
Long-read genome sequencing and variant reanalysis increase diagnostic yield in neurodevelopmental disorders
- Susan M. Hiatt1,
- James M.J. Lawlor1,
- Lori H. Handley1,
- Donald R. Latner1,
- Zachary T. Bonnstetter1,
- Candice R. Finnila1,
- Michelle L. Thompson1,
- Lori Beth Boston1,
- Melissa Williams1,
- Ivan Rodriguez Nunez1,
- Jerry Jenkins1,
- Whitley V. Kelley1,
- E. Martina Bebin2,
- Michael A. Lopez2,3,4,
- Anna C.E. Hurst4,
- Bruce R. Korf4,
- Jeremy Schmutz1,
- Jane Grimwood1 and
- Gregory M. Cooper1
- 1HudsonAlpha Institute for Biotechnology, Huntsville, Alabama 35806, USA;
- 2Department of Neurology, University of Alabama at Birmingham, Birmingham, Alabama 35924, USA;
- 3Department of Pediatrics, University of Alabama at Birmingham, Birmingham, Alabama 35924, USA;
- 4Department of Genetics, University of Alabama at Birmingham, Birmingham, Alabama 35924, USA
Abstract
Variant detection from long-read genome sequencing (lrGS) has proven to be more accurate and comprehensive than variant detection from short-read genome sequencing (srGS). However, the rate at which lrGS can increase molecular diagnostic yield for rare disease is not yet precisely characterized. We performed lrGS using Pacific Biosciences “HiFi” technology on 96 short-read-negative probands with rare diseases that were suspected to be genetic. We generated hg38-aligned variants and de novo phased genome assemblies, and subsequently annotated, filtered, and curated variants using clinical standards. New disease-relevant or potentially relevant genetic findings were identified in 16/96 (16.7%) probands, nine of which (8/96, ∼9.4%) harbored pathogenic or likely pathogenic variants. Nine probands (∼9.4%) had variants that were accurately called in both srGS and lrGS and represent changes to clinical interpretation, mostly from recently published gene-disease associations. Seven cases included variants that were only correctly interpreted in lrGS, including copy-number variants (CNVs), an inversion, a mobile element insertion, two low-complexity repeat expansions, and a 1 bp deletion. While evidence for each of these variants is, in retrospect, visible in srGS, they were either not called within srGS data, were represented by calls with incorrect sizes or structures, or failed quality control and filtration. Thus, while reanalysis of older srGS data clearly increases diagnostic yield, we find that lrGS allows for substantial additional yield (7/96, 7.3%) beyond srGS. We anticipate that as lrGS analysis improves, and as lrGS data sets grow allowing for better variant-frequency annotation, the additional lrGS-only rare disease yield will grow over time.
Although genome and exome sequencing (GS/ES) are increasingly used to identify molecular diagnoses for rare diseases, reported diagnostic rates range from 20% to 60% (Srivastava et al. 2019; Baxter et al. 2022), indicating that many conditions suspected to be genetic remain refractory to genomic testing. While some tested individuals may have phenotypes resulting from polygenic and/or environmental risk factors (e.g., Niemi et al. 2018), a subset of undiagnosed cases likely result from genetic factors that we are as-yet unable to identify. It is well-known that short-read genome sequencing (srGS) has poor sensitivity to many types of variants, especially structural variants (SVs) and variants affecting repetitive sequences (Sanghvi et al. 2018; Wenger et al. 2019; Mahmoud et al. 2024). Long-read genome sequencing (lrGS), in contrast, has been shown to greatly improve sensitivity to many of the variants missed by srGS (Logsdon et al. 2020), in addition to facilitating de novo assemblies to allow for more effective evaluation of structural variation (Cheng et al. 2021). Accordingly, lrGS has great potential to improve rare disease diagnostic testing and has been applied to several rare disease cohorts (Hiatt et al. 2021; Miller et al. 2021; Cohen et al. 2022).
In addition to changes in sequencing technology, the scope of knowledge about genes and our ability to annotate genetic variants has steadily increased. As such, systematic reanalysis of GS/ES data can also lead to the discovery of previously overlooked clinically relevant variants. Diagnostic yield increases from reanalysis have been reported to range from 4% to 31% depending on a variety of factors, most notably time since the previous analysis (Hiatt et al. 2018; Liu et al. 2019; Schobers et al. 2022; Hartley et al. 2023). While a variety of factors contribute to reanalysis discoveries, they often result from the discovery of new disease genes, which contributes to 42%–75% of reanalysis findings (Hiatt et al. 2018; Liu et al. 2019; Schobers et al. 2022; Hartley et al. 2023). This reflects the rapid pace of discovery of new disease genes in the rare disease research community, which has been facilitated by data sharing via the Matchmaker Exchange and GeneMatcher (Philippakis et al. 2015; Sobreira et al. 2017).
Here, we discuss findings from lrGS on a cohort of 96 short-read-negative cases, drawn from several studies focused on rare, suspected genetic diseases, especially early onset neurodevelopmental disorders (NDDs). We describe 19 relevant or potentially clinically relevant variants not previously evaluated or considered in 16 cases. We show that both more comprehensive variant detection from lrGS and variant reanalysis contribute to these discoveries. However, lrGS clearly provides unique advantages, and these advantages are likely to grow in the future, to maximize the rate of discovery of highly penetrant variation in any given individual with rare disease.
Results
We selected individuals with rare diseases who had undergone short-read exome sequencing (srES; n = 2) or srGS (n = 94) in previous research studies yet had no pathogenic or likely pathogenic variants (P/LP) nor variants of uncertain significance (VUS) identified (Bowling et al. 2017, 2022; East et al. 2021). Most of our cohort consisted of children (89% were <18 years of age at the time of enrollment) with an NDD (70%), multiple congenital anomalies (MCA, 22%), or a suspected genetic myopathy (8%). Probands consisted of 66% males (63/96); genetically inferred ancestries for probands revealed 72% European (69/96), 21% African/African American (20/96), 3% Admixed American (3/96), 1% Southeast Asian (1/96), and 3% unspecified ancestry admixture (Table 1). For these 96 cases, we performed lrGS using Pacific Biosciences “HiFi” sequencing to a median depth of 27× (Table 1; Supplemental Table S1). For a subset (10/96), we also performed lrGS on parents (median parental HiFi depth of 22×) (Supplemental Table S2). We also generated de novo assemblies for each proband using hifiasm (Cheng et al. 2021), with parental srGS used for k-mer-based binning and phasing when available. While the assemblies were not used for SV calling in this study, they have proven useful in visualizing and evaluating complex structural variation (Hiatt et al. 2021). The median N50 for assembled proband contigs was 29.05 Mb (Table 1).
Demographics, and sequencing and assembly metrics for the cohort
HiFi reads were aligned to hg38, and variant calling was performed using DeepVariant (Poplin et al. 2018) and pbsv (https://github.com/PacificBiosciences/pbsv, see Methods). A median of 4.4 million single nucleotide variants (SNVs) and 970,031 indels were called in each proband (Table 1). We detected a median of 55,586 SVs of varying classes across the 96 probands using pbsv, of which a median of 25,218 are >50 bp in length (Table 1; Supplemental Table S3). These counts do not include any variant-quality filtration metrics and are expected to include an undetermined fraction of false positive calls; only variant calls of potential disease interest were evaluated for read support and quality (see Methods). Given that SVs are commonly arbitrarily defined as >50 bp in length, our counts generally are consistent with previous studies that have detected a range of 22,000–33,000 SVs per genome (Pauper et al. 2021; Cohen et al. 2022; Groza et al. 2024).
Findings from lrGS
lrGS SNVs/indels were annotated with features such as gene overlaps, coding consequences, computational impact scores, and allele frequencies. They were then filtered and analyzed using in-house software that is also used for srGS data (Hiatt et al. 2021). Rare SVs (see Methods) were assessed by visualization of reads in Integrative Genomics Viewer (IGV) and prioritization and analysis using SvAnna (Danis et al. 2022). All variants of interest were subject to curation using the American College of Medical Genetics and Genomics and Association for Molecular Pathology (ACMG/AMP) and ClinGen criteria to identify potentially clinically relevant variation (Richards et al. 2015; Riggs et al. 2020). We ultimately identified 19 potentially “clinically relevant” variants, defined here as being pathogenic, likely pathogenic, or variants of uncertain significance (P/LP/VUS), in 16 of the 96 cases (Table 2). Seven of these have a case-level classification of Definitive Diagnostic or Likely Diagnostic, which we define as P/LP variants that likely fully explain the reason for testing (Bowling et al. 2022). The remaining nine cases have Uncertain case-level classifications, either due to the variants being VUSs or being P/LP variants in genes whose associated phenotypes do not closely match the observed phenotype. Findings in seven probands exemplify the unique benefits of lrGS and are highlighted below.
Variants identified by long-read sequencing
lrGS-informed SVs
lrGS uncovered a de novo, 4 Mb, copy-neutral, paracentric inversion on Chromosome 3 (NC_000003.12:g.110477273_114639202_inv) in proband 1 (Fig. 1). This inversion spans about 35 protein-coding genes, and one breakpoint of this inversion lies within an intron of ZBTB20 (MIM: 606025), between exons 6 and 7 in the 5′ UTR of NM_001348800.3. The event breakpoints are visible in both long and short reads for this proband, but not in short reads for either parent, suggesting it arose de novo. Phasing in long-read data indicates the inversion is on the proband's paternal allele (Fig. 1A). While evidence of the breakpoints is visible in the proband's srGS data (Fig. 1A,B), and the breakpoints are called by manta (v1.6.0) (Chen et al. 2016), the event only survived filtration and curation in lrGS (see Discussion; Supplemental Fig. S1, which compares inversion and breakend calls between srGS and lrGS for this proband). The variant is private to the proband and is predicted to disrupt ZBTB20. While the breakpoint lies between two UTR exons, it moves and flips the transcript's promoter and the first six exons away from the remaining exons (Fig. 1C). This inversion is easily visualized in alignment of the proband's paternal contig versus the reference genome in this region (Fig. 1D). Loss-of-function (LOF) variation in ZBTB20 is associated with Primrose Syndrome (MIM: 259050) and 3q13.31 Microdeletion Syndrome (Juven et al. 2020). Proband 1's reported features include moderate intellectual disability (ID), delayed speech and language development, muscular hypotonia, strabismus, and hypoplastic corpus callosum. She is also nonambulatory. Several of these features overlap Primrose Syndrome. We have classified this variant as likely pathogenic and the case-level designation is Likely Diagnostic (Table 2).
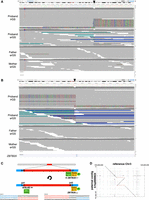
A de novo, 4 Mb paracentric inversion in proband 1, affecting ZBTB20. (A,B) Visualization of a subset of proband and parent reads in IGV at the 5′ (A, Chr 3:110,477,108–110,477,357) and 3′ (B, Chr 3:114,639,120–114,639,284) breakpoints (black arrowheads) indicate a de novo event. Reads in A show the SV is present on the proband's paternal allele. (C) Schematic of the proband's inversion. Section B–C is inverted, and junctions 1 and 2 (jct1, jct2) are displayed. The inversion disrupts ZBTB20, with a promoter (P) and exons 1–6 of NM_001348800.3 (yellow boxes) remaining at the distal end of the chromosome near jct2/D and exons 7–12 of NM_001348800.3 (green boxes) moving upstream to the breakpoint at the more proximal end of the chromosome (near jct1/A). Sequence level resolution of the inversion is also shown. (D) Alignment of the proband's assembled paternal contig versus the reference genome supports the inversion.
Proband 2 was originally enrolled for sequencing at the age of ∼20 years, and presented with spasticity, ataxia, and leukodystrophy. srGS was negative, but lrGS identified two SVs in trans in ALS2 (MIM: 606352). These include a maternally inherited 4.65 kb deletion (Chr 2:201720435-201725085_del) that removes exons 21–23 of NM_020919.4, and a paternally inherited ∼1.6 Mb deletion (Chr 2:200115181-201739349_del) that spans several genes, including the 3′ end of ALS2 (deletion of exons 12–34 of NM_020919.4) (Table 2; Fig. 2A; Supplemental Figs. S2–S5). These deletions are easily visualized in the alignment of the proband's maternal (Fig. 2B) and paternal (Fig. 2C) contigs versus the reference genome in this region. While these variants were called in short-read data, the smaller deletion was called as a heterozygous deletion in the mother and as a homozygous deletion in the proband, obfuscating the nature of the variation and raising quality control concerns (e.g., nested and conflicting copy-number states and Mendelian inconsistency) that prevented effective curation (Supplemental Figs. S6, S7). Given the results of the lrGS, the srGS variant calls logically resulted from the small maternal deletion intersecting with the larger, overlapping paternal deletion. This case highlights the difficulties in the identification and analysis of overlapping SVs of unknown phase. Variation in ALS2 is associated with several AR conditions (Juvenile amyotrophic lateral sclerosis 2, MIM: 205100; Juvenile Primary lateral sclerosis, MIM: 606353; and infantile-onset ascending Spastic paralysis, MIM: 607225), each of which has features that overlap the proband's presentation. These variants are classified as P/LP and given the degree of overlap with expected phenotypes, the case-level designation is Definitive Diagnostic.
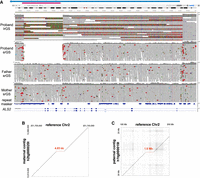
Two ALS2 deletions in trans in proband 2. (A) Visualization of proband and parent reads in IGV (Chr 2:201,719,400–201,741,000) indicate two overlapping deletions in ALS2; a smaller maternal deletion (pink bar) and a larger paternal deletion (blue bar/arrow). Note that the three breakpoints shown here overlap Alu sequence in the RepeatMasker track. Alignment of the proband's assembled maternal (B) or paternal contig (C) versus the reference genome supports the two deletions (red dashed lines).
Proband 3 is a male, enrolled in our research study at age ∼50 years, and has a strong X-linked family history of ID (Fig. 3A). srES and srGS were both negative, although in srES, two neighboring SNVs of uncertain significance were called, and manually curated, in the 3′ UTR of HCFC1 (MIM: 300019). While srGS resulted in no calls in this region, visualization of reads in IGV suggested an insertion of unknown length and consequence (Fig. 3C). Variation (mostly missense and proposed regulatory variation) in HCFC1 has been associated with X-linked recessive Methylmalonic aciduria and homocysteinemia, cblX type (MIM: 309541). However, most affected individuals present with severely delayed psychomotor development, seizures, and methylmalonic aciduria. Proband 3's family reported neither of the latter two features. Given the uncertainties in the identity, structure, and consequence of the potential variants seen in srES and srGS, and the lack of clear phenotypic relevance for the gene, this region was curated but not originally considered to be a strong candidate for clinical relevance. HiFi sequencing identified a 4902 bp mobile element insertion (MEI) in the 3′ UTR of HCFC1 (Chr X:153948602_ins4902), consisting of both SVA and L1 sequence (Fig. 3B). This variant was likely inherited from a heterozygous carrier mother, as indicated by srGS reads at the breakpoints (Fig. 3C). While this insertion does not affect the protein-coding sequence, it is predicted to increase the length of the 3′ UTR from 1791 to 6693 nt. We subsequently performed 3′-end RNA-seq on blood samples from both the proband and his father, generating ∼8 million reads for each sample (see Methods). HCFC1 shows the greatest expression decrease in the proband relative to his father, an ∼8.8-fold reduction, among all genes with at least 10 counts in each sample (Supplemental Fig. S8). While these results are consistent with the hypothesis that the insertion has a large effect on HCFC1 expression and potential activity, they are not definitive. Further, expression or segregation analyses in additional family members could not be assessed. Given the uncertainty of the molecular consequence, the differences between observed phenotypic features and those reported to associate with HCFC1, and the lack of additional segregation data, we classified this as a VUS, with a case-level designation of Uncertain.
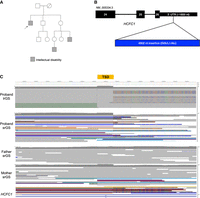
Proband 3 has a 4 kb insertion in the 3′ UTR of HCFC1. (A) The proband's family has a history of X-linked ID, as the proband (arrow) and two other male relatives (gray squares) are affected. (B) Model of the relative length of the insertion in the 3′ UTR NM_005334.3. Only exons 24–26 are shown. (C) The hemizygous insertion is likely inherited from a heterozygous carrier mother, as indicated by srGS reads. Note that some reads contain unaligned ends (multicolored bases) while some span the entire insertion (purple line at the 5′ end of the target site duplication [TSD]); all the proband's reads support the insertion. A TSD is a hallmark of an L1-mediated insertion.
Proband 4 has a complex de novo SV affecting 16p13.2 (NC_000016.9:g.(8742452_9220783)dup_ins[(8742452_8879961)_(9000190_9220783)]) (Table 2; Supplemental Figs. S9–S18). Proband 4 first had trio srES and no variants were returned, and this SV was not called. Trio lrGS identified the breakpoints, de novo status, phase, probable order, orientation, and copy number of segments in this SV. However, there remains some uncertainty about the SV, as neither manual curation nor the proband's de novo assembly could definitively resolve the full, exact structure of the locus. Two possible structures are shown in Supplemental Figure S9. We believe that this is likely a limitation due to the length of the reads, but it may also be influenced by low coverage in this proband (∼17×), lack of parental srGS data for hifiasm input, and a small number of informative variants near the breakpoints to facilitate phasing.
Overlapping duplications in this region have been reported in gnomAD but are rare (Lek et al. 2016). One individual in Decipher (Patient: 251349) has also been reported with a very similar duplication of uncertain consequence (Deciphering Developmental Disorders Study 2015). This region spans seven genes, three of which are associated with disease: ABAT, PMM2, and USP7. The first (ABAT) is intersected by a duplication breakpoint in proband 4, but all other breakpoints lie within intergenic regions (Supplemental Figs. S9, S10). USP7 is the only gene associated with autosomal dominant disease (Hao-Fountain Syndrome, MIM:616863), but this gene is expected to remain at a copy number of two in this proband, whereas LOF is generally the mechanism associated with disease (Hao et al. 2015). Some general features of Hao-Fountain Syndrome overlap this proband, but it is not a strong phenotypic fit. The proband is reported to have moderate ID, seizures, microcephaly, and facial dysmorphisms. Based on the uncertain global structure and molecular consequence of the SV in this proband, in addition to the clinical significance of variation in this region, we have classified this variant as a VUS, with a case-level designation of Uncertain. Note that this variant is pending orthogonal validation. Proband 4 was one of the two individuals who only previously had srES rather than srGS, and this duplication is not easily visualized in srES data (Supplemental Fig. S19).
lrGS-informed repeat expansions
Improved variant calling in repeat regions is also a benefit of lrGS (Nurk et al. 2022). In addition to the analysis of SNVs and SVs in our standard pipeline, we assessed variant calls in 66 tandem repeat expansion (TRE) regions, including both known disease-associated and candidate disease-associated loci (Supplemental Table S4). We intersected TRE regions of interest with pbsv-called variants in each individual and compared these calls to known pathogenic expansion sizes from the literature. We identified several large heterozygous insertions in repeat regions that, while longer than the expected pathogenicity threshold, were predicted after manual curation to be benign based on their sequence content (Supplemental Fig. S20; Nakamura et al. 2020). In two probands, we identified large heterozygous insertions in RFC1 (MIM: 102579), one of which is benign based on sequence content and one of which is expected to be a pathogenic insertion. However, RFC1-associated disease (CANVAS, MIM: 614575) is caused by biallelic expansions, which we did not observe (Supplemental Fig. S20), suggesting the latter proband is merely a heterozygous carrier.
In proband 5, we observed a de novo 18 bp alanine tract expansion in PHOX2B (MIM: 603851, NM_003924.4:c.741_758dup, p.(Ala255_Ala260dup)) (Table 2; Supplemental Fig. S21), associated with Central Hypoventilation Syndrome, with or without Hirschsprung Disease (MIM: 209880). This disorder was clinically suspected, but variation in PHOX2B was missed by initial clinical genetic testing and srGS, which was performed on a PCR+ srGS library. As ExpansionHunter is intended to run on PCR-free srGS data, it was not run on this sample (Dolzhenko et al. 2019). This variant is reported as pathogenic in the literature (Amiel et al. 2003), is a strong match to the proband's observed symptoms, and was confirmed independently by additional clinical testing. We have classified this variant as pathogenic, with a case-level classification of Definitive Diagnostic.
In proband 6, we identified a 270 bp insertion in AFF3 (MIM:601464, NM_001386135.1:c.-64-281_-64-280insGGC[90]) (Table 2; Supplemental Fig. S22). While missense variants in AFF3 have been associated with KINSSHIP Syndrome (MIM:619297), an expansion of a CGG-repeat in the promoter of this gene and subsequent hypermethylation of the promoter has recently been reported to be associated with NDDs (Jadhav et al. 2023). Most probands in our cohort (77/96) had both alleles matching hg38 in this region (Supplemental Fig. S23). Among the 19 probands harboring nonreference alleles, proband 6 had a 270 bp insertion, and the remaining 18 probands had insertions ranging from 24 to 84 bp in length (8–28 triplet repeats). Similarly, when comparing to a larger database of in-house HiFi genomes (n = 266, “set 2,” see Methods), only 53/266 individuals harbor a nonreference allele; the longest insertions outside of proband 6 are 105 bp (35 repeats) and 93 bp (31 repeats), each in different individuals, while the median nonreference insertion is 36 bp (12 repeats). Jadhav et al. suggest that normal variation ranges to up to ∼38 repeat units, with ≥61 repeats being a likely pathogenic threshold. Thus, the 270 bp insertion (90 repeat units) in proband 6 is well above the normal range and pathogenicity threshold reported by Jadhav et al. and more than twice as long as the second longest insertion in our sample of 266 individuals, which is in the normal range reported by Jadhav et al. (Supplemental Table S4; Supplemental Fig. S23). Proband 6 was sequenced as a neonate and presented with intrauterine growth restriction (IUGR) and hypoplastic left heart (HLH); Jadhav et al. reported a wide range of symptoms, including ID/global developmental delays, seizures, behavioral disturbances, and generalized hypotonia, but the specificity of phenotypic overlap with proband 6 is unclear and neither supports nor refutes pathogenicity. While these results are consistent with the pathogenicity of the insertion in proband 6, there remains a need to further replicate and confirm the spectrum of normal and pathogenic variation in AFF3 repeat lengths. Given this uncertainty and the uncertainty regarding the proband's cognitive development, we have classified this TRE as a VUS, and the case-level classification is Uncertain. Also, note that this variant is pending orthogonal validation.
lrGS-informed SNV/indels
A de novo SHANK3 single-base deletion, predicted to lead to a frameshift (NM_033517.1:c.3161delT, p.(Leu1054Argfs*10)), was identified by lrGS in proband 7. While two reads in the srGS data support this deletion, the variant was not called by our srGS variant calling pipeline (Supplemental Fig. S24). LOF variation in SHANK3 (MIM: 606230) is associated with Phelan–McDermid syndrome (MIM: 606232). Features of this syndrome are consistent with the proband's features, and we classified this variant as pathogenic (case-level Definitive Diagnostic). Coverage of this region in short-read data does not indicate a systematic coverage deficiency, as mean coverage within 50 bp of this variant in the srGS data for the cohort is 27.9× (n = 94), while it is 15.1× for proband 7. Further, this gene is not present in the list of medically relevant genes that tend to be poorly covered by srGS (Wagner et al. 2022). These observations suggest the no-call may have resulted from a stochastic loss of alternative allele reads in the srGS data in this proband. Nevertheless, lrGS has been shown to provide better overall sensitivity and specificity to SNVs and indels in Genome-in-a-Bottle (GIAB) gold-standard data sets compared to srGS (Logsdon et al. 2020; Hiatt et al. 2021) and thus detection failures to variants such as this SHANK3 event are more likely in srGS data in general.
Reinterpretation of SNVs
The remaining nine cases had relevant variation identified following lrGS that were equally well detected through reanalysis of existing srGS variant data (Table 2; Supplemental Case Reports). In four cases (probands 8–11, HNRNPU, CSNK2B, GNB2, MCF2), we identified variants in genes that had additional published support for the association of the gene or the variant with the disease since the time of the most recent analysis. In another four cases (probands 12–15, NOTCH3, AFF4, KCNT2, KIF21A, NRXN1) we identified variants in established disease genes that conflicted with the published data regarding molecular mechanisms or expected mode of inheritance. Lastly, we identified a variant of interest in SCN1A that resulted from a targeted analysis of candidate “poison exon” variants (proband 16) (Felker et al. 2023). We note that in seven of these nine cases, variants were identified in an unaffected or mildly affected parent, which was somewhat unexpected in these cases due to suspicion of high penetrance (also see Supplemental Case Reports).
Cohort analysis of SVs
Individual SV case analyses were performed on a rolling basis and variants were filtered and prioritized using the HiFi variant data generated up to that point in time (see Methods; Supplemental Fig. S25). However, we also sought to characterize how filtering SVs by frequency could reduce the manual curation burden for future analyses by considering five allele frequency resources (Supplemental Fig. S26). First, we created a set of “cohort” SVs by performing SV call merging across all 96 probands using Jasmine (Kirsche et al. 2023) and generating allele counts from the merged set (set 1). We then used a second Jasmine merge step to match cohort SVs with SV frequencies from: an in-house set of 266 HiFi genomes including all cohort probands and parents, samples from other internal projects, and public HiFi data (set 2, see Methods); gnomAD v4 SV frequencies from 63,046 short-read samples (set 3, Collins et al. 2020); Human Genome Structural Variant Consortium phase 2 (HGSVC2) assembly-based calls from 18 HiFi samples (set 4, Ebert et al. 2021, https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC2/release/v2.0/integrated_callset/variants_freeze4_sv_insdel_alt.vcf.gz); and a PacBio-provided set of pbsv calls from 103 HiFi samples from the Human Pangenome Reference Consortium (HPRC) and Genome in a Bottle (GIAB) consortia (set 5, Zook et al. 2019; Liao et al. 2023; https://zenodo.org/records/8415406). We note that these data are partially redundant with one another (e.g., set 1 is a partial subset of set 2, and some individual samples such as NA12878 are shared between sets 2–5). We also note that gnomAD allele frequencies should be cautiously applied in the context of lrGS SV annotation, as the srGS artifacts inherent in gnomAD variant calls may inappropriately mask legitimate lrGS variants of interest, particularly given that gnomAD sample size is orders of magnitude larger than the other data sets included in this analysis. However, our goal in this analysis was to maximize filtering impact by including calls from as many data sets and discovery methods as possible. Jasmine merging parameters were heuristically tuned until further increased stringency demonstrated an inflection point in the unique variant count (see Methods; Supplemental Fig. S26).
When filtering SVs using these SV frequency data sets, we found that probands had a median of 1721 “rare” SVs, defined as having an allele frequency <1% in each of the public SV databases (sets 3–5) and an allele count <4 in each internal cohort (sets 1 or 2). A small subset of these, a median of 87 SVs per proband, overlapped a genomic position within 50 bp of a RefSeq exon (Supplemental Table S3). We also filtered to identify “private” SVs as those absent from sets 3–5, having a set 1 allele count of 1, and having a set 2 allele count of ≤2 (allowing for uniparental inheritance). This filtering results in a median of 733 SVs per proband, only 40 of which overlap a position 50 bp of a RefSeq exon (Supplemental Table S3). This analysis includes all pbsv calls, and does not include quality, length, read-depth, or other filters that might further reduce the number of variants needing curation.
Discussion
There remains a substantial fraction of rare disease suspected to have genetic causes that is refractory to genomic testing, a finding that has been repeatedly shown across many clinical and research projects (e.g., Srivastava et al. 2019; Baxter et al. 2022). Several nonmutually exclusive hypotheses exist to explain these observations. Environmental risk factors, such as teratogenic exposure or infectious disease, may be relevant to some phenotypes. Multigenic contributors are also likely to explain at least some cases. For example, a small but appreciable fraction of “double diagnoses,” in which an affected individual is observed to harbor two distinct diseases resulting from highly penetrant variation in two distinct genes, has been observed in clinical genomic studies (e.g., Posey et al. 2017). Notably, such discoveries are typically only made when the variation in both genes is independently amenable to a pathogenicity determination (i.e., would be P/LP regardless of P/LP variation in the other gene), and it is possible if not probable that at least some conditions result from combinations of variants in different genes that are not pathogenic in isolation (Papadimitriou et al. 2019). At the further end of this spectrum is a polygenic accumulation of many risk-factor alleles, which is known to be relevant to many common, complex diseases and which may contribute to some rare conditions, as has been suggested for at least a subset of NDDs (Niemi et al. 2018).
We find it likely that a substantial fraction of unexplained rare disease arises from highly penetrant variation that we have not yet been able to precisely identify or confidently interpret. The results from this study are consistent with that hypothesis, with over 15% of probands with previously negative testing being now found to harbor a relevant or potentially relevant genetic variant. Further, these observations are consistent with the general picture of rare disease testing in recent years. With the advent of exome capture and sequencing ∼15 years ago (Ng et al. 2009, 2010) and subsequent improvements in cost and efficiency, genome-wide detection of highly penetrant variation has greatly accelerated in recent years (Bamshad et al. 2019; Baxter et al. 2022; Boycott et al. 2022; Hamosh et al. 2022). lrGS represents the next phase of that acceleration by facilitating a substantial increase in variant comprehensiveness and accuracy.
We note that the benefits of lrGS for detection of highly penetrant variation derive not just from improved variant sensitivity, but specificity gains as well. Indeed, effective analysis requires not just the ability to detect a variant but to do so accurately and with sufficient specificity for routine and scalable use. For example, effectively all the variation we describe in this study as being newly found within lrGS is, in fact, visible in srGS data, at least at the breakpoint levels. However, being retrospectively visible, once the location and structure of a variant are known to exist, is a much lower bar than the ability to prospectively detect, define, filter, and curate such variation. For example, we describe here a 4.9 kb insertion of SVA and L1 sequence into the 3′ UTR of HCFC1. In retrospective analysis, the Mobile Element Locator Tool (MELT) (Gardner et al. 2017) detected a 1.2 kb SVA mobile element at this location in srGS. However, this call is incorrect with respect to size and sequence composition and has only one read flagged as supporting the right breakpoint. As another example, the breakpoints of the inversion observed to overlap ZBTB20 were correctly called as breakends by manta (Chen et al. 2016). However, each of these was retrospective analyses since MELT calls and breakend calls produced by manta are too numerous for manual curation and thus not used as part of our standard variant calling pipelines (Supplemental Fig. S1; Hiatt et al. 2021). Related to this, srGS copy-number variant (CNV)/SV callers are known to produce many false calls and require strict filtering to reduce calls to a reasonable number for curation. For example, among the 94 samples that had srGS in this study, the raw output from the four different CNV/SV callers that we currently run (see Methods) produces a mean of 9944 total calls per sample, of which only ∼50 are subject to manual curation.
Our results are also consistent with other studies of the benefits of lrGS for discovering genetic contributors to disease. In 2021, for example, we showed in a small pilot project that two of six previously srGS-negative probands harbored clinically relevant variation uniquely interpretable by lrGS (Hiatt et al. 2021). Since that time, several other studies have also used lrGS for molecular diagnosis of rare disease. For example, Cohen and colleagues showed an increased yield of GS (both lrGS and srGS) in exome-negative cases (Cohen et al. 2022). Unique discoveries for lrGS included the detection of novel repeat expansions of STARD7 and a compound heterozygous SNV/deletion that was easier to detect in lrGS. However, the increase in diagnostic yield (∼13%) was mainly from variants interpretable in either lrGS or srGS. Other studies have also shown the diagnostic value of lrGS, especially in small, well-phenotyped cohorts or families (Miller et al. 2021; Del Gobbo et al. 2023; Fukuda et al. 2023; Audet et al. 2024; Kilich et al. 2024; Sakamoto et al. 2024). Our results are similar to these studies at a high-level. However, some important differences are worth noting. One difference is that we have shown the value of lrGS in singletons. While srGS data was available for most of the probands’ parents (79/96, ∼82%), only 10/96 probands had parent lrGS data. Our lrGS variant filtering strategies were sufficient to allow the curation of proband variants without parental lrGS data. However, once flagged for interest, inheritance data could often be assessed by looking at parental srGS reads. Based on this experience, prioritization of proband lrGS with targeted validation in parents is an effective way to increase diagnostic yield with lrGS while reducing lrGS sequencing needs.
We have also provided a direct, systematic comparison of lrGS to contemporaneously analyzed srGS in previously negative cases. Our results thus allow for the separation and description of clinically relevant variants that are “new” by virtue of benefitting from the unique advantages of lrGS versus those that are “new” by virtue of reanalysis, and which could be detected and accurately interpreted via lrGS or srGS-reanalysis. In that context, we note that the benefits of reanalysis of older srGS data remain considerable. In the results described here, the “new” discoveries in 9 of 16 cases in lrGS were called correctly and interpretable within srGS data. These observations reflect the rapid pace of gene discovery in rare disease (Baxter et al. 2022; Boycott et al. 2022; Hamosh et al. 2022), and are consistent with other studies. For example, we previously showed that the probability of a negative srGS data set harboring a clinically relevant variant increased from 1% within 1 year of a previous analysis to ∼22% if more than 3 years have passed since a previous analysis (Hiatt et al. 2018). Several other studies found similar results, with many reanalysis findings being due to recent publications of new gene-disease associations (Liu et al. 2019; Schobers et al. 2022; Hartley et al. 2023).
One possible optimal path to maximizing overall yield in previously srGS-negative individuals is to include srGS-reanalysis before lrGS. However, this reflects a cost/benefit ratio that depends on the cost of the analysis step in relation to the costs of sequencing. While lrGS costs currently remain higher than srGS, as lrGS costs decrease there may reach a point where the cost of variant analysis alone (which is nontrivial and requires both software and computer resources as well as manual curation) is substantial relative to sequencing costs per se and which might favor a process of simply performing lrGS. Further, evaluation of a study design or testing process needs to consider sequencing as only one of several parts of the overall process (e.g., participant recruitment and phenotyping). Again, especially as lrGS costs decline, these factors are likely to increasingly favor the use of lrGS.
One caveat to this study is that the results were generated over a period of time with considerable change in lrGS protocols. For example, most probands in this study were sequenced on Sequel IIE machines (n = 64) before the Revio (n = 32) became available. Given the costs of data production, the 64 Sequel IIE samples were covered at lower-depth (median coverage 24.65×) than the 32 Revio-sequenced samples (median coverage 30.09×) (Supplemental Fig. S27), which may have reduced our sensitivity to variants in the earlier samples. Further, methylation data were not available in the early period of this study, although methylation calls are now being generated and are available for the most recent 44 probands, which may also impact diagnostic yield. For example, evaluation of the AFF3 expansion in proband 6 (Supplemental Figs. S22, S23) would benefit from an assessment of methylation levels at this locus, as hypermethylation, in addition to insertion length, is likely associated with disease risk (Jadhav et al. 2023). Additionally, some probands may harbor methylation alterations that are clinically relevant even in the absence of a pathogenic genetic variant (Aref-Eshghi et al. 2021).
In addition to changes in sequencing, informatic changes have also been considerable over the course of the data generation for this study. While we present and describe a uniform set of variant calls, assemblies, and annotations (see Methods), analysis of individual samples took place simultaneously with the optimization of variant calling and annotation pipelines. One particularly relevant change is variant-frequency annotations. While the key strength of lrGS is its ability to see variants that are invisible or poorly detected in srGS, the ability to discriminate genuine highly penetrant variation from the background of benign alleles depends on the ability to annotate and remove alleles that are common in the general population. As the main allele frequency resources are built from srGS data (e.g., Lek et al. 2016), we have limited ability to filter away likely benign alleles among the variants uniquely detected by lrGS. This ability, however, improved as more lrGS data sets were produced over the course of this study (Ebert et al. 2021; Nurk et al. 2022). Projects like the Consortium of Long Read Sequencing (CoLoRS) (https://colorsdb.org/, currently with lrGS data from almost 1400 individuals) are likely to improve frequency annotation in the future and continue to improve variant curation efficacy. Accumulating lrGS data from as many samples and studies as possible is critical for the long-term maximization of lrGS benefits. Further, assembly-based SV calling is an exciting opportunity that will likely improve SV detection and prioritization (Groza et al. 2024).
In sum, rare disease genetics continues to be a rapidly advancing field. With data sharing (Philippakis et al. 2015; Sobreira et al. 2015; Muenzen et al. 2022) and technology improvement (Wenger et al. 2019), the overall diagnostic yield for individuals with rare disease is increasing at a considerable pace each year. In that context, lrGS has clear benefits over srGS, providing substantial gains to variant specificity and sensitivity, especially for complex and repeat-associated variants (Schuy et al. 2022). We anticipate that the degree of improvement will widen over time, as sequencing and analysis pipelines mature and as lrGS data sets grow.
Methods
Short-read sequencing and variant calling
Probands, their parents, and, when appropriate, affected siblings, were enrolled in one of several research studies aimed at identifying genetic causes of rare disease (Bowling et al. 2017, 2022; East et al. 2021). These studies were monitored by Western IRB and UAB IRB (WIRB 0071, UAB IRB protocols 170303004, 300000328, and 130201001). srES or srGS was performed as described (Bowling et al. 2017, 2022; East et al. 2021; Hiatt et al. 2021). Briefly, whole blood genomic DNA was isolated using the QIAsymphony (Qiagen), and sequencing libraries were constructed by the HudsonAlpha Genomic Services Lab or the Clinical Services Laboratory, LLC, using a standard protocol that generally included PCR amplification (86/96). Genomes were sequenced at an approximate mean depth of 30×, with at least 80% of base positions reaching 20× coverage. Exomes were sequenced to a mean depth of 71×. For short-read reanalysis, srES and srGS reads were aligned to hg38 and small variants and CNVs were called with a uniform pipeline. SNVs/indels and CNVs were curated using an in-house software tool, as previously described (Hiatt et al. 2021). Expected sample relatedness was confirmed with Somalier (v. 0.2.10) (Pedersen et al. 2020) and predicted major genetic ancestries were calculated with peddy (v. 0.4.1) (Pedersen and Quinlan 2017). srGS data for participants who consented to controlled-access sharing in NIH-funded studies are available via dbGaP/AnVIL (CSER1: https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001089.v4.p1, dbGaP accession phs001089; SouthSeq: https://anvilproject.org/data/studies/phs002307, dbGaP accession phs002307). Supplemental Table S6 provides the applicable srGS sample ID and dbGaP accession of all individuals who consented to sharing, and the dbGaP/AnVIL metadata may be used to locate relevant parent and familial srGS data.
Long-read sequencing, variant calling, analysis, and de novo assemblies
Long-read sequencing was performed using HiFi (CCS) mode on either a PacBio Sequel II or Revio instrument (Pacific Biosciences of California, Inc.). Libraries were constructed using a SMRTbell Template Prep Kit (V1.0, 2.0, or 3.0) and tightly sized on a SageELF or BluePippin instrument (Sage Science). Sequencing was performed using a 2 h preextension with either 24 or 30 h movie times. The resulting raw data were processed using either the CCS3.4 or CCS4 algorithm, as the latter was released during the course of the study. Comparison of the number of high-quality indel events in a read versus the number of passes confirmed that these algorithms produced comparable results. Probands were sequenced on 2–3 Sequel II or one Revio SMRT cell. Top off sequencing was performed if the sequencing did not meet the desired coverage (>20×). This resulted in an average estimated HiFi read-depth of 26.1× (range 16.7–41) of raw, unaligned sequence data for probands. For 10 families, parents were also sequenced on 2–3 Sequel II SMRT cells, with an average estimated depth of 21.6× (range 14–28) of raw, unaligned sequence data for parents. Aligned sequencing metrics are shown in Supplemental Tables S1 and S2. HiFi reads were aligned to the hg38 no-alt analysis set (https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz) using pbmm2 (v. 1.10.0, https://github.com/PacificBiosciences/pbmm2). SNVs and small indels were called with DeepVariant (v. 1.5.0) and used to haplotag the aligned reads with WhatsHap (v. 1.7) (Martin et al. 2016). SVs were called using pbsv v2.9.0 (https://github.com/PacificBiosciences/pbsv), which discovers SVs based on read alignment and split reads. Read-depth-based SV discovery was not performed. For the 10 cases with parent lrGS, candidate de novo SVs required a proband genotype of 0/1 and parent genotypes of 0/0, with ≥6 alternate reads in the proband along with 0 alternate reads and ≥5 reference reads in the parents.
De novo assemblies were generated for all probands using hifiasm (v. 0.15.2 [r334 or v.0.16.1-r375]) (Cheng et al. 2021). Hifiasm was used to create two assemblies. First, the default parameters were used, followed by two rounds of Racon (v1.4.10) polishing of contigs (Vaser et al. 2017). For cases with parent GS data, trio-binned assemblies were built using k-mers (srGS). The k-mers were generated using yak (v0.1; https://github.com/lh3/yak) using the suggested parameters for running a hifiasm trio assembly (k-mer size = 31 and Bloom filter size of 2**37). Maternal and paternal contigs went through two rounds of Racon (v1.4.10) polishing. Individual parent assemblies were also built with hifiasm (v0.15.2) using default parameters. The resulting contigs went through two rounds of Racon (v1.4.10) polishing.
Coordinates of breakpoints were defined by a combination of assembly–assembly alignments using minimap2 (Li 2018) followed by the use of bedtools bamToBed (Quinlan and Hall 2010), visual inspection of CCS read alignments, and BLAT. Dot plots illustrating sequence differences were created using Gepard (Krumsiek et al. 2007).
Structural variant merging and filtering for case analysis
Overall SV counts in Supplemental Table S1 were generated from the proband pbsv VCFs using BCFtools (v1.15.1) (Danecek et al. 2021) and awk (e.g., bcftools filter -i 'SVTYPE=="DEL" <input.vcf>| {a[i++]=$1; sum+=$1} END{asort(a); min=a[1]; max=a[i]; if(i%2==1) median=a[int(i/2)+1]; else median=(a[i/2]+a[i/2+1])/2; mean=sum/i; print mean, median, min, max;). Counts of SVs > 50 bp in length in Table 1 were created by the addition of the BCFtools filters -e "SVLEN>−50 && SVLEN<50". No quality filtration or read support filtration was used.
An internal allele frequency catalog was constructed and periodically updated using all available internally sequenced HiFi pbsv variant calls and pbsv calls generated from public HiFi sequencing data (n = 266 at the end of this analysis, “set 2”). Samples included: a majority of participants from this cohort and our previous NDD pilot cohort (Hiatt et al. 2021) (120/266); samples from HudsonAlpha non-NDD projects (35/266); HudsonAlpha-sequenced data from HG001, HG003, HG004, HG006, and HG007 (5/266); HG00514, HG00731, HG00732, NA19240 from HGSVC2 (Ebert et al. 2021) (4/266); CHM13 (1/266) (Nurk et al. 2022, https://www.ncbi.nlm.nih.gov/sra/?term=SRX789768*+CHM13); and the HPRC Year 1 and HPRC_PLUS releases (101/266) (Liao et al. 2023). pbsv calls were merged naively using bcftools merge (v. 1.15.1), i.e., merging only variants that were identical in terms of location, reference sequence, and alternate sequence. The internal pbsv allele frequency catalog included affected probands as well as parent/child trios. Supplemental Figures S25 and S26 detail the pipeline for the creation and use of this catalog.
Structural variant annotation and curation
For individual case SV analysis, a frequency-filtered subset of the proband's pbsv calls was generated using bcftools annotate and bcftools filter, requiring that calls overlap a genomic position within 50 bp of a RefSeq exon and have an allele count of <4 in the HudsonAlpha internal allele frequency catalog (set 2, described above). Exon regions were defined as RefSeq exons ±50 bp (calculated from bedtools slop) and were used to restrict output from BCFtools to only calls overlapping those regions (the -R command line option). For SVs that span multiple bases of the reference genome (duplications, deletions, and inversions), this filter requires only at least 1 bp of overlap between the SV span and the target regions. For SVs that exist at a single base of the reference genome (insertions and breakends), the position must be within the target regions. Each call was visualized using a custom pipeline to automatically generate IGV screenshots (see Supplemental Code for implementation, https://github.com/HudsonAlpha/igv-grapher, Robinson et al. 2011). Additionally, these filtered pbsv variants were annotated, prioritized, and visualized with SvAnna (v1.0.4, annotations v.2204 or v.2304) (Danis et al. 2022) based on manually curated HPO terms for each case. Supplemental Figure S25 details the SV case analysis pipeline from variant calling through curation.
Variant interpretation and orthogonal confirmation
Variant interpretation was performed using ACMG and ClinGen recommendations (Richards et al. 2015; Riggs et al. 2020). Variants of interest were either clinically confirmed by the HudsonAlpha Clinical Services Lab, confirmed within a research laboratory, and/or were supported by short-read data, except where noted in the text (Table 2).
Structural variant merging and filtering for cohort-level analysis
To provide a cohort-level descriptive analysis of SVs and assess the filtering efficacy of combining multiple allele frequency resources, a more robustly merged cohort catalog (n = 96) was constructed using Jasmine. Jasmine allows for merging of similar SVs that may have nonidentical representation in terms of genomic position or variant sequence via spanning a SV proximity graph. First, heuristic trials were conducted to determine a set of stringent Jasmine merge options that would still effectively reduce the resulting count of distinct SVs. The centroid merging strategy was chosen instead of the Jasmine default to lessen the risk of overmerging two dissimilar SVs connected through a third SV of intermediate position and length. In a single trial of Jasmine merging strategies, we observed that using the centroid merging strategy increased the unique SV count by ∼8000 while using the most-stringent clique merging increased the unique SV count by ∼99,000. Supplemental Figure S28 shows the effects of the minimum overlap percentage threshold on the unique deletion count and the minimum sequence identity threshold on the unique insertion count. Jasmine was ultimately run with options ‐‐centroid_merging ‐‐min_overlap=0.65 ‐‐min_sequence_id=0.75 ‐‐output_genotypes to create the merged catalog. This cohort catalog was then annotated with five sets of SV frequency annotation. Cohort allele frequencies were generated from the merged set with bcftools+fill-tags (set 1, n = 96). A second Jasmine merge was used to combine the cohort catalog allele count with additional allele frequency resources: the HudsonAlpha internal pbsv callset (described above) (set 2, n = 266); gnomAD SVs (Collins et al. 2020, v4.0, https://gnomad.broadinstitute.org/news/2023-11-v4-structural-variants) (set 3, n = 63,046); HGSVC2 SVs (Ebert et al. 2021, https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC2/release/v2.0/integrated_callset/variants_freeze4_sv_insdel_alt.vcf.gz) (set 4, n = 18); pbsv calls for individuals from the Human Pangenome Reference Consortium and Genome in a Bottle (from PacBio, Zook et al. 2019; Liao et al. 2023, https://zenodo.org/records/8415406) (set 5, n = 103). The second Jasmine merge was run with the prior settings but without the ‐‐output_genotypes option and records without genotypes were discarded. A custom Python script was used to transfer allele frequency annotations from the annotation source VCFs to the merged VCF based on unique variant identifiers (which were created for each allele frequency resource as needed) and the IDLIST record from Jasmine (see Supplemental Code, https://github.com/HudsonAlpha/jasmine_sv_annotations). The annotated catalog was split into individual VCFs with BCFtools (bcftools view -I -s <id>) which were filtered with BCFtools to generate the counts in Supplemental Table S2. The “rare” filter described in Supplemental Table S3 was defined as excluding variants with any of the following: within-cohort allele count >3, in-house allele count >3, gnomAD maximum population allele frequency (POPMAX_AF) ≥1%, HPRC_GIAB allele frequency ≥1%, or HGSVC2 allele frequency ≥1%. The “proband-exclusive” filter described in Supplemental Table S3 was defined as excluding variants with any of the following: within-cohort allele count >1, in-house allele count >2, gnomAD allele count >0, HPRC_GIAB allele count >0, or HGSVC2 allele count >0. The in-house allele count cutoff was set at 2 in order to account for the fact that some parental samples were included in the in-house frequency database. Exon regions were defined as RefSeq exons ±50 bp (described above). Variant filtering and counting was performed with BCFtools and command-line tools, e.g., bcftools view -R refseq_exons_plus50 bp.bed.gz -e "AC>3 | inhouse_pbsv_AC>3 | gnomad4_POPMAX_AF>=0.01 | hgsvc2_AF>=0.01 | hprc_giab_pbsv_AF>=0.01" -H <input_file>| wc -l for the rare filter and bcftools view -R refseq_exons_plus50 bp.bed.gz -e "AC>1 | inhouse_pbsv_AC>2 | gnomad4_AC>0 | hgsvc2_AC>0 | hprc_giab_pbsv_AC>0" -H <input_file>| wc -l for the exclusive filter. No filtering based on SV length was used; counts include insertions and deletions <50 bp in length that were called by pbsv. Supplemental Figure S26 details the cohort SV merging and analysis pipeline.
Sequencing metrics
Sequencing metrics were generated from the aligned BAMs using the Sentieon implementations of Picard sequencing metrics (Kendig et al. 2019). Sentieon algorithms QualityYield, AlignmentStat were run with default settings. Sentieon WgsMetricsAlgo was run with settings ‐‐include_unpaired true ‐‐min_map_qual 0 ‐‐min_base_qual 0 ‐‐coverage_cap 5000. Further coverage metrics were generated with cramino using the ‐‐phased option (De Coster and Rademakers 2023). Supplemental Table S5 maps each tool or command and output field name to the corresponding entry in Supplemental Tables S1 and S2. SNV and indel counts and ratios were calculated with rtg vcfstats (https://github.com/RealTimeGenomics/rtg-tools). Summary statistics and graphs were calculated with R (v4.3.1), RStudio (v2023.9.1 build 494), and ggplot2 (v3.4.3) (Wickham 2011; Team RStudio 2020; R Core Team 2023). Coverage across regions of interest, such as SHANK3, was calculated using samtools bedcov with default parameters.
Repeat region detection and analysis
We curated a BED file of disease-associated low-complexity repeat regions in 66 genes from previous studies (Hiatt et al. 2021; Cohen et al. 2022 and references therein). Variant calls from pbsv were extracted from these regions ±30 bp (Supplemental Table S4). Reads were also visualized using the IGV. Coverage across low-complexity repeat regions was calculated using samtools bedcov with default parameters. A coverage of at least 8× across the low-complexity region was required for inclusion in Supplemental Table S4 and Supplemental Figure S23. We also used TRGT (Dolzhenko et al. 2024) and companion tool TRVZ for visualization of a subset of calls including those for display in Supplemental Figure S3. TRGT was fed an hg38 reference genome FASTA, BED catalog of tandem repeats, and a sample's BAM to generate a VCF containing genotypes for each tandem repeat from the provided catalog in the given sample and a BAM containing only reads that span the repeat sequences. Output VCFs and BAMs were sorted, indexed, and fed into TRVZ with the same hg38 reference FASTA and BED catalog of tandem repeats to generate pileup plots for any desired variants.
3′ mRNA-seq
Total RNA was isolated from blood samples in PAXgene RNA tubes (PreAnalytiX 762165) according to the manufacturer's instructions and stored short-term at −20°C. RNA was isolated using the PAX gene Blood RNA Kit (Qiagen 762164) according to the manufacturer's instructions. Isolated RNA was quantified by the Qubit RNA HS Assay Kit (Thermo Q32855). 425 ng of RNA was used as input for the QuantSeq 3′ mRNA-Seq Library Prep Kit FWD for Illumina and UMI Second Strand Synthesis Module for QuantSeq FWD (Illumina, Read 1) from Lexogen (015.96 and 081.96, respectively). Libraries were quantified using the Qubit DNA HS Assay Kit (Invitrogen Q32854) and visualized with the Bioanalyzer High Sensitivity DNA Analysis kit (Agilent 5067–4626) and 2100 Bioanalyzer Instrument (Agilent). Sequencing was carried out using Illumina NextSeq 75 bp single-end. UMIs were first extracted from the reads with UMI-tools extract with regex. Reads were then trimmed with bbduk (https://sourceforge.net/projects/bbmap/) and aligned to hg38-GENCODEv42 using STAR (Dobin et al. 2013) with the Lexogen recommended parameters for QuantSeq. BAMs were deduplicated by UMI and mapping coordinates using UMI-tools dedup (Smith et al. 2017). Count tables were generated using htseq-count with the intersection-nonempty method (Anders et al. 2015).
Data access
Out of 96 families, 82 (85%) consented to controlled-access genomic data sharing, including all probands described in detail in the manuscript and listed in Table 2. Consented samples include 100 lrGS genomes (82 probands and 18 parents, comprising 9 full trios and 73 singletons). For participants who consented to controlled-access sharing, the lrGS data generated in this study will be available through AnVIL (https://anvilproject.org/data/studies) under accession number phs003537. Researchers may apply for access via the database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap/). Supplemental Table S6 describes the samples and de-identified file identifiers and, when applicable, their corresponding short-read dbGaP identifiers and other metadata. Custom scripts for automated IGV plot generation and Jasmine structural variant merging and annotation are available in Supplemental Code and on GitHub (https://github.com/HudsonAlpha/igv-grapher and https://github.com/HudsonAlpha/jasmine_sv_annotations).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the families who have contributed to our studies and our collaborating physicians and clinical staff for recruitment and enrollment for these research studies. Some reagents were provided by PacBio as part of an early access testing program. The CSER1 project was supported by a grant from the US National Human Genome Research Institute (NHGRI; UM1HG007301). The SouthSeq project (U01HG007301) was supported by the Clinical Sequencing Evidence-Generating Research (CSER2) consortium, which was funded by the National Human Genome Research Institute with co-funding from the National Institute on Minority Health and Health Disparities and the National Cancer Institute. The Alabama Genomic Health Initiative is an Alabama-State earmarked project (F170303004) through the University of Alabama in Birmingham. One sample from the cohort was funded by the Alabama Pediatric Genomics Initiative. lrGS of some samples was supported by a Research Grant from the Muscular Dystrophy Association (MDA 963255, https://doi.org/10.55762/pc.gr.157020).
Author contributions: S.M.H., J.M.J.L., L.H.H., D.R.L., Z.T.B., C.R.F., M.L.T., L.B.B., M.W., I.R.N., and J.J. generated, analyzed, and/or interpreted data. J.M.J.L. and C.R.F. coordinated data sharing to AnVIL. W.V.K., E.M.B., M.A.L., A.C.E.H., and B.R.K. recruited and enrolled patients and provided clinical assessment. S.M.H., J.M.J.L., J.S., J.G., and G.M.C. designed the study and oversaw the interpretation of data. S.M.H., J.M.J.L., and G.M.C. drafted and revised the manuscript. All authors read and approved the final version of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279227.124.
- Received February 29, 2024.
- Accepted August 19, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








