
Understanding isoform expression by pairing long-read sequencing with single-cell and spatial transcriptomics
- Natan Belchikov1,2,3,
- Justine Hsu1,2,
- Xiang Jennie Li1,2,4,
- Julien Jarroux1,2,
- Wen Hu1,2,
- Anoushka Joglekar5,6 and
- Hagen U. Tilgner1,2
- 1Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, New York, New York 10065, USA;
- 2Center for Neurogenetics, Weill Cornell Medicine, New York, New York 10021, USA;
- 3Physiology, Biophysics, and Systems Biology Program, Weill Cornell Medicine, New York, New York 10065, USA;
- 4Computational Biology Master's Program, Weill Cornell Medicine, New York, New York 10065, USA;
- 5New York Genome Center, New York, New York 10013, USA;
- 6Department of Biomedical Informatics, Columbia University, New York, New York 10032, USA
Abstract
RNA isoform diversity, produced via alternative splicing, and alternative usage of transcription start and poly(A) sites, results in varied transcripts being derived from the same gene. Distinct isoforms can play important biological roles, including by changing the sequences or expression levels of protein products. The first single-cell approaches to RNA sequencing—and later, spatial approaches—which are now widely used for the identification of differentially expressed genes, rely on short reads and offer the ability to transcriptomically compare different cell types but are limited in their ability to measure differential isoform expression. More recently, long-read sequencing methods have been combined with single-cell and spatial technologies in order to characterize isoform expression. In this review, we provide an overview of the emergence of single-cell and spatial long-read sequencing and discuss the challenges associated with the implementation of these technologies and interpretation of these data. We discuss the opportunities they offer for understanding the relationships between the distinct variable elements of transcript molecules and highlight some of the ways in which they have been used to characterize isoforms’ roles in development and pathology. Single-nucleus long-read sequencing, a special case of the single-cell approach, is also discussed. We attempt to cover both the limitations of these technologies and their significant potential for expanding our still-limited understanding of the biological roles of RNA isoforms.
Over the past decade, the availability and capabilities of two transformative sequencing technologies, microfluidics-based single-cell sequencing and long-read sequencing, have greatly expanded. Both have been used in transcriptomics to characterize the mRNA expression profiles of cells. Single-cell sequencing allows transcript molecules to be traced to individual cells, thus preserving cell-type-specific information about expression. Long-read sequencing offers a less fragmented (or even unfragmented) view of individual transcripts compared to short-read sequencing, providing information on relationships between transcript elements. Later, spatial sequencing emerged as an alternative to single-cell sequencing that preserved information on the spatial origins of sequenced molecules, though it was at first too low-resolution to separate molecules from different neighboring cells. Important advances have been made by combining long-read RNA sequencing with either single-cell or spatial approaches. This becomes especially useful for characterizing RNA isoform expression, which will be the focus of this review.
Alternative exons and other RNA variables
To understand how single-cell and spatial long-read sequencing has been applied in studies of isoform expression, it will be helpful to briefly discuss the phenomena that contribute to the formation of distinct isoforms from the same underlying gene. Alternative splicing (AS) allows genes to generate multiple RNA isoforms from a single genomic region (Johnson et al. 2003; Harrow et al. 2006; Pan et al. 2008; Wang et al. 2008). The key phenomenon here is the inclusion or exclusion of alternative exons—those that are present in only some transcripts, in contrast to constitutive exons, which are always included. In addition to affecting the inclusion of entire exons, AS can manifest as alternative splice site usage, where an exon is altered via a shift in its boundary with a neighboring intron, thus shortening or lengthening the exon. AS appears to be especially common in mammals, where it occurs in the vast majority of genes (Johnson et al. 2003; Pan et al. 2008; Wang et al. 2008), but is seen throughout the multicellular eukaryotes, with around half of plant genes affected (Filichkin et al. 2010; Lu et al. 2010; Shen et al. 2014). In addition to AS, several other phenomena lead to the production of different varieties, or isoforms, of RNA molecules from the same underlying gene. In alternative transcription start site (TSS) usage and alternative polyadenylation (poly(A)) site usage, the start and end positions, respectively, of a transcript shift. Finally, in intron retention, an intronic sequence can remain within an otherwise fully spliced transcript (Fig. 1). Additional transcriptome diversity can also arise from RNA modification, where individual nucleotides are altered after transcription, with methylation of adenosine (m6A) being the most common.
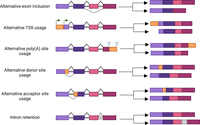
Multiple RNA variables contribute to the production of distinct isoforms from the same gene. In alternative exon inclusion, an exon can be either included or spliced out of a transcript. In alternative TSS usage, transcription can start at two different positions. In alternative poly(A) site usage, the 3′ untranslated region can vary in length. In alternative donor or acceptor site usage, the boundary between an exon and an intron can shift. In intron retention, an intron can be included in the mature mRNA.
Such variations in the sequence present in mature RNA molecules—which we term “RNA variables” (Foord et al. 2023)—contribute to the complexity of isoform expression, which can broaden the diversity of phenotype across cells, cell types, tissues, organs, and individuals. For instance, a shift toward the more upstream poly(A) sites among genes with multiple sites within the last exon has been associated with the activation of CD4+ T cells (Sandberg et al. 2008). Alternative poly(A) usage has also been identified as a mechanism for differential-expression regulation in ubiquitously transcribed genes—here defined as genes that are expressed in a wide variety of human tissues but that are not the classical housekeeping genes associated with protein biosynthesis and energy metabolism (Lianoglou et al. 2013). In neuronal differentiation, a combination of AS, alternative TSS usage, and alternative poly(A) usage contributed to isoform changes observed in vitro, with 45% of isoforms being attributable to TSS or poly(A) site usage in particular (Ulicevic et al. 2024). Importantly, AS and other isoform changes can serve as specifiers of cell type separately from differential expression, allowing for the fine-tuning of differences between cell populations. For instance, we found that in the brain, the astrocyte and oligodendrocyte lineages clearly differ by gene expression, but at the splicing level, oligodendrocyte precursor cells cluster with astrocytes rather than committed and mature oligodendrocytes (Joglekar et al. 2024).
The resulting isoforms can lead to the expression of multiple distinct protein isoforms, although they may also be invisible at the protein level (Nilsen and Graveley 2010; Ward and Cooper 2010). A protein–protein interaction scan suggests that many protein isoform pairs have distinct functions (Yang et al. 2016), though others are expected to lack functional consequences (Pickrell et al. 2010; Wright et al. 2022). Some isoforms result in the appearance of a premature stop codon, usually due to a frameshift, leading to nonsense-mediated decay, which can serve as a regulator of expression (Lewis et al. 2003) but can also result in a variety of genetic diseases (Scotti and Swanson 2016; Zhang et al. 2022; Kurolap et al. 2023). In other cases, the resulting changes in sequence lead to protein products with distinct functions. For example, in human embryonic stem cells, an AS event in the FOXP1 transcription factor that alters its DNA-binding properties was found to increase the expression of other transcription factors that facilitate continued pluripotency and suppress differentiation (Gabut et al. 2011). In one of many phenotypic AS effects identified in insects, 16% of multi-isoform genes in the bumblebee Bombus terrestris were observed to display isoform expression specific to a particular developmental stage, sex, or caste (Price et al. 2018). Further examples of AS functional consequences have been reviewed (Wright et al. 2022). Given its wide array of physiological consequences and the multiplicity of variables involved, isoform expression deserves careful attention and further efforts to characterize its complexity. There are likely to be many more as-yet-undiscovered effects. And since it is a crucial means of deriving varied functionality from the same genome, it is critical to understand how it changes within a single organism, tissue, or cell type.
The emergence of single-cell approaches
Initial transcriptomic single-cell research focused on the heterogeneity of single cells (Kamme et al. 2003; Subkhankulova et al. 2008; Shalek et al. 2013; Fan et al. 2015). Droplet-based single-cell barcoding has enabled the advent of large-scale single-cell sequencing (Klein et al. 2015; Macosko et al. 2015; Zeisel et al. 2015) and coupling this with single-cell suspension production enabled its application to tissues with diverse cell types such as the blood (Villani et al. 2017), brain (Macosko et al. 2015; Zeisel et al. 2015), and embryos (Yan et al. 2013). By clustering cells into groups or clusters of cells using their gene expression profiles, a variety of approaches now allow the definition of cell types or cell states (Trapnell et al. 2014; Satija et al. 2015; Stuart et al. 2021). Thus, at the time of writing, single-cell short-read RNA sequencing is a widely employed technology that has significantly contributed to our understanding of physiology and disease in human, mouse, and other model organisms (Fig. 2A). Many consortium-led research programs now seek to comprehensively catalog the diversity of cell types using single-cell methods. The Human Cell Atlas project focuses on cataloging all human cells, while more specialized efforts exist in neuroscience, including both the PsychENCODE Project and the BRAIN Initiative's Cell Census Network (BICCN) and Cell Atlas Network (BICAN) (Bhaduri et al. 2021; Di Bella et al. 2021; Yao et al. 2021).
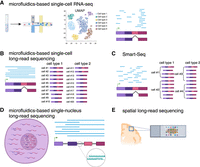
Several key features differentiate the commonly used single-cell and spatial sequencing methods. (A) In the standard single-cell RNA-seq approach, a single-cell suspension is created and passed through a microfluidic system that captures each individual cell in an oil droplet with a bead containing a unique barcode. After short-read sequencing, cells can be clustered based on gene expression into groups representing distinct cell types. Each read covers only a small section of a transcript, and there is a bias toward the ends of a transcript (most often the 3′ end). Relatively few reads cover an exon junction. (B) With long-read sequencing, a read covers all or most of a transcript, including multiple exon junctions. This enables comparisons between cell types in terms of the relative abundance of particular isoforms or RNA variables such as alternative exons. Many cells can be sequenced, but relatively few transcripts are sequenced per cell. (C) Smart-seq approaches allow for better coverage over the full length of a transcript, including over exon junctions, than with standard single-cell RNA-seq, while still relying on short reads. More molecules are sequenced per cell than in single-cell long-read sequencing. (D) When the entire cell is used as the source of RNA, most sequenced molecules are cytoplasmic and thus fully spliced. However, in single-nucleus sequencing, where only nuclear RNA is available, many molecules will be unspliced or partially spliced. Also, because some of the introns present in sequenced transcripts will have “decoy” poly(A) stretches, some reads will start there rather than at the 3′ end of the transcript. (E) In spatial methods, a tissue slice is tagged with a grid of barcodes that encode each transcript's position within the slice rather than pointing to an individual cell of origin.
It is worth mentioning here that an alternative approach to single-cell RNA sequencing that does not make use of microfluidics technology exists. In split pool ligation-based transcriptome sequencing (SPLiT-seq), the single-cell (or -nucleus) suspension is split across a 96-well plate, unique barcodes are applied in each well, and then all wells’ contents are mixed back together and split again, with the process typically being done for four or more rounds to produce a unique combination of barcodes for each cell by random chance (Rosenberg et al. 2018). This combinatorial barcoding approach obviates the need for specialized laboratory equipment, though it requires its own computational processing for accurate barcode identification (Kuijpers et al. 2024). The random hexamer priming that has been incorporated into SPLiT-seq reduces the 3′ bias compared to standard single-cell approaches, thus offering a better view of the full length of a transcript molecule. SPLiT-seq has recently been commercialized by Parse Biosciences.
Long-read single-cell methods
The first long-read sequencing platform to be commercialized was the single-molecule real-time (SMRT) sequencing method from Pacific Biosciences (PacBio), where a zero-mode waveguide confines the fluorescent signal generated as DNA polymerase synthesizes a new strand while using the original strand as a template (Eid et al. 2009). This was followed by the release of the Oxford Nanopore Technologies (ONT) platform, which measures changes in current induced by the movement of a nucleic acid molecule through a nanopore. Since then, both technologies’ capabilities have found numerous applications spanning genomics, epigenomics, transcriptomics, and epitranscriptomics. For a more detailed discussion, several excellent reviews of the mechanisms and applications are available (Rhoads and Au 2015; Wang et al. 2021).
From an RNA perspective, long-read sequencing appeared on the scene in the early 2010s, with multiple studies devoted to plant (Koren et al. 2012) and human isoform research (Au et al. 2013; Sharon et al. 2013). With potentially thousands of bases per read, associations between the various RNA variables can be analyzed even when the variables are separated by distances greater than the typical length of short reads, which tends to be just several hundred bases (Tilgner et al. 2015, 2018). After the advent of long-read sequencing approaches, the first applications to single cells focused on isoform diversity among 6, 7, and 96 single cells (Byrne et al. 2017; Karlsson and Linnarsson 2017; Volden et al. 2018). We and our colleagues then engineered an approach to work with barcoded cDNAs from thousands of individual cells through microfluidics (Gupta et al. 2018; Singh et al. 2019; Lebrigand et al. 2020). In these approaches, one usually sequences fewer individual molecules per cell—but for thousands of cells (Fig. 2B). Thus, cell-type clusters can be formed from single-cell data through the analysis of gene expression patterns just like in the previously mentioned short-read single-cell studies (Klein et al. 2015; Macosko et al. 2015; Zeisel et al. 2015).
Alternative exon inclusion is usually quantified by calculating a percent-spliced-in (PSI, or Ψ) value for each exon based on the number of reads that include the exon and the number that skip it. For whole isoforms, we have used an extension of this concept, calculating percent-isoform (PI, or Π) values, which are based on the percentage of reads supporting a specific isoform among all isoforms of a gene (Joglekar et al. 2021). Because a TSS is located the furthest from the 3′ end of a transcript, its accurate quantification represents a challenge, given that the absence of a more upstream site in a read could represent either true alternative usage or simply a truncation during sequencing or cDNA synthesis during reverse transcription.
Both major long-read platforms offer bioinformatic pipelines for processing single-cell transcriptomic data. On ONT's EPI2ME open-source platform, the wf-single-cell pipeline allows the user to map reads, identify genes and transcripts, and extract cell barcodes and unique molecular identifiers (UMIs) in order to generate count matrices from single-cell ONT data. The transcript assignment function adapts the procedure from FLAMES, which is also a standalone tool for isoform detection for both bulk and single-cell long-read data (Tian et al. 2021). Similarly, the Iso-Seq tool from PacBio allows users to process and map single-cell PacBio data, handle barcodes and UMIs, and produce gene and isoform count matrices.
Novel isoform detection—and comparing platforms
Since the beginning of long-read RNA sequencing, detecting novel isoforms and novel genes was a primary goal. In our case, we did so by considering the ordered list of all introns in a read (the intron chain) and asked whether this was identical to the intron chain of an annotated transcript or to a truncated version of one (Sharon et al. 2013; Tilgner et al. 2014, 2015; Gupta et al. 2018). If not, we considered the read to represent a novel transcript. This definition of “novel” versus “known” has the advantage of including many types of novelty: novel exons, novel exon-skipping events, novel acceptor sites, novel donor sites, and novel combinations of known splice sites. However, teasing apart which of these apply is not necessarily easy. Also, a potential drawback is that a downstream TSS, located slightly upstream of or inside exon 2 of the “canonical” transcript, would be considered a truncated version of a known transcript—and thus would only be considered possibly novel at best. In recent years, the SQANTI categories have become a popular approach to classifying distinct types of novelty (Tardaguila et al. 2018). The approach also relies on a comparison to a reference genome annotation. If a sequenced transcript has a novel intron chain but still uses only annotated donor or acceptor sites, it is categorized as “Novel in Catalog” (NIC), referring to the fact that all these splice sites are included in a given catalog or annotation. If, however, at least one of the donor or acceptor sites used is not annotated, the transcript is termed “Novel Not in Catalog” (NNC). Additional categories include “Genic Intron” for transcripts mapping entirely within an annotated intron, “Intergenic” for transcripts mapping entirely outside of annotated genes, and “Genic Genomic” for transcripts that partially belong in one of the two prior categories but also overlap an annotated exon. In the “Fusion” category, a transcript maps to more than one annotated gene. The final novel-transcript category is “Antisense,” where a transcript maps to the complementary strand of an annotated transcript and contains a poly(A) region (verifying that it is not merely the reverse complement of the true transcript). Known transcripts, where the intron chain matches one found in the annotation, are labeled “Full Splice Match” (FSM) if all splice junctions of an annotated transcript are covered and “Incomplete Splice Match” (ISM) in the case of a truncated read.
Isoforms originating from novel genes constitute an important category of novel isoforms. For instance, a set of such isoforms identified using PacBio long reads has been associated with pluripotency in human cells (Au et al. 2013). This subject has been further reviewed in detail (Au and Sebastiano 2014). In a somewhat similar manner, ∼5000 unannotated, largely noncoding, genes were discovered in human embryos (Torre et al. 2023).
A fundamental question regarding the discovery of novel isoforms is whether they should be included in state-of-the-art annotations, such as GENCODE (Harrow et al. 2012; Frankish et al. 2023), RefSeq (Pruitt et al. 2005), or AceView (Thierry-Mieg and Thierry-Mieg 2006). If one were to include every observed isoform, these annotations would grow dramatically, potentially even exponentially, eventually including every mistake the cellular machinery ever makes. Furthermore, single-cell methods themselves produce PCR artifacts and molecules formed as a result of reverse-transcriptase template switching, which can create the appearance of additional novel isoforms. Direct RNA methods, in which RNA is sequenced directly with ONT sequencing without first being converted to cDNA, avoid these pitfalls, but eliminate the cell-type information provided by single-cell sequencing. On the other hand, extreme conservatism might lead to missing biologically important isoforms. An important point, which has been repeatedly noted, is that novel isoforms are—on average—more lowly expressed than known ones (Au et al. 2013; Sharon et al. 2013; Tilgner et al. 2014, 2015; Gupta et al. 2018). While not every highly expressed isoform is necessarily functional, high expression is nevertheless potentially suggestive of functionality and thus should not be disregarded. Single-cell and spatial isoform data sets can add an important layer of information here: What may be a lowly expressed isoform overall (e.g., 5 out of 1000 molecules) may be highly expressed in a lowly abundant cell type or a particular location (e.g., 5 out of 5 molecules). Such a situation warrants at least a more careful analysis of the isoform in question.
Intrinsically linked to novel- (as well as known-) isoform identification is the issue of the differences between the various isoform-sequencing strategies, including PacBio, ONT, and other approaches, in this regard. The earliest work on long-read RNA-seq using PacBio included detailed comparisons to Illumina (Au et al. 2013; Sharon et al. 2013), as well as to other “almost-long-read” approaches such as 454 sequencing (Sharon et al. 2013), which of course demonstrated the advantages of long-read sequencing in connecting splice sites and exons into full-length isoforms. A detailed comparison of PacBio and ONT sequencing found that both protocols were suitable for isoform biology, though PacBio's error rates were lower (Weirather et al. 2017). This work also used hybrid sequencing, i.e., using Illumina reads to remove the sequencing errors from high-error-rate ONT long reads. A recent multilaboratory effort to compare many long-read protocols and software tools found that for the detection of lowly expressed isoforms, using orthogonal data and replicates is advisable (Pardo-Palacios et al. 2024a,b), and assessed the performance of multiple long-read software packages. Another benchmarking study by an independent group of researchers compared nine long-read isoform detection software tools using simulated data (Su et al. 2024), highlighting IsoQuant as the top performer, with Bambu and StringTie2 also performing well (Kovaka et al. 2019; Chen et al. 2023; Prjibelski et al. 2023). An intriguing point to consider about such comparisons of PacBio and ONT is that one rarely sequences cDNAs derived from the same RNA molecule on both platforms—and if one does, one would not know it. Here again, single-cell and spatial barcoding technologies can help: Using barcodes and UMIs as well as mappings to the genome, one can tell whether a cDNA sequenced on PacBio and a cDNA sequenced on ONT originate from the same original RNA. In this situation, any divergence in the mapping of the two cDNAs necessarily represents an error in one of the platforms. This approach, comparing the same RNA molecule's cDNAs between PacBio and ONT, allowed us to recognize important error patterns in ONT sequencing (Mikheenko et al. 2022), which contributed to the development of accurate isoform interpretation software (Prjibelski et al. 2023). It is crucial to note that there continues to be a need for improvements in accurate UMI identification methods, and that approaches that rely on obtaining multiple reads per UMI will require increased sequencing depth and therefore incur higher costs. A surprising and important finding has been the recent discovery that Illumina sequencing can detect some junctions that are missing from long-read sequencing, pointing to an issue for the long-read community to understand and overcome (Han et al. 2024).
Elucidating coordination of RNA variables
One of the key values of long-read transcriptomics is the analysis of coordination that becomes possible. If each RNA variable in a transcript were entirely independent of all others, the number of possible isoforms would grow exponentially with the number of variables. Yet patterns of association between different exons and between promoters and exons have been identified for many genes even before the advent of high-throughput sequencing (Cramer et al. 1997; Tasic et al. 2002; Fededa et al. 2005). Coordination patterns involving alternative exons have been observed both in terms of associated and mutually exclusive pairs within the gene, and some of these appear to be evolutionarily conserved (Tilgner et al. 2015) and to play an important role in protein-coding sequences in particular (Tilgner et al. 2018). For example, a mutually exclusive relationship was observed in healthy human brain between two distant alternative exons in MAPT, the gene that codes for tau. Multiple neurodegenerative diseases involve the pathological aggregation of tau in the brain, but Alzheimer's disease (AD) and progressive supranuclear palsy were found to display distinct coordination patterns between one of these exons and another distant exon (Bowles et al. 2022). In addition, the usage of different promoters within the same transcript, which constitutes a subtype of alternative TSS usage with typically longer inter-TSS distance, has been associated with distinct splicing patterns in those transcripts (Xin et al. 2008). Long-read sequencing allows for more powerful statistical analysis of such associations, as well as associations between exons and other variables (Fig. 3). It appears that the choice of TSS during the initiation of transcription can often be closely associated with AS and alternative poly(A) site choices, with these processes occurring in close spatial and temporal proximity. Long-read studies have been instrumental in identifying these associations in the human and Drosophila transcriptomes (Anvar et al. 2018; Alfonso-Gonzalez et al. 2023; Calvo-Roitberg et al. 2024). For instance, we found coordinated inclusion to be much more common with pairs of adjacent exons than with pairs of nonadjacent exons in mouse cerebellum (Gupta et al. 2018). In human brain, we reported that patterns associating TSS and exons, poly(A) sites and exons, and nonadjacent exons serve to distinguish neural cell types, whereas adjacent exon coordination is seen more often within a given cell type in human brain (Hardwick et al. 2022). In Drosophila, coordination between alternative poly(A) usage and alternative exon inclusion was identified in 23 genes (with ∼14,000 total protein-coding genes in the genome) (Zhang et al. 2023b).
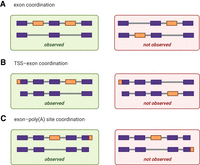
The full-transcript view offered by long-read sequencing allows for the observation of coordination between RNA variables. (A) When two alternative exons are coordinated, only molecules with either both exons included or excluded are observed in long-read sequencing data. (B) In TSS–exon coordination, an alternative exon may only be present in molecules using one of two alternative TSS. (C) In exon–poly(A) site coordination, an alternative exon may only be present in molecules using one of two alternative poly(A) sites.
Differences between long-read and Smart-seq methods
A distinct single-cell RNA-seq technology that is important to understand in relation to long reads is Smart-seq, which was developed to improve coverage over the entire lengths of transcripts compared to earlier methods, while still relying on short-read sequencing (Ramsköld et al. 2012; Picelli et al. 2013; Hagemann-Jensen et al. 2020). While this approach does not extend the size of individual reads beyond the capabilities of Illumina sequencing, it provides a fuller picture of the transcriptome than 3′ end sequencing by generating short reads over the entire length of an RNA molecule, rather than just its ends, thus enabling the assembly of a long-range view of each transcript. As a result, Smart-seq and its successors (Smart-seq2, Smart-seq3) could be seen as alternatives to microfluidics-based single-cell long-read approaches as a way to analyze full-length transcripts and therefore study isoform expression. An important advantage of Smart-seq methods is that they capture a much larger proportion of the transcripts present in each cell compared to microfluidics-based methods, which are sparse at the level of individual genes within individual cells (Fig. 2C). Smart-seq3 could detect ∼8000 molecules of at least 500 bases in length per cell, compared to several hundred to 1000 UMIs per cell for long-read single-cell methods. While estimates vary, the number of mRNA molecules per cell has been assessed to be as high as 300,000 in some cases (Marinov et al. 2014), though there is wide variation between cell types. The median number of mRNA molecules per gene per cell was found to be 17 in one study (Schwanhäusser et al. 2011). Furthermore, the phenomenon of transcriptional bursting, where only one of the two alleles of a gene is transcribed exclusively at any given period of time, contributes to transcript heterogeneity (Deng et al. 2014). Thus, there is substantial value in being able to sequence a representative sample of transcripts from each cell. On the other hand, by eschewing microfluidic technology, Smart-seq results in much lower throughput in terms of the number of cells sequenced per experiment and a higher error rate. This can pose a disadvantage when seeking to draw statistical inferences about isoform expression patterns across many genes, cell types (especially rare ones), tissues, and regions.
Single-nucleus processing for frozen tissue
As described above, making a single-cell suspension has enabled important research advances, such as in understanding the central nervous system of model organisms. For the splicing and long-read field, this has allowed for the characterization of full-length cDNAs, which are mostly derived from cytosolic mRNAs. However, producing a similar single-cell suspension from frozen brain tissue has proven difficult. This motivated the usage of single-nucleus suspensions (Lake et al. 2016; Hu et al. 2017), which can be readily produced from frozen tissue—and therefore from postmortem human samples and others that must be stored before library preparation. Earlier work on single-nucleus suspension coupled with transcriptomics (Grindberg et al. 2013) predated the advent of high-throughput single-cell sequencing, but this approach has then also enabled single-cell characterization of the human brain (Lake et al. 2016; Zhou et al. 2020; Tran et al. 2021; Yang et al. 2022). This coupling of microfluidics with single-nucleus work has proven highly successful for the definition of cell types for the human brain as well as in other species. For example, analyses of multiple brain regions have been conducted in various mammals, including mice, humans, and other primates, helping to define regionally specific cell types (Krienen et al. 2020; Kozareva et al. 2021).
In contrast to single-nucleus approaches, cDNAs generated from single-cell studies are dominated by cytosolic RNA molecules, which are dramatically more completely spliced than nuclear RNAs, given that splicing has been found to promote the export of mRNA from the nucleus (Valencia et al. 2008). As a result, “decoy” poly(A) tails—here loosely defined as somewhat A-rich regions in interior, often intronic, parts of the RNA molecule—are much less abundant with whole-cell approaches than with single-nucleus protocols (Fig. 2D). These decoys are used as alternative priming sites for poly(dT) reverse transcription and generate cDNAs with lower coverage of the transcript's 3′ end. Of note, cDNAs deriving from decoy poly(A) tails are not useless: In fact, short reads representing these molecules have been highly successfully used to detail dynamic transcriptional identities using what is called “RNA velocity” (La Manno et al. 2018). However, these decoy poly(A) tail-derived cDNAs are largely uninformative for the description of alternative exon inclusion, combinatorial exon usage, and to some extent also for poly(A)-site and TSS usage. While methods to de-enrich for purely intronic cDNAs now exist (Hardwick et al. 2022), these observations demonstrate that methods providing cDNAs generated from cytosolic RNAs generally do better than those relying on nuclear RNAs when it comes to describing complete and mature isoforms. Some single-nucleus studies have found an increased proportion of antisense reads. The larger presence of intronic sequence within nuclear transcripts compared to that seen in whole-cell experiments may be a contributing factor.
Studying disease with single-cell long-read approaches
We will now discuss several applications of single-cell long-read sequencing. Combining the resolution of single-cell technology with the long-range information offered by long-read sequencing has resulted in new insights into the transcriptomes of disease-relevant systems with heterogeneous cell populations. In cancer, transcriptomic variation among cells can be layered on top of genomic variation. Short-read sequencing can be inadequate for understanding this complexity, as was evident when ovarian cancer samples with a genomic-origin IGF2BP2::TESPA1 gene fusion appeared as TESPA1 overexpression in short-read single-cell RNA-seq data while being correctly identified with PacBio sequencing. Furthermore, the abundance of noncoding transcripts of protein-coding genes visible in the long-read data would have resulted in an ∼20% overestimation of protein expression based on short-read data (Dondi et al. 2023). Associations between genotype and splicing changes using ONT single-cell RNA-seq were likewise observed in kidney cancer (Shiau et al. 2023).
Single-cell long-read RNA-seq has also proven valuable in understanding infectious diseases. The parasite Plasmodium vivax, one of the species that causes malaria, had been difficult to accurately profile transcriptionally with bulk methods because of the variety of developmental stages present in any given infection, and with short-read methods because of the heterogeneity in isoform expression. Single-cell sequencing with PacBio enabled comprehensive characterization of isoforms specific to particular P. vivax developmental stages (Hazzard et al. 2022).
Viral infections result in the hijacking of cellular transcriptional processes by the virus. Variations in the output of this process constitute another promising target for single-cell long-read study. For instance, the high mutation rate of the influenza virus results in changes in the sequence of expressed viral transcripts in a set of cells. By combining standard Illumina-based single-cell RNA-seq quantification of cultured cells’ transcriptomes with PacBio sequencing of the viral transcripts, it was found that this viral genetic variation explained part, but not all, of the heterogeneity in viral gene expression and the cells’ innate immune response (Russell et al. 2019). The value of long reads for both DNA and RNA sequencing was demonstrated by a study of the human adaptive immune system's response to MMR vaccination. ONT sequencing was used to characterize both the transcriptome of single B cells and relevant loci in the donor's germline genome in a haplotype-resolved manner in order to investigate the complex genetic recombination patterns that are responsible for antibody production, demonstrating a powerful use case for combining the two modalities (Beaulaurier et al. 2024). A subject where virology intersects with neuroscience (another isoform-rich domain) is the issue of HIV persistence in the brain, where the virus has been found in replication-competent form despite antiretroviral therapy (Tang et al. 2023); since microglia appear to play an outsize role here, a cell-type-specific long-read perspective could be valuable. Notably, the SCORCH consortium is currently undertaking an investigation of this phenomenon as it relates to opioid use disorder (Ament et al. 2024).
Understanding development
As previously seen in the study of P. vivax, changes in isoform expression are often characteristic of the development of an organism, necessitating the use of long-read methods for a more exhaustive view of individual transcript molecules. At the same time, this developmentally important isoform diversity is often also cell-type specific, requiring the use of single-cell approaches. Mouse embryonic development has been investigated in this way, leading to the observation of a general trend toward decreasing isoform diversity over preimplantation development, an enrichment in 3′ partial transcripts that lack stop codons in the oocyte and zygote, and the identification of dynamic changes in transposable-element expression (Wang et al. 2024).
The central nervous system features multiple cell types arranged in complex networks that vary and even migrate during development. Our analysis of the prefrontal cortex and hippocampus of the postnatal mouse revealed the predominance of cell-type specificity in driving isoform variation between the regions (Joglekar et al. 2021). By sequencing samples from multiple developmental time points and brain regions, we demonstrated a complex interplay between age, brain region, and cell type in influencing isoform expression in the mouse brain (Joglekar et al. 2024). Development and isoform variation in the human brain have been investigated using induced pluripotent stem cell (iPSC)–derived organoids, revealing increased intron retention in neurons compared to progenitors and an association between differential exon usage and autism-associated exons (Yang et al. 2023).
Clustering approaches
Clustering of single-cell RNA-seq data enables the grouping of the individual single cells of similar expression profiles represented in a data set into biologically meaningful categories, allowing useful conclusions about gene and isoform expression to be drawn. High cell counts are important for efficient clustering and identification of rare cell types, given within-group heterogeneity. The lower throughput of long-read sequencing approaches thus presents a challenge for effective clustering, as does the higher error rate of long-read platforms, which causes distortions in the relatively short cell barcodes, generating some fictitious ones. Error rates for ONT and PacBio have been dropping, and are currently approaching 1% and 0.1%, respectively, compared to ∼0.1% for Illumina; throughput per run varies by instrument, but for Illumina and ONT can be as high as several terabases, whereas it is only a few hundred gigabases at most for PacBio. A common solution is the parallel use of both short-read and long-read sequencing, where the same single-cell barcodes are used on both platforms. Clustering is performed on the short-read data, allowing the long reads to be categorized without clustering them directly. Our software package scisorseqR facilitates this kind of approach, performing barcode deconvolution and selection of full-length spliced reads, among other functions (Table 1; Joglekar et al. 2021). Alternative approaches have been developed, however, for performing clustering directly on long-read data. The computational tool scNanoGPS algorithmically filters cell barcodes from ONT sequencing in order to retain only high-confidence barcodes that are then clustered, demonstrating high concordance with short-read results (Table 1; Shiau et al. 2023). Two other software tools for ONT sequencing, ScNaUmi-seq and BLAZE, perform algorithmic correction of both cell barcodes and UMIs (Table 1; Lebrigand et al. 2020; You et al. 2023). The latest version of ScNaUmi-seq offers both the option to incorporate parallel sequencing with Illumina into the error-correction process or to rely solely on the ONT data. All three tools allow the user to derive a more accurate set of cell barcodes (and UMIs in the case of ScNaUmi-seq and BLAZE) from ONT sequencing output. Finally, MAS-ISO-seq avoids short reads by relying on the concatenation of cDNA molecules to improve the efficiency of circularized consensus sequencing with PacBio (Table 1; Al'Khafaji et al. 2024).
Comparison of clustering methods for single-cell long-read sequencing
Spatial sequencing
A key drawback of creating a single-cell or -nucleus suspension is the resulting loss of information about the spatial organization of the constituent cells. The first high-throughput RNA-seq method that preserved spatial information was applied to mouse olfactory bulb and human breast cancer biopsy tissue. Instead of barcodes encoding individual-cell identity, as with single-cell RNA-seq, positional barcodes were used to indicate the physical locations of captured transcripts within a tissue slice, with each location having a diameter of 100 µm and a center-to-center distance of 200 µm, resulting in a coarser-than-single-cell level of resolution (Ståhl et al. 2016). However, spatial resolution continued to grow with subsequent methods, approaching the sizes of individual cells (Rodriques et al. 2019; Vickovic et al. 2019; Chen et al. 2022; Zhang et al. 2023a) and now reaching subcellular resolution (Cho et al. 2021; Schott et al. 2024).
Methods that combine spatial approaches with ONT long-read sequencing have yielded biological insights into multiple structurally complex biological tissues (Fig. 2E). In mouse brain, we analyzed isoform usage in the hippocampus and prefrontal cortex using a combination of ONT with the 10x Genomics Visium platform, yielding examples of region-specific isoform diversity (Joglekar et al. 2021). Analysis of two forms of glioma revealed the role of isoform diversity in brain cancer, also utilizing the Visium platform (Ren et al. 2023). In both studies, spatial transcriptomics was supplemented by single-cell transcriptomics to provide a broader picture of the relevant biology. A study of postinfarction mouse myocardium—revealing increased intron retention in injured areas—was used to demonstrate the efficacy of a new method of spatial barcode assignment (Boileau et al. 2022). A spatial ONT study of mouse brain identified regional patterns of both isoform expression and RNA editing (Lebrigand et al. 2023), relying on the abovementioned ScNaUmi method of barcode and UMI correction that was originally developed for single-cell ONT sequencing (Lebrigand et al. 2020). As with many single-cell long-read approaches, the method leveraged the higher depth of short-read sequencing of the same samples for barcode clustering.
It is worth noting that imaging approaches that do not involve sequencing have found widespread use in spatial transcriptomics. Among the most prominent is multiplexed error-robust fluorescence in situ hybridization (MERFISH), where the sample is exposed to multiple rounds of fluorescent probes designed to be complementary to various portions of a set of RNA sequences of interest. Each tested RNA sequence gets assigned a unique binary code during probe design, and the pattern of fluorescent points seen over multiple rounds spells out one of these codes at each location. In this way, the expression of several hundred to 1000 genes can be measured simultaneously and quantitatively (Chen et al. 2015). MERFISH and related nonsequencing-based methods provide a high degree of spatial resolution but lack the capability to detect the expression of genes beyond the limited set that is chosen for targeting. Furthermore, they identify sequences based on overall complementarity between probes and targets rather than a base-by-base correspondence, making them less suited for detecting differences between isoforms of the same gene.
Spatial versus single-nucleus approaches
Spatial sequencing approaches offer a number of advantages over single-nucleus and even single-cell approaches, especially when it comes to isoforms. Most obviously, spatial approaches provide information on the exact position of a cell within a tissue slice. Secondly, as a cell's cytosol covers a larger area than its nucleus, approaches that cover spatially distributed spots, such as current spatial approaches, will tend to be dominated by cytosolic RNAs, implying more mature RNA molecules (see above). Moreover, with rising spatial resolution (Cho et al. 2021), spatial approaches will increasingly be able to distinguish between cytosolic and nuclear RNAs—and even RNAs of other organelles. Additionally, spatial approaches have an advantage when using antibody-based targeting: Single-cell and single-nucleus approaches require the target to be present within the cell or nucleus, or on the cell or nuclear membrane (Stoeckius et al. 2017; Mimitou et al. 2019). Spatial approaches, however, offer the additional ability to detect these targets of interest in extracellular space. Another advantage of spatial approaches is that cells are not isolated from each other in a disruptive fashion. In the nervous system, this could be an important advantage, as neuronal axons and synapses can remain in place, preserving cell–cell interactions and enabling short-range connectome studies. In cancer studies, information about microenvironments could be preserved.
However, the above considerations come with a catch: Single-nucleus (and for some of the following points, also single-cell) approaches do offer some important upsides. First, single-cell and single-nucleus approaches offer opportunities to enrich or de-enrich for important but possibly rare cell populations, using prior cell-sorting experiments. Such rare populations can include widely recognized cell types, such as blood vessel-associated cells, which tend to be rare in single-cell, single-nucleus, or even spatial experiments. Thus, single-nucleus experiments have shed light on important aspects of the brain's vasculature (Garcia et al. 2022).
In the brain, other examples might include rare subtypes of inhibitory or excitatory neurons. Moreover, cells with a certain disease-associated trait, such as protein aggregates in neurodegeneration, could also be sorted for, given the clinically meaningful heterogeneity being identified in this domain (Handsaker et al. 2024). An equivalent solution for spatial approaches would require the dynamic placing of physical spots at nonregular intervals on tissue slices—a technology that does not appear obvious at the time of writing. Thus, while spatial approaches can also be used for antibody-based methods, for now, they will divert most sequencing resources to cells that do not belong to the rare cell population of interest.
Another drawback of spatial sequencing is the fact that, as long as the spots are placed either at regular intervals or randomly, larger cells will tend to dominate the signal. In the human or nonhuman primate hippocampus, for example, pyramidal neurons are, therefore, likely to occupy a much larger fraction of the signal, while smaller cells, unless extremely abundant, would yield smaller fractions of reads in any sequencing experiment. Again, the abovementioned sorting approaches can help focus sequencing depth on the desired cell type. Finally, it is worth noting that while the prospect of being able to separately barcode distinct subcellular components—thanks to growing spatial resolution—is appealing, the issue of how to assign barcodes to particular components will be nontrivial and may require new computational approaches.
Discussion
The various features of a transcript molecule that can differentiate it from others derived from the same gene play an important role in determining the characteristics of cells and tissues. A detailed understanding of the processes involved is still lacking, especially in comparison to the better-characterized effects of differential gene expression.
The development of single-cell RNA sequencing was initially limited to short reads—offering limited coverage of RNA variables within a transcript and the correlations between them—but it introduced the ability to analyze transcriptomic data at the level of distinct cell types. With the advent of long-read technologies, researchers gained an improved view of the TSS, exons, exon junctions, and poly(A) sites whose variability contributes to isoform diversity. A broad array of technologies, both molecular and computational, have been deployed around single-cell and later spatial long-read RNA sequencing over the past decade. These have produced novel tissue-, disease-, and species-specific observations of the role of RNA variables in physiology and pathology. However, many unanswered questions remain regarding the precise functional consequences of this isoform diversity and regulation. Indeed, since the induced phenotypic changes may in many cases be subtle, they can be difficult to identify. Conversely, they may also be catastrophic and therefore fatal to a cell or organism, posing a further observational challenge. But with new discoveries and technological improvements continuing to be made, we are entering an era where we will be learning a great deal more about the organs or cell types experiencing changes in isoform expression, allowing us to assemble a detailed picture of the role of RNA isoform diversity in both disease processes and homeostasis maintenance. Such investigations now have the potential to be further enhanced by emerging advances in multimodal measurements, allowing for the simultaneous measurement of RNA variables and other related factors such as chromatin accessibility. Another route toward an expanded understanding of isoform regulation could come from more detailed long-read studies investigating the interaction between splicing and processes like somatic mutation, RNA editing, and RNA modification, something we have previously commented on (Joglekar et al. 2023).
We and others have continued to rely on short reads in order to reach sufficient coverage for effective cell-type identification. We have also enriched our long-read libraries for genes of interest to improve depth for isoform analysis (Hu et al. 2024). As long-read sequencing costs continue to decrease, leading to larger read numbers, these strategies may become less necessary, especially with the development of computational approaches designed to efficiently extract barcodes while taking higher error rates into account and without short-read sequencing in parallel. On the other hand, approaches emphasizing high numbers of reads per cell are continuing to be developed and in some cases commercialized, such as Smart-seq (Ramsköld et al. 2012) and MAS-ISO-seq (Al'Khafaji et al. 2024). This may require continued supplementation with short reads if statistically meaningful numbers of cells are to be sequenced for analyses of differences between cell types or conditions.
For spatial sequencing, the continued movement toward higher spatial resolution promises to unlock new abilities to analyze the influence of the hyperlocal environment—for example, extracellular protein aggregates such as amyloid beta in AD—on cells’ isoform expression. Even subcellular structures like nuclei or synapses could one day be analyzed separately. Indeed, recent advances in the development of subcellular resolution and three-dimensional spatial short-read sequencing with the Open-ST experimental and computational methods make the prospect of a long-read approach with similar capabilities all the more likely and promising (Schott et al. 2024).
As we have sought to demonstrate in this review, the single-cell and spatial long-read RNA sequencing field sits at the confluence of several novel technologies and has already provided insight into the biological importance of isoform diversity. There continues to be a need for a deeper, more isoform-focused understanding of transcriptomics that goes beyond the quantification of gene expression—a need that is likely to further spur the development of methods that can meet the challenges posed by these questions.
Long-read sequencing, including in a single-cell and spatial context, is an increasingly used and innovative technology. However, as is the case with many new technologies, there are still challenges the community needs to overcome. For instance, long reads may miss some splice junctions that are detectable by short-read methods at the same sequencing depth (Han et al. 2024). Long-read-derived mRNA sequences show inconsistencies at 5′ and 3′ ends and often do not match annotated coordinates (Calvo-Roitberg et al. 2024). Furthermore, the accuracy of novel-transcript discovery with long reads may still be lagging behind the accuracy of annotated-transcript identification (Pardo-Palacios et al. 2024b). The relative strengths and weaknesses of ONT and PacBio also deserve further attention and study—for instance, our direct comparison suggests that ONT, with its higher error rate but also higher data yield per run, is best suited for quantification of the expression of annotated-transcript features, while PacBio is preferable for identifying novel features (Mikheenko et al. 2022).
Competing interest statement
H.U.T. has presented at user meetings of 10x Genomics, Oxford Nanopore Technologies, and Pacific Biosciences, which in some cases included payment for travel and accommodations. The other authors declare no competing interests.
Acknowledgments
This work was supported by National Institutes of Health grants R35GM152101 (to H.U.T.), U01DA053625 (to H.U.T. among others), and T32DA039080 (to N.B. and J.H.). Figures were generated with BioRender (https://www.biorender.com/).
Footnotes
-
Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279640.124.
-
Freely available online through the Genome Research Open Access option.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








