (***) P-value < 0.001.
There are several class-I pseudogenes. Among them, HLA-U had the highest diversity with pairwise sequence divergence similar to HLA-A’s. Despite being a pseudogene, HLA-U is also expressed in tissues such as whole blood, lung, and EBV-transformed lymphocytes (Supplemental Fig. S3). On the other hand, HLA-Y and HLA-N had the lowest diversity. HLA-Y is a pseudogene possibly derived from HLA-A (Williams et al. 1999). It was in 28.7% of HPRC haplotypes and 12.7% of CPC haplotypes, and the CDS diversity was 0.0% in HPRC and 0.017% in CPC among all carriers. HLA-N was first reported by Geraghty et al. (1992a). It is annotated with one exon, and the CDS region has only 161 bp. All HPRC and CPC samples were found to carry identical CDS, and the estimated CDS diversity is 0.002% among high-coverage WGS samples from The 1000 Genomes Project (n = 3202) (Supplemental Table S8; Byrska-Bishop et al. 2022).
In most genomic regions outside HLA, African populations have higher diversity than non-African populations owing to the out-of-Africa bottleneck in non-African populations (The 1000 Genomes Project Consortium 2015; Mallick et al. 2016; Bergström et al. 2020). Nevertheless, African samples in HPRC (n = 40) and Chinese samples in CPC (n = 57) have similar diversity for most HLA genes (Fig. 3B). The pairwise CDS sequence divergence between the two populations is also indistinguishable from the within-population pairwise divergence based on a permutation test (Methods). This observation is probably caused by balancing selection that tends to maintain the high diversity in MHC.
HLA gene deletions
Deletion of HLA-Y (Y-del) is the most common gene deletion among HLA genes (71.3% in HPRC and 87.3% in CPC). It is rarely studied in large cohorts because the human reference genome GRCh38 does not contain this gene. With long-read assembled haplotypes, we found that HLA-Y was highly associated with specific HLA-A allele groups: A*29, A*30, A*31, A*33, and A*68 (Fig. 4A). This association was especially strong in Han Chinese samples in that all A*29, A*30, A*31, and A*33 alleles are connected to the HLA-Y gene (Supplemental Fig. S4B).
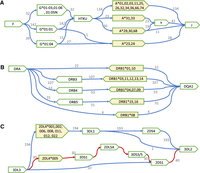
Local haplotype structures and their linkage to HLA/KIR alleles. (A) Haplotypes with the deletion of the HLA-H, -T, -K, and -U genes (HTKU-del) and the deletion of the HLA-Y gene. (B) DR haplotypes. (C) Haplotypes in the KIR telomere region.
The second most common HLA gene deletion we observed was the deletion of four pseudogenes: HLA-H, -T, -K and -U (HTKU-del), which was reported to be in linkage disequilibrium with HLA-A*23/24 and HLA-G*01:04 (Geraghty et al. 1992a,b; Carlini et al. 2016; Paganini et al. 2019). In our data, HTKU-del happened in 9.6% of HPRC assemblies and 16.4% of CPC assemblies, and it was highly linked to A*23/24. Different from earlier observations, we also found 13 G*01:04 haplotypes had HTKU genes, and 27 G*01:04 haplotypes had HTKU-del (Fig. 4A). We observed that all HTKU-del haplotypes had Y-del, but some Y-del haplotypes still carried the four pseudogenes, suggesting that the HTKU-del event might be more recent than Y-del. Using the read depth information, we genotyped HTKU-del events for samples from The 1000 Genomes Project (Supplemental Fig. S5; Byrska-Bishop et al. 2022). The estimated allele frequency of HTKU-del was 39.4% in the Japanese population (JPT), 18.8% in Chinese (CHS + CHB), and 14.3% in African and American together.
Protein-coding gene MICA may also be deleted. MICA functions as a stress-induced ligand for integral membrane protein receptor NKG2D (Bauer et al. 1999). The deletion of MICA has low frequency in HPRC (3.2%) and CPC (1.9%) but has higher frequency in a Mexican population (MXL; 11.9%) and in a Peruvian population (PEL; 5.7%) (Supplemental Fig. S6). Additionally, we also found that ∼22.1% of all our MICA calls have incomplete ORF (Supplemental Table S7).
HLA DR haplotypes
The HLA class II region is highly structured (Klitz et al. 2003; Norman et al. 2017; Chin et al. 2023), which is commonly characterized by DR haplotype. Following earlier work (Houwaart et al. 2023), the HLA DR haplotype is defined by the combination of HLA-DRB1 allele groups and the presence or absence of DRB3/4/5 genes. All 217 HLA haplotypes in our analysis are in agreement with the DR haplotype definition: DRB1*03, DRB1*11, DRB1*12, DRB1*13, and DRB1*14 are always linked to DRB3 (DR3); DRB1*04, DRB1*07, and DRB1*09 are linked to DRB4 (DR4); DRB1*15 and DRB1*16 are linked to DRB5 (DR5); and DRB1*01, DRB1*10, or DRB1*08 never carry DRB3/4/5 alleles (DR1 + DR8) (Fig. 4B).
We further explored the polygenetic relationship of DRB1/3/4/5 alleles based on CDS differences (see Methods). The DRB1 alleles from each of the DR3, DR4, and DR5 groups formed a separate clade, but the DRB1 alleles from the DR1 group were split into two different clades, suggesting two different evolutionary paths leading to the formation of the DR1 haplotypes in human (Supplemental Fig. S7).
C4 genotypes
Immuannot also annotates one MHC class III gene, complement C4. A C4 gene consists of 41 exons, encoding a transcript of 5.4 kb in length. The human reference genome GRCh38 has two C4 genes, C4A and C4B, which have different chemical reactivities and binding affinities in interacting with antigens (Yu et al. 1986; Blanchong et al. 2001; Wang and Liu 2021). Both C4A and C4B may have two forms: a long form (L) of 20.6 kb in length on GRCh38 or a short form (S) of 14.6 kb. The long-form C4 gene harbors a 6.2 kb insertion of endogenous retrovirus sequences in the ninth intron, whereas the short-form C4 gene does not (Yu et al. 1986; Blanchong et al. 2001). Therefore, there are four possible types of C4 genes: C4AL, C4AS, C4BL, and C4BS. Their copy numbers vary in different individuals, and the copy-number changes may result in different disease risks (Dodds et al. 1996; Jaatinen et al. 1999; Blanchong et al. 2001; Yang et al. 2004; Breukink et al. 2015; Sekar et al. 2016; Jüptner et al. 2018). Immuannot annotates all four types of C4 genes.
In our data, C4A achieved a frequency of 57%. Every haplotype had at least one copy of C4A gene, and no haplotypes had more than two C4A genes or more than two C4B genes. The frequencies of monomodular (one copy of C4), bimodular (two copies of C4), and trimodular (three copies) were 13.9%, 79.8%, and 6.3%, respectively, for HPRC and were 8.8%, 71.9%, and 19.3% for CPC. No quadrimodular case (four copies of C4) was detected (Supplemental Table S9).
Human C4A and C4B genes are >99% identical. In the phylogenetic tree constructed from the CDS of all C4 genes sequences in our data, C4A and C4B were clearly separated, but the long and short forms were mixed in both the C4A and C4B clades (Supplemental Fig. S8). In addition to the five SNPs in exon 26 that are often used to distinguish C4A and C4B, four SNPs in exon 28 can also separate most, although not all, C4A and C4B genes. The imperfect linkage suggests possible gene conversions or recombination between exon 26 and exon 28 (Supplemental Table S10).
KIR haplotypes
We collected 234 full-length KIR haplotypes that are defined by including both 3DL3 and 3DL2 in one contig. We observed 25 unique KIR haplotypes, including the most common A-haplotype at 66.2% frequency and 24 B-haplotypes. The B-haplotypes may contain five to 15 KIR genes, including one to five activator genes in addition to 2DL4. We also observed the deletion (n = 10) and duplication (n = 6) of two framework genes, 3DP1 and 2DL4 (Fig. 5). Among the 44 samples with both KIR haplotypes fully assembled in HPRC, 18 carry two A-haplotypes (AA carriers), 21 were AB carriers, and only five were BB carriers.
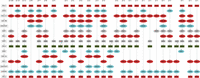
KIR haplotypes. Boxes indicate framework genes; ellipses, nonframework genes. Red indicates inhibitor genes; blue, activator genes; gray, pseudogenes; and green, genes that can be both inhibitor and activator.
Because of frequent recombination between genes 3DP1 and 2DL4 (Cisneros et al. 2020), we further split the KIR region into a centromere region (c-region; from 3DL3 to the first 3DP1) and telomere region (t-region; from the last 2DL4 to 3DL2). We observed 15 unique haplotypes in the c-region, and 11 of them were rare (two or fewer copies); we observed five unique haplotypes in the t-region, and one of them was rare (two or fewer copies). The common c-region haplotypes and common t-region haplotypes (more than two copies) explained >95% of all KIR haplotypes, as we observed (Supplemental Fig. S9). Interestingly, the gene 2DL4 in B-haplotypes always had the allele 2DL4*005 (Fig. 4C), in line with previous studies (Amorim et al. 2021).
Evaluating T1K genotyping with short reads
There are 40 HPRC samples that also have short-read WGS data from The 1000 Genomes Project. We took the Immuannot annotation as the ground truth to evaluate the performance of genotypers that use short reads only. We in particular focused on the results from T1K (Song et al. 2023). We did not evaluate other genotypers because most of them only work for classical HLA genes, could not genotype KIR genes, and have been evaluated in other benchmark studies (Klasberg et al. 2019; Liu et al. 2021; Thuesen et al. 2022; Yu et al. 2023).
We carefully evaluated T1K for each of HLA and KIR genes (Fig. 6) and also summarized in different categories (Supplemental Table S11). For classical HLA genes (HLA-A, -B, -C, -DRB1, -DQB1, -DPB1) that were well-curated in IPD-IMGT/HLA, T1K achieved an average sensitivity of 97.3% and a precision of 98.1–98.5%, in which the precision had a range of values owing to different evaluation strategies for novel alleles (see Fig. 6 caption). For all HLA genes, the average sensitivity of T1K is 95.5%, and the precision is 87.0%–97.5%. The decreasing precision was probably related to novel alleles not in the IPD database. For KIR genes, T1K achieved a sensitivity of 91.2% and precision of 83.4%–89.0%, lower than the accuracy for HLA genes. This could be caused by the high copy-number variations in KIRs that were not directly modeled in T1K's genotyping algorithm. When ignoring the copy numbers (i.e., just match the allele prediction), the sensitivity and precision increased to 93.8% and 88.0%–93.2%, respectively. In sum, our benchmark suggested that genotyping with short reads yielded highly accurate results for classical HLA genes but remained a challenge for other highly polymorphic genes.
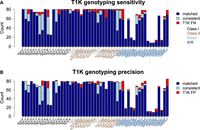
T1K evaluation for HLA/KIR genes. (A) Sensitivity of T1K on HPRC samples. Immuannot HPRC annotation is taken as the ground truth. An Immuannot allele and a T1K allele are “matched” if both alleles correspond to the same protein sequence. Synonymous or noncoding differences are ignored. An Immuannot allele and a T1K allele are “consistent” if one allele leads to a novel protein sequence absent from the IPD databases and the two alleles come from the same allele group (in case of HLA genes) or are the same gene (in case of KIR genes). A “T1K FN” is an allele called by Immuannot that is not matched by or consistent with a T1K allele. The copy number of homozygous calls is considered in this comparison: If Immuannot annotates a gene to have two copies but T1K only finds one, this is also considered a FN call. (B) Precision of T1K. A “T1K FP” is an allele called by T1K that is not matched by or consistent with an Immuannot allele.
Discussion
Immuannot is a convenient and efficient computational tool to annotate both HLA and KIR genes on assembled contigs at full resolution. It provides the coordinates and structures of these genes, types known as gene alleles in the IPD-IMGT/HLA and the IPD-KIR databases, and also reports novel alleles. The Immuannot annotations are highly consistent with existing HLA and KIR annotations in previous work.
Immuannot relies on high-quality phased assemblies that can only be produced with PacBio HiFi reads for the time being. It also does not report incomplete genes caused by assembly gaps. Nonetheless, with reduced sequencing cost, improved Nanopore read accuracy, and refined assembly algorithms, we anticipate more phased assemblies to be produced in the near future. Relatedly, Immuannot may misidentify a novel allele caused by base errors in contigs. Although, based on the analysis of error rates, we inferred that the great majority of novel CDS alleles should be real, independent experimental validation would still be preferred.
The Immuannot algorithm also has room for improvement. For example, it may miss a gene if a long insertion/deletion on a novel allele fragments minimap2 alignment. Although this issue can be naturally addressed with more complete IPD databases, algorithm improvement, such as jointly considering CDS alignment at the template finding step, may help to alleviate the problem as well.
Immuannot can only annotate HLA and KIR genes for now. Nonetheless, it provides an efficient framework to conduct assembly-based annotation/genotyping when multiple reference gene sequences are presented. For example, PharmGKB (Hewett et al. 2002) collected 13 clinically important genes covering 1906 unique sequences representing different alleles. It would be interesting to know which alleles are carried by the target genome assembly for pharmacogenetics studies.
Even though HLA and KIR regions have been studied for decades, many HLA or KIR genes may not be well-understood owing to challenges in allele typing, and they may benefit from further study in large cohorts. For example, many South Americans lack MICA genes but few Europeans have this deletion. It is important to further check whether it is an adaptation process or random drift in the South Americans. Thanks to new long-read data types and more sophisticated assembly algorithms, high-quality phased assembly will become a norm. We believe that Immuannot will provide high-fidelity HLA and KIR annotations to improve the completeness of IPD-IMGT/HLA and IPD-KIR and to enhance the research on these genes in general.
Methods
Identifying candidate genes
Immuannot aligns known gene sequences in existing databases (which are called “query sequences”) to assembled contigs (called “target sequences”) with minimap2. As some genes, such as KIR2DL4 and C4, may have copy-number variations, Immuannot allows one query sequence to be aligned to multiple locations on the target sequences. For each alignment, Immuannot calculates a gap-compressed identity as m/(m + d), where m is the number of matching bases in the alignment, and d is the number of mismatching bases and gaps, regardless of gap length. Immuannot ranks alignments by the gap-compressed identity and then by the alignment length. It creates clusters of alignment by iterating alignments from the best to the worst. For a given alignment, Immuannot adds it to existing clusters if it overlaps with existing ones, or Immuannot creates a new cluster. Alignments that overlap with more than one existing cluster will be skipped because we only use full-length gene sequences, and we also require a high mapping rate (>90%). In the end, each cluster ideally represents a candidate gene. This greedy procedure assumes genes are well-separated on the genome, which has not been an issue for HLA and KIR genes.
Typing genes in the IPD-IMGT/HLA and IPD-KIR databases
At present, Immuannot uses IPD-IMGT/HLA Database v3.52.0 and IPD-KIR v2.12 for typing. The two databases included 20,393 gene sequences and 38,684 partial/full CDS sequences. To fully leverage the IPD resources, only full-length gene sequences are used for gene structure identification, and all available CDSs are used for allele calling. Within each alignment cluster, Immuannot annotates gene structures and type alleles in the following three steps.
-
Annotating gene structures. Immuannot filters a gene sequence if <90% of the sequence is mapped or its gap-compressed identity is <97%. The remaining gene sequences are classified into four groups in decreasing priority: with perfect alignment to the target contig, with all CDS aligned and without gaps in CDS, with all CDS aligned but with frameshifts, and with some CDS unaligned. If there are multiple sequences in one priority group, the gene sequence with fewest gap-compressed differences and then with longest alignment length is chosen as the template allele. The gene structure of template allele is lifted to the target contig to define the gene structure on the contig.
-
Allele typing. If the template allele in step 1 perfectly matches the target contig in full length, the template allele is concluded as the final allele call. Otherwise, CDS on the target contig is extracted based on the annotated gene structures in step 1. The known CDS alleles in the IPD databases are aligned to the target CDS and are ranked by edit distance and then by alignment length. The partial CDS alleles are used only when the alignments cover the full length of query CDS and have no mismatches. Immuannot may report multiple closest alleles for a gene if their alignments to the contig have identical edit distance and alignment length.
-
Naming novel alleles. For a novel allele that is not present in the IPD databases, Immuannot may name it with a “new” keyword. For an HLA gene, if the best-matching alleles identified in step 2 come from multiple allele groups (i.e., the first fields in their names are different), Immuannot will call the allele as “HLA-X*new,” where “HLA-X” is the gene name. Otherwise, Immuannot finds the longest prefix among names of the best-matching alleles and assigns “new” to the second/third/fourth field, if the protein/CDS/noncoding sequence is affected, respectively.
Typing C4 genes
Typing C4 gene is easier than typing other HLA and KIR genes because Immuannot only attempts to distinguish between C4A and C4B and between the long form and the short form. It extracts the C4A transcript sequence on NG_011638.1 and aligns the sequence to assembled contigs in the splice mode to annotate C4 gene structures. Immuannot distinguishes C4A and C4B based on four amino acid differences between the two genes on exon 26 (Blanchong et al. 2001; Sekar et al. 2016; Wang and Liu 2021). It considers the gene to have a long form if the length of intron 9 is >5 kb; otherwise Immuannot denotes the gene to have a short form.
Population CDS divergence calculation
Gene divergence and diversity are calculated based on the CDS of each gene per sample. CDS sequences are extracted by gffread (Pertea and Pertea 2020), with the assemblies and our annotation GTF file as input. Then for each gene, CDSs are aligned by kalign3 (Lassmann 2019) for each data panel (HPRC and CPC). Based on the aligned CDS, the pairwise difference per sample is calculated as the average Hamming distance to the remained samples. The per gene diversity is calculated as the average of all pairwise differences of that gene.
Construct HLA-DRB polygenetic tree
Full-length CDS of DRB1, DRB3, DRB4, and DRB5 are collected from HPRC, CPC, CHM13.0, GRCh38.p14, CN-T2T, IPD-IMGT/HLA, and IPD-IMGT/MHC, including 198 HLA-DRB1, seven HLA-DRB3, four HLA-DRB4, and six HLA-DRB5 unique CDSs in total. DRB CDSs from non-human primate species, chimpanzee (patr), crab-eating macaque (mafa), and rhesus monkey (mamu), are used as the outgroup for tree construction; the most recent common ancestry of all outgroup sequences is set as the root in the phylogenetic tree. Kalign3 is used for the multiple sequence's alignment, and IQ-TREE 2 (Kalyaanamoorthy et al. 2017) is used for the tree construction with the option “-m MFP” to select the best-fit model. Tree plot is generated with jsTree view.
Estimate HLA gene deletion frequency in The 1000 Genome Project data
To detect the deletion of HLA genes in The 1000 Genome Project data populations, we calculated the median read depth of the target gene region and 2000 randomly selected regions within Chr 6:20 Mb–50 Mb as the controls that were supposed to be diploid. The read depth of a particular gene is normalized in two steps to reduce within- and between-sample variance. In step 1, the read depth of the gene is divided by half of the median read depth in the control regions of that individual. For a diploid individual without copy-number changes, this procedure would normalize the original read depth to a number close to 2. In step 2, the normalized copy number from step 1 is divided by the median normalized copy number across all samples, assuming that the majority of the samples carry two copies of the targeted gene. This method turns out to work well to distinguish the copy-number variation for the four-pseudogene deletion (HTKU-del) and the MICA deletion (Supplemental Fig. S10).
Copy-number estimation of T1K's call
The copy number of a gene is inferred from the abundance estimate outputted by T1K. To infer the copy number, we first apply
the square root to the abundance such that the resulting values approximately follow a normal distribution. We then use the
smallest 30% of the transformed allele abundances to infer the mean and standard variance, denoted as μ and σ, respectively.
This step can also incorporate the user-provided genes that are expected to have two copies. In this work, we assumed three
HLA genes (A, B, and C) for HLA allele calling and four KIR framework genes (3DL3, 2DL4, 3DP1, and 3DL2) for KIR allele calling. After obtaining the parameters of the normal distribution, our script will calculate the likelihood
for each allele's copy number c based on the formula
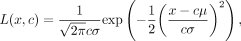 where x is the transformed abundance of an allele and c is the copy number; this formula is based on the additive property of normal distribution. Then, the copy number of this
allele will be the value c that maximizes the likelihood—for example, argmaxc L(x, c).
where x is the transformed abundance of an allele and c is the copy number; this formula is based on the additive property of normal distribution. Then, the copy number of this
allele will be the value c that maximizes the likelihood—for example, argmaxc L(x, c).
Data sets
The 94 HPRC assemblies were downloaded from https://github.com/human-pangenomics/HPP_Year1_Assemblies; the 114 CPC assemblies were obtained from the National Genomics Data Center (https://ngdc.cncb.ac.cn) under accession number PRJCA011422. Fifty KIR contigs were also available from GenBank (accessed February 28, 2023); the contig names are NT_113949.2, NW_003571054.1, NW_003571055.2, NW_003571056.2, NW_003571057.2, NW_003571058.2, NW_003571059.2, NW_003571060.1, NW_003571061.2, NT_187636.1, NT_187637.1, NT_187638.1, NT_187639.1, NT_187640.1, NT_187641.1, NT_187642.1, NT_187643.1, NT_187644.1, NT_187645.1, NT_187668.1, NT_187669.1, NT_187670.1, NT_187671.1, NT_187672.1, NT_187673.1, NT_187674.1, NT_187675.1, NT_187676.1, NT_187677.1, NT_187683.1, NT_187684.1, NT_187685.1, NT_187686.1, NT_187687.1, NT_187693.1, NW_016107300.1, NW_016107301.1, NW_016107302.1, NW_016107314.1, NW_016107313.1, NW_016107312.1, NW_016107311.1, NW_016107310.1, NW_016107309.1, NW_016107308.1, NW_016107307.1, NW_016107306.1, NW_016107305.1, NW_016107304.1, and NW_016107303.1. The eight MHC reference contigs (Houwaart et al.’s data) (Houwaart et al. 2023) were obtained from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA764575. NA12878 HIFI reads were downloaded from the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number SRP194450.
Software availability
Immuannot is available at Github under an MIT license (https://github.com/YingZhou001/Immuannot). The software source code and scripts to reproduce the major results can be found in the Supplemental Code.
Data access
HLA and KIR gene annotations generated in this work can be found at Zenodo (https://zenodo.org/records/8372992) and as Supplemental Data; extended results and detailed information can be found in the Supplemental Material.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This project is supported by National Human Genome Research Institute (NHGRI) grant R01HG010040 and U01HG010961 to H.L. and by National Institute of General Medical Sciences (NIGMS) grant P20GM130454 to Dartmouth (L.S).
Author contributions: H.L. conceived and designed the project. Y.Z. implemented the software Immuannot. L.S. provided the script for copy-number estimation of T1K's call. Y.Z. conducted the analyses with input from L.S. and H.L. Y.Z., L.S., and H.L. wrote the manuscript. All the authors agreed on the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278985.124.
- Received January 12, 2024.
- Accepted May 22, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








