
Complete genomes of Asgard archaea reveal diverse integrated and mobile genetic elements
- Luis E. Valentin-Alvarado1,2,
- Ling-Dong Shi1,
- Kathryn E. Appler3,
- Alexander Crits-Christoph1,2,11,
- Valerie De Anda3,4,
- Benjamin A. Adler1,
- Michael L. Cui1,
- Lynn Ly5,
- Pedro Leão3,12,
- Richard J. Roberts6,
- Rohan Sachdeva1,
- Brett J. Baker3,4,
- David F. Savage1,7,8 and
- Jillian F. Banfield1,4,9,10
- 1Innovative Genomics Institute, University of California, Berkeley, California 94720, USA;
- 2Department of Plant and Microbial Biology, University of California, Berkeley, California 94720, USA;
- 3Department of Marine Science, University of Texas at Austin, Marine Science Institute, Port Aransas, Texas 78373, USA;
- 4Department of Integrative Biology, University of Texas at Austin, Austin, Texas 78712, USA;
- 5Oxford Nanopore Technologies Incorporated, New York, New York 10013, USA;
- 6New England Biolabs, Ipswich, Massachusetts 01938, USA;
- 7Howard Hughes Medical Institute, University of California, Berkeley, California 94720, USA;
- 8Department of Molecular and Cell Biology, University of California, Berkeley, California 94720, USA;
- 9Earth and Planetary Science, University of California, Berkeley, California 94720, USA;
- 10Department of Microbiology, Biomedicine Discovery Institute, Monash University, Victoria 3168, Australia
Abstract
Asgard archaea are of great interest as the progenitors of Eukaryotes, but little is known about the mobile genetic elements (MGEs) that may shape their ongoing evolution. Here, we describe MGEs that replicate in Atabeyarchaeia, a wetland Asgard archaea lineage represented by two complete genomes. We used soil depth–resolved population metagenomic data sets to track 18 MGEs for which genome structures were defined and precise chromosome integration sites could be identified for confident host linkage. Additionally, we identified a complete 20.67 kbp circular plasmid and two family-level groups of viruses linked to Atabeyarchaeia, via CRISPR spacer targeting. Closely related 40 kbp viruses possess a hypervariable genomic region encoding combinations of specific genes for small cysteine-rich proteins structurally similar to restriction-homing endonucleases. One 10.9 kbp integrative conjugative element (ICE) integrates genomically into the Atabeyarchaeum deiterrae-1 chromosome and has a 2.5 kbp circularizable element integrated within it. The 10.9 kbp ICE encodes an expressed Type IIG restriction-modification system with a sequence specificity matching an active methylation motif identified by Pacific Biosciences (PacBio) high-accuracy long-read (HiFi) metagenomic sequencing. Restriction-modification of Atabeyarchaeia differs from that of another coexisting Asgard archaea, Freyarchaeia, which has few identified MGEs but possesses diverse defense mechanisms, including DISARM and Hachiman, not found in Atabeyarchaeia. Overall, defense systems and methylation mechanisms of Asgard archaea likely modulate their interactions with MGEs, and integration/excision and copy number variation of MGEs in turn enable host genetic versatility.
Asgard archaea, including Loki-, Hermod-, Thor-, Odin-, Baldr-, Freya/Jord-, Sif-, Heimdall-, Atabey-, Asgard-, and Wukongarchaeia, bridge our understanding of the evolution of eukaryotes and prokaryotes (Spang et al. 2015; Seitz et al. 2016; Zaremba-Niedzwiedzka et al. 2017; Imachi et al. 2020; Farag Ibrahim et al. 2021; Sun et al. 2021; Zhang et al. 2021; Tamarit et al. 2022, 2024; Eme et al. 2023; Valentin-Alvarado et al. 2024). Their genomic features, particularly the presence of eukaryotic signature proteins (ESPs), provide insights into the steps leading to eukaryotic cellular complexity. Recent phylogenetic analyses place eukaryotes within Asgard archaea (Spang et al. 2015; Zaremba-Niedzwiedzka et al. 2017), most closely related to Hodarchaeales (Eme et al. 2023). Despite intense interest in their functionality and evolutionary relationships, little has been reported regarding Asgardarchaeota mobile genetic elements (MGEs) that may shape their population diversity, contribute to genome divergence, and facilitate cross-domain horizontal gene transfer (Ghaly et al. 2022). Recent studies identified viruses of Loki-, Odin-, Thor-, and Heimdallarchaeia (Medvedeva et al. 2022; Rambo et al. 2022; Tamarit et al. 2022; Wu et al. 2022), as well as putative transposons carrying cargo genes that replicate within Heimdallarchaeia (Wu et al. 2022), primarily based on CRISPR spacer targeting. However, the limitations of CRISPR-based approaches and the lack of complete genomes in archaea make it challenging to comprehensively identify viruses and other MGEs in this domain. To our knowledge, no plasmids or integrative conjugative elements (ICEs) have been described for Asgard archaea.
Recently, we reported two complete and three near-complete genomes for Atabeyarchaeia, a new group of Asgard archaea, and a complete genome for Freyarchaeia (Valentin-Alvarado et al. 2024). Here, we present a comprehensive analysis of MGEs in Atabeyarchaeia and Freyarchaeia, two groups of Asgard archaea, using a combination of metagenomic, metatranscriptomic, and epigenetic approaches. Leveraging complete metagenome-assembled genomes and read-based population analyses, we track subtle strain variations of integrated MGEs over a soil depth profile. This approach allows us to establish the host, precisely define MGE insertion sites, and determine MGE lengths.
Our study aims to expand the repertoire of known MGEs in Asgard archaea and provide new insights into their potential roles in archaeal evolution and ecology. Additionally, we investigate MGE integration and excision events in natural populations of Atabeyarchaeia and genomically define groups of circular viruses and unclassified MGEs in Asgard archaea. We also describe genomically encoded defense systems of both Atabeyarchaeia and Freyarchaeia, confirming their expression using metatranscriptomic data, and report methylation patterns that distinguish these archaea, including the identification of transcriptionally active MGE-encoded methylases.
This study demonstrates the power of combining complete metagenome-assembled genomes, population-level analyses, and multiomic approaches to uncover the diversity and functionality of MGEs in uncultured microorganisms from complex environmental samples.
Results
Complete Atabeyarchaeia genomes contain integrated genetic mobile elements
Short- and long-read metagenomic data sets were generated from wetland soil sampled at a single local site (for details, see Methods). We mapped reads from 28 samples collected from soil depths of 60 to 175 cm to the Atabeyarchaeum deiterrae group 1 (GCA_037308085.1), A. deiterrae group 2 (GCA_037310415.1), and Freyarchaeum deiterrae (GCA_037305845.1) genomes previously assembled from this environment and used mapped read details to uncover evidence for integrated, excised, and coexisting circularized MGEs (Fig. 1). The absence of MGEs in some cells can lead to lower-than-average coverage over the integrated region, whereas higher read depth of coverage is likely owing to coexisting extrachromosomal versions of the MGE. The sequences identified occur only once in the genome and do not occur in other genomes from the same sample. By manual inspection of sequencing depth and read alignment discrepancies, we identified 14 chromosomally integrated MGEs in the A. deiterrae group 1 (Atabeya-1) genome (Fig. 2A) and four in the A. deiterrae group 2 (Atabeya-2) genome (Fig. 2B), ranging from 1.3 to 40 kbp in length. No integrated elements were identified in the F. deiterrae genome using this approach (Fig. 2C). Comparison of the sizes of these Atabeyarchaeia MGEs to other known archaeal MGEs reveals that they fall within the typical size range for archaeal plasmids and viruses (Supplemental Fig. S1). The identified MGEs range from 1.3 to 40 kbp, which is consistent with the size distribution observed in other archaeal lineages, including Euryarchaeota and Asgardarchaeota.

Read mapping to the reference genome provides evidence for integration and excision, illustrated for the case of one mobile genetic element (MGE). The central region of the integrated sequence of Yucahu-i (between black bars) has been deleted to focus on details of reads mapped to the start and end of the region. Read sequences that match the genome sequences are shown as gray bars; small vertical colored bars adjacent to read portions in agreement with the reference indicate bases that disagree with the reference. The yellow, purple, blue, green, and orange arrows indicate distinct nucleotide sequences (same color, same sequence). In the panel demonstrating that some cells that lack the integrated MGE, one read has been split (the black line links the two parts of a single read) to illustrate agreement with the flanking sequence at both ends of the integrated region. Note that the sequence designated by the purple arrow occurs twice when the MGE is integrated. The vertical colored bars indicate single-nucleotide polymorphisms (SNPs) relative to the reference genome sequence and thus do not represent the sequence itself.
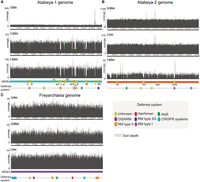
Chromosomally integrated MGEs and defense systems in soil Asgard Archaea genomes. Each panel (A–C) depicts the coverage across each complete genome, as determined by mapping to metagenome reads derived from three different soil depth profiles. Regions exhibiting low coverage suggest strain variations associated with specific soil depths and may indicate the presence of integrated MGEs in only a subset of cells. Notably, the Freyarchaeia genome exhibits even coverage using reads from all sampling depths, with no discernible integrated MGEs identified. Some of the low-coverage regions are not labeled as potential MGEs; these regions are strain variants with sequences so divergent that read mapping is precluded. Oval symbols indicate predicted defense systems.
These 18 integrated MGEs in Atabeyarchaeia genomes were classified based on their genomic content, size, and phylogenetic analysis of proteins associated to MGEs (for details, see Methods). The classification resulted in five insertion sequence-like transposons (ISs), three putative ICEs (7.9–12 kbp in length) carrying integration machinery and cargo genes, two defense islands, six provirus, and two elements that could not be definitively classified owing to their unique gene contents (Supplemental Table S1). To date, the only Asgard nonviral integrated MGEs reported are Heimdallarchaeia “aloposons” (Wu et al. 2022), which are transposons that carry cargo genes. These previously reported MGEs do not display any similarity at the nucleotide level with those found in Atabeyarchaeia. However, some Atabeyarchaeia integrated MGEs and these aloposons encode partition proteins (ParB-like) that are distantly related (Supplemental Fig. S2A). The Atabeyarchaeia tyrosine-like integrases are most closely related to those found in genomes of Njordarchaeales, Bathyarchaeia, and Aenigmarchaeota, which share a similar ecological distribution in terrestrial wetlands and also occur in deep ocean sediments (Supplemental Figs. S2B, S3; Seitz et al. 2019).
Ten of the integrated MGEs coexist in circularized forms with their integrated versions in the same metagenomic samples (e.g., Fig. 1). One of these of particular interest is Atabeya-1 MGE-i (Yucahu-i, in homage to the son of the Taíno goddess Atabey, reflecting our previous designation of the host archaeon as Atabeyarchaeia), for which the coexisting circularized version in 60 cm deep soil is four times more abundant than the integrated version (Fig. 3A). Some of the reads span the genome, which indicates that a subset of Atabeya-1 genomes lack or have excised this integrated element (Figs. 1, 3B), enabling us to determine the exact length of the Yucahu-i to be 10,867 bp. The MGE is inserted following an AATTAACTTAT sequence that is also present at the end of the integrated Yucahu-i and occurs within the excised, circularized version. This region likely represents the attachment (att) site, a unique location within the genome. The low GC content (9%) compared with the genome-wide average (∼50%) suggests that the DNA in this area may exhibit increased susceptibility to cleavage during processes such as excision or integration.

Integration and excision of Yucahu. (A) For the 60 cm sample, elevated coverage and paired reads indicate that Yucahu-i is integrated into the genome, excised from some genomes (blue lines), and coexists in circularized form (yellow lines). The red box indicates elevated coverage from a related gene from another genome. (B) For the 165 cm sample, low coverage over the MGE and read sequence discrepancies indicate that most cells in this sample lack the MGE. (C) For the 70 cm sample, coverage and paired read information indicate that Yucahu-i is integrated into essentially all cells. The circularized Yucahu-i is present but rare. Paired reads pointing out internal to the MGE indicate that a 2644 bp element has integrated into the plasmid and coexists in circularized form.
The Yucahu-i element includes 11 open reading frames (ORFs) (Fig. 3A). Some of the gene products could be functionally annotated using protein homology and in silico structural prediction. The first gene encodes a tyrosine recombinase/integrase that likely recognizes and cuts at the AATTAACTTAT motif in the genome and in the circularized version (resulting in Yucahu-i linearization) and may be involved in integration of the linear sequence. The subsequent gene is a Holliday junction resolvase, which likely acts in conjunction with the integrase. We are uncertain if a host integration factor is required, but it is possible that two of the following genes predicted to encode DNA-binding proteins, based on their HTH-domains, may have this function. Yucahu-i also encodes a superfamily 3 (SF3) helicase that may unwind the DNA and initiate plasmid replication (Guo and Huang 2010), and a novel Type IIG restriction-modification (IIG RM) protein fusion that combines endonuclease and methyltransferase (MTase) activities. Phylogenetic analyses suggest the IIG RM sequence shares its most recent common ancestor with sequences found in DPANN archaeal genomes (>60% amino acid identity). Basal to this clade are many sequences from bacteria, which supports the inference that the origin of the sequences in question is likely archaeal, potentially acquired via horizontal gene transfer (Supplemental Fig. S4). Metatranscriptomic data indicate that the IIG RM gene is transcribed (Supplemental Table S2). Based on predicted protein functions and presence of the excised, circular (copy ratio up to 4×) MGE, Yucahu-i is likely an ICE (Wozniak and Waldor 2010).
We investigated how frequently Yucahu-i was integrated in, or coexisted in circular form with, the Atabeya-1 genome by systematically analyzing reads from the 20 soil metagenomes that contained this archaeon (Supplemental Fig. S5; Supplemental Table S3). In 11 samples, Yucahu-i is integrated into essentially all Atabeya-1 cells; however, the data indicate substantial variation in presence/absence of the integrated version and in the copy number of the circularized version (Fig. 3A; Supplemental Fig. S5; Supplemental Table S3). A few reads from the 70 cm deep soil revealed evidence for the circularization of a 2644 bp element that is integrated within Yucahu-i. We refer to this as mini-Yucahu-I (Fig. 3C). Its presence highlights the genetic plasticity of the plasmid. The mini-Yucahu-i carries a putative ParG, a hypothetical protein, and Holliday junction ATP-dependent DNA helicase RuvB. Interestingly, the identical 11 bp Yucahu-i putative attachment motif is also present adjacent to, and within, a 7848 bp integrated element in the Atabeya-2 genome (iMGE-xvi). However, the genomes share no detectable similarity; the percentage of identity of the tyrosine integrases are <25%; and they are phylogenetically unrelated (Supplemental Fig. S6).
Viruses and plasmids targeted by CRISPR systems
To explore exogenous MGEs of Atabeyarchaeia and Freyarchaeia, we mined CRISPR spacers from their genomes and matched them to unbinned metagenomic scaffolds from the same wetland soil. More than 30 putative MGE scaffolds are confidently targeted by CRISPR spacers and thus are predicted to have once replicated within Atabeyarchaeia (Supplemental Table S4). We manually curated them and obtained one complete 20.8 kbp circular plasmid genome; two circular, complete 40.1 kbp genomes for a pair of closely related viruses; and a circular complete 26.7 kbp genome for an unclassified MGE.
The 20.8 kbp plasmid has 24 ORFs, primarily encoding hypothetical proteins (Supplemental Fig. S7; Supplemental Tables S5, S6). It also encodes plasmid proteins such as protein repressor ribbon–helix–helix protein from the CopG family (Gomis-Rüth et al. 1998), usually present in bacterial conjugative plasmids. Other predicted proteins are implicated in autonomous replication, such as a DNA primase-helicase, mini-topoisomerase VI, and a tyrosine integrase, as well as other genes, involved in nucleic acid processing. Seven of these proteins contain transmembrane domains, suggesting the presence of a putative conjugative system or a secretion-like system (Supplemental Fig. S7). A protein with a Glu–Glu motif was annotated as an integral membrane CAAX-like protease self-immunity, based on structural modeling and phylogeny (Supplemental Fig. S8). It encodes a NTPase with similar function to ParB, a protein typically associated with plasmid chromosome partitioning during replication. Phylogenetic analysis places this protein within a clade that contains MGEs recently discovered in Heimdallarchaeia (Supplemental Fig. S2A). Interestingly, this clade also contains ParB-like proteins from publicly available draft genomes of Lokiarchaeia and Thorarchaeia, along with other archaeal genomes (e.g., Sulfolobales), suggesting that this plasmid lineage is widespread in other Asgard archaea. Also included are sequences from Streptomyces plasmids. Therefore, these plasmid partitioning genes may have undergone inter-domain horizontal gene transfer.
We identified a circular 40,094 bp novel virus predicted to infect Atabeyarchaeia-2 based on CRISPR spacer matches. A search against the IMG/VR v4 (Camargo et al. 2023) database revealed no matches for these spacers to any known viral sequence, highlighting the novelty of both the virus and the host. Genomic analysis of the virus with geNomad (Camargo et al. 2024) classified this group of viruses within the class Caudoviricetes (realm Duplodnaviria) (Supplemental Table S7). We named this virus “Opia” after a mythical creature associated with the Taíno goddess Atabey. Interestingly, we found this virus integrated into the end of a 2.58 Mbp PacBio-derived genome fragment from a related Atabeya-2 strain (Atabeya-2′), confirming its host association and a putatively temperate replication cycle.
The Opia virus genome encodes structural proteins typical of archaeal tailed viruses, including capsid-like, phage head morphogenesis, portal, and tail-like proteins, as well as a large terminase subunit (KEGG: K06909). Phylogenetic analysis of capsid and terminase proteins groups Opia with other Asgard viruses, notably Nidhogg virus (Fig. 4A,B). The genome also contains genes for nucleic acid processing, including a Mu-like prophage protein, putative transposase, tyrosine recombinase-like enzyme, site-specific DNA-MTase, and a ParB-like NTPase.
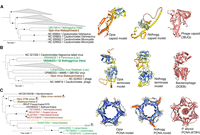
Evolutionary relationships and structural conservation of Opia virus hallmark proteins. (A–C) Maximum-likelihood phylogenetic trees of capsid (A), terminase large subunit (B), and proliferating cell nuclear antigen (PCNA; C). Trees are midpoint-rooted; circles indicate bootstrap support >70%. Insets show structural models: Opia virus (infecting Atabeyarchaeia, this study) and Nidhogg virus (infecting Helarchaeales) colored by AlphaFold3 confidence (blue to red, high to low), with best FoldSeek match in coral. Reference structures: bacteriophage capsid (PDB: 3BJQ) for A, large terminase from thermophilic bacteriophage D6E (PDB: 5OEB) for B, and P. abyssi PCNA (PDB: 6T8H) for C. Scale bars represent one substitution per site. Opia virus variants cluster with other Asgardarchaeota MGEs, highlighting their evolutionary relationships within this archaeal phylum.
Structural models of Opia virus hallmark proteins, generated using AlphaFold 3, reveal conservation with known viral and archaeal structures (Fig. 4). The capsid and terminase models show similarity to Nidhogg virus and reference bacteriophage structures, suggesting potentially functional conservation across diverse viral lineages. The PCNA-like protein model closely resembles that of Nidhogg virus and Pyrococcus abyssi PCNA, indicating a conserved role in DNA replication.
A DNA polymerase sliding clamp subunit (PCNA-like protein) was identified (Fig. 4C), which may promote viral DNA synthesis and manipulate host pathways. Phylogenetic analysis places the Opia PCNA-like sequence within an Asgard archaea clade, with its closest homolog in Njordarchaeales (MCD6165036.1) from the Auka vent field (Speth et al. 2022), suggesting that viruses related to Opia virus integrate into other Asgard genomes. Similar proteins are found in the Sköll viral genome infecting Lokiarchaeia and other archaeal viruses (Raymann et al. 2014; Mizuno et al. 2019; Medvedeva et al. 2022; Rambo et al. 2022; Tamarit et al. 2022).
At the whole-proteome level, the Opia virus clusters with other Atabeyarchaeia MGEs and has similarity to the Ratatoskr, Nidhogg, Skoll, and Fenrir viruses (Rambo et al. 2022), known to infect various Asgard archaea (Fig. 5A). Analysis of genome sizes among reported Asgard viruses, including Opia virus from this study, reveals a diverse range of genomic lengths (Fig. 5B; Supplemental Table S8). The Opia virus, with its 40,094 bp genome, falls within the mid-range of Asgard viral genome sizes, which span from ∼15 kb to >100 kb. This variation in genome size likely reflects the diverse replication strategies and host interactions among Asgard-infecting viruses, with Opia representing an intermediate complexity within this spectrum.
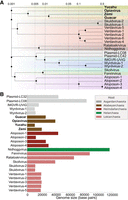
Comparative genomics of Asgardarchaeota MGEs. (A) Whole-proteome similarity network of MGEs from Atabeyarchaeia and other Asgardarchaeota (Medvedeva et al. 2022; Rambo et al. 2022; Wu et al. 2022). Nodes represent individual MGEs, with edge thickness indicating similarity strength. Yucahu, Opia virus, Zemi, and Guacar form a distinct cluster, separate from other Asgard MGEs. Analysis based on NCBI database entries. (B) Genome size distribution of reported Asgard viruses/MGEs, including those from this study.
We identified at least seven distinct Opia virus variant genotypes from different samples. The sequences align near-perfectly over >85% of the genomes (Fig. 6A). All Opia variants (and their identifiable fragments) are exactly targeted by one CRISPR spacer present in loci of both Atabeya-2 and Atabeya-2′ (two identical sequential spacers in Atabeya-2′), despite the presence of the Opia provirus in the Atabeya-2′ genome. The provirus was only partially recovered, so it is impossible to say whether the integrated version differs in the targeted region. A hotspot in the Opia virus genomes encodes a series of genes that are distinctly different in some variants. Included are up to six small cysteine-rich proteins, many with predicted double Zn-binding domains (Fig. 6A). The genes for specific cysteine-rich proteins occur in different combinations from different genotypes (e.g., one has sequence types A, B, D; another has A, C, D; and another has C, E) (Fig. 6B). In addition, a three-gene block (one of which has sequence variants) and adjacent intergenic sequences are variably present/absent. Finally, different versions of ParB-like partition proteins occur in the variable region, and some lack a C-terminal endonuclease domain (Fig. 6A).
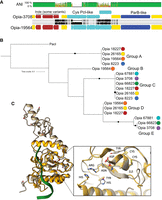
Sequence variation in the cysteine-rich proteins of Opia viruses and comparison to Pacl, a rare-cutting HNH restriction endonuclease. (A) The aligned ∼5.6 kbp variable region of two Opia viral genotypes. Light gray bars indicate perfect nucleotide identity, and thin vertical black lines indicate SNPs. The three genes labeled in brown are present in both Opia-3708 (top) and Opia-19564 (bottom) but are absent in some other genotypes. Light blue genes are cysteine-rich (For 3708L, 10, five, nine, five, 10; for Opia-19564, three, 14, five, nine, four, 10 cysteines per protein). Dark blue genes encode ParB-like proteins that are very divergent between some genotypes (Opia-3708 vs. Opia-66823, 67881). Before the three-gene indel and after the ParB-like gene, the ∼35 kb regions of all genomes are essentially identical. (B) Phylogenetic tree including the 17 larger cysteine-rich proteins from the variable regions of seven Opia genomes (numbers represent genome names) (for context, see Supplemental Material). Proteins with identical sequences occur in five different combinations across the genotypes. (C) Comparison of the active site region of PacI (with nine cysteines) and the structure of Opia-19564 protein 16 (with 10 cysteines; silver). Active site residues of PDB 3m7k (gold) are displayed based on figure 1 of Shen et al. (2010). The critical ββα-metal motifs are well aligned. Tyrosine and histidine residues exist in proximity to the active site, although their locations differ somewhat, possibly owing to fold inaccuracy.
We predicted the structures of the largest cysteine-rich protein from Opia-3708 (gene 46) and two Opia-19564 proteins (genes 16, 18). All three represent different protein sequence clusters, but they share core structural components, including two sets of four cysteines, and an alpha helix in proximity to paired antiparallel beta strands (i.e., ɑ, ββ,-metal; Fig. 6C). HHpred predicts the Opia-3708 protein (197 aa) to be related to an HNH endonuclease, and the best match for the three-dimensional structure (PDB 3M7K) is the Rare-Cutting HNH Restriction Endonuclease PacI, a homing endonuclease that is one of the smallest restriction endonucleases known (142 aa) (Shen et al. 2010). Zinc bound by the four cysteines is required for the DNA cleavage by PacI endonucleases. The H, DR, and CxxCN catalytic residues of 3M7K HNH endonuclease are generally conserved in the Opia proteins (e.g., Opia-19564_16) (Fig. 6C). However, the expected tyrosine residue precedes rather than follows the DR motif, and its placement in the predicted structure is offset from that in 3M7K. The histidine active site residue is also slightly differently positioned (Fig. 6C). These discrepancies may be attributed to uncertainties in protein folding. However, the positioning of histidine in the location typically occupied by tyrosine suggests its potential involvement in DNA cleavage, as occurs in other HNH endonucleases. Elsewhere, the predicted structures have large regions of positively charged surface, likely involved in DNA binding. These findings suggest that the Opia proteins share characteristics with PacI restriction endonucleases, yet they may represent a novel class of enzymes, likely with homing endonuclease function (Fig. 6B). The biochemically characterized PacI homodimer has a target recognition sequence of 5′-TTAATTAA-3′ and cleaves between the internal thymine residues. Pacl endonucleases rely on the absence of the recognition site elsewhere in the host genome. We could not determine the recognition sequence for the Opia Pacl-like homing restriction endonuclease, but apparently it was possible for different combinations of six variants to insert in the same region of a series of Opia genotypes.
We further reconstructed a circular, complete 26,349 bp genome (MGE-9917) for another circularized element that is targeted by three CRISPR spacers from Atabeya-1. This element, named “Guacar” after the twin son of Atabey, could not be definitively classified as either a virus or plasmid based on its predicted protein functions. All Guacar genes are encoded on the same strand. The genome features a CT-rich intergenic tandem repeat region with six, seven, eight, or nine units of 8 bp in length (variants identified using mapped reads). MGE-9917 encodes at least 14 proteins with transmembrane domains, two of which are 1402 and 1202 amino acids in length and lack related sequences in the NCBI database (Supplemental Tables S5, S6). The genome contains a protein that combines an N-terminal ParB-like nuclease domain with a C-terminal tRNA G10 N-methylase Trm11. Additionally, Guacar-9917 includes genes for a tyrosine recombinase, a transposase, and a Type IV methyl-directed restriction enzyme featuring an HNH motif. A family of related elements occurs in virtually all of the deep soil samples. One version differs owing to the presence of a transposase that is related to those found in the Opia viruses.
Defense systems and epigenetic regulations in Atabeyarchaeia and Freyarchaeia
We used DefenseFinder (Tesson et al. 2022) and PADLOC (Payne et al. 2022) to identify 10 defense systems in Freyarchaeia, five in Atabeyarchaeia-1, and six in Atabeyarchaeia-2. Freyarchaeia harbored at least four different defense system classes, including type I-B, III-A, and III-D CRISPR-Cas systems; IIG RM systems; the Hachiman antiphage defense system; and the antiphage system, defense island system associated with restriction-modification (DISARM) (Supplemental Fig. S9A; Supplemental Tables S9, S10). The identification of DISARM in Freyarchaeia adds to the growing list of defense systems recently reported in Asgard archaea (Leão et al. 2024). The DISARM system comprises drmABC, a MTase (drmMI, N6 adenine-specific MTase or drmMII, C5 cytosine-specific DNA MTase), and drmD or drmE (Ofir et al. 2018). The Freyarchaeia system includes drmA, drmB, drmC, drmMII, drmE, and drmD (helicase similar to the RNA polymerase [RNAP]-associated SWI2/SNF2 protein), classifying this system as a DISARM class II (DISARM-II). Interestingly, drmD is a homolog typically found in DISARM class I. The DISARM methylase modifies host CCWGG motifs to distinguish its own DNA from foreign DNA. A specific conformation of the DrmAB complex (trigger loop) inhibits the complex to prevent an autoimmune response (Bravo et al. 2022). DrmA is responsible for DNA targeting in DISARM through multiple nonspecific interactions with the DNA backbone (Bravo et al. 2022). By not requiring a specific sequence for DNA binding, DrmA distinguishes this defense system from other common restriction-modification systems, endowing DISARM with a broad spectrum of action against viruses (Tesson et al. 2022). Phylogenetic analysis of the helicase DrmA places the gene within the Euryarchaeota and Chloroflexota, suggesting that this system has been laterally transferred (Supplemental Fig. S9B). The Hachiman system is encoded by hamA, a DNA endonuclease (DUF1837), and hamB, a ski2-family helicase (pfam00271) bearing relation to archaeal Hel308 (Doron et al. 2018; Tuck et al. 2024). Recently, a model for Hachiman-mediated defense was reported (Tuck et al. 2024). Helicase HamB senses DNA structures from DNA damage (or structurally related replication intermediates), unleashes further DNA degradation through HamA endonuclease activity (Tuck et al. 2024), and confers antiphage immunity through abortive infection. Whether Hachiman-mediated immunity performs a similar role in archaea and synergizes with other immune systems remain unknown. Potentially, an ATP-dependent endonuclease is an overcoming lysogenization defect (OLD) upstream of the hamAB locus, providing synergistic protection against invading MGEs.
We used DNA polymerase kinetics from Pacific Biosciences (PacBio) metagenomic sequencing data and the Restriction Enzyme database (REBASE) to illuminate the DNA methylation patterns and methylases in the genomes of Freyarchaeia, Atabeya-1, and Atabeya-2. In the genome of Atabeya-1, we identified 13 methylation sequence motifs, of which seven were directly linked to a specific methylase gene. Similarly, Atabeya-2 has 11 methylation motifs, five of which could be linked to a methylase. Freyarchaeia has only five detectable methylation motifs (Supplemental Table S11A,B). Interestingly, one of those motifs is CCWGG, which has been characterized as a motif targeted by DISARM class II.
Atabeya-1 and Atabeya-2 both have 4-methylcytosine (m4C) and 6-methyladenosine (m6A) methylation. Atabeya-1 Yucahu-i MGE encodes a IIG RM system that targets an m6A methylation motif and is the only candidate enzyme that could methylate the Atabeya-1 genome. The Yucahu-i system is analogous to the MmeI family, which typically recognizes a 6–7 bp motif with adenine as the penultimate base (Morgan et al. 2009). The motif GYATGAG (m6A) was methylated at 66% of sites within the Atabeya-1 genome and could represent the active methylation motif of this RM system. In contrast, Freyarchaeia has five m4C motifs but no m6A methylation motifs.
To validate these findings, we analyzed previously sequenced Oxford Nanopore soil data (Schoelmerich et al. 2024). Both technologies support the m6A motifs. Although there was no Nanopore model for calling m4C, overextended versions of the m4C motifs were also detectable as m6A motifs (i.e., GCGm4C was detected as GCGCm6A) (Supplemental Table S11B,C). Additionally, two potential 5mC motifs were called: GATm5C in Freyarchaeia-1 and RG5mCWGCY in Atabeya-1.
Integrated mobile-like regions encode ESPs
We identified two small GTPases in a region of the Atabeya-1 genome that appears to be enriched in genes often associated with MGEs. This region is absent in some Atabeya-1 strains. The classification of these proteins as GTPases is supported by sequence and structural homology, as well as structural predictions in comparison to reference eukaryotic sequences (Supplemental Text). Based on phylogenetic analysis, these proteins cluster with Arf GTPases from other Asgardarchaeota and eukaryotes (Supplemental Fig. S10).
Discussion
Here, we describe chromosomally integrated and coexisting MGEs that replicate in Atabeyarchaeia by leveraging extensive sequencing of a series of soil samples in which soil depth and biogeochemical conditions select for different strain variant populations. By examining read mappings across multiple samples from the same environment, we established a larger repertoire of MGEs than could be found from the analysis of any single metagenome. Integrated elements and coexisting circularized MGEs range from 2.5 to 40 kb in length; all complete MGEs were circular, and at least some replicate bidirectionally. We could not confidently identify MGEs by changes in read mapping abundances in the Freyarchaeia genome. This might indicate stable integration into the Freyarchaeia genome across the entire population studied (thus excision sites and coexisting versions of circular elements were not detected by our methods). Assuming that Freyarchaeia do carry MGEs that excise, the best approach to finding them would be to analyze populations from a larger set of samples. The presence of coexisting integrated and free, circularized MGEs, likely mostly plasmids, as well as variation in copy number of circularized elements and in the fraction of cells with integrated elements, suggests regular movement of MGEs into and out of the Atabeyarchaeia chromosomes. Insertion/excision and variation in copy number may enable Atabeyarchaeia to respond to changes in their environment. For example, MGEs may behave synergistically, and increase in MGE copy number (thus gene content) serves as a response to increased pressure from other MGEs (Krupovic et al. 2019). The tiny mini-Yucahu indicates another layer of genomic variability, as just this portion of the host MGE can excise.
Interestingly, the attachment motifs for Yucahu-i plasmid-like Atabeya-1 MGE and for a circular, unclassified and essentially unrelated MGE linked to Atabeya-2 are exactly the same, implying that the very different integrases of each (25% aa ID) recognize and cut at the same motif. Protein sequence divergence may enable host chromosome specificity, yet the active site apparently evolved to target the same motif.
The novel cluster of genomically similar Opia viruses of Atabeya-2 and Atabeya-2′ encode sequential cysteine-rich proteins inferred to have nuclease activity owing to their distant homology with PacI. A set of two or three of five protein types occurs in the seven genomes, but the set present is generally different, except in the case of yellow and orange genotypes (Fig. 5B), which both have group A, B, and D protein types. These involve combinatorial patterns (Fig. 5B) that may have arisen via recent recombination events in which these putative endonucleases may have played a role. The multiple variants might form heterodimers rather than the normal homodimers expected for Pacl, possibly extending target recognition. The results suggest the importance of diverse nuclease activity for these viruses.
The Opia virus proteomes exhibit similarities (e.g., capsid, tube and tail proteins) to those of tailed viruses, which commonly replicate in bacterial and archaeal hosts from hypersaline environments (Senčilo and Roine 2014). They are quite distinct from those of eukaryotic viruses, supporting the suggestion that, despite the evolutionary relationship between Asgard archaea and eukaryotes, their viruses display no obvious evolutionary relationships (Medvedeva et al. 2022; Rambo et al. 2022; Tamarit et al. 2022).
Genome context (dominated by MGE-associated genes) and apparent excision of the region encoding the GTPases from some strain genotypes supports the inference that some Atabeyarchaeia MGEs encode ESPs. ARF GTPases are involved in membrane trafficking in eukaryotes and have been previously described as ESPs in Asgard archaea (Spang et al. 2015; Eme et al. 2023). If it is established that this is an active MGE that can excise, the presence of these GTPases provides the first indication that increase in cellular complexity could be associated with the transfer of ESPs via MGEs (Supplemental Figs. S10–S12; Supplemental Tables S5, S11).
Our results suggest several examples of genes integrated into Atabeyarchaeia genomes or in their coexisting MGEs that have nearest homologs in the bacterial domain (e.g., ParB, DrmA, and IIG RM enzymes). These findings are consistent with recent work on cross-domain gene transfer via movement of integrons associated with diverse MGEs (Ghaly et al. 2022) and extend earlier work inferring the acquisition of archaeal genes by bacteria (e.g., Hug et al. 2013). As we discover new Asgard archaea from genome-resolved metagenomes, we can expect to find further parallels between bacterial and archaeal immune systems. These associations could have implications for the evolution of eukaryotic immune systems (Wein and Sorek 2022).
IIG RM systems, to our knowledge, have not previously been associated with archaeal MGEs. The observation that the only methylase seemingly able to methylate at the host genome's well-represented m6A motif is carried by Yucahu-i, as well as that it is transcriptionally active, suggests that the Yucahu-i Type IIG plays a significant role in host genome epigenetic modification. This MGE-encoded system may protect its host Atabeyarchaeia against infection by other MGEs. This behavior aligns with the emerging perspective that defense systems themselves can serve as MGEs (Rocha and Bikard 2022; Wu et al. 2022).
To our knowledge, these are the first metagenome-derived Asgard archaeal complete genomes for which methylation patterns have been reported. PacBio sequences corresponding to these complete, manually curated genomes (Valentin-Alvarado et al. 2024) were used to infer the methylation motifs and to determine the fraction of sites that were methylated, as well as the methylation patterns of their newly reported MGEs. Using REBASE, which features all biochemically characterized methylases, it was possible to link methylated sites with likely methylases encoded on both genomes and MGEs. Relatively little is known about genome methylation in archaea, especially in Asgard archaea (Anton and Roberts 2021). These genomes and their methylation motif data presented here provide a starting point for detailed biochemical studies to expand the known inventory of archaeal methylases. The higher number of methylation motifs in Atabeyarchaeia compared with the Freyarchaeia genome could be an evolutionary response to the larger inventory of MGEs associated with Atabeyarchaeia.
We leveraged the read diversity inherent to population genomic data, long-read sequencing, methylation pattern analysis, comparative genomics, and functional and structural prediction to explore integrated and coexisting MGEs of one group of Asgard archaea. These analyses brought to light an extensive landscape of MGEs that associate with Atabeyarchaeia, including viruses, plasmids, and as-yet-unclassified entities (Fig. 7). The excision, insertion, and changes in copy number of these MGEs may enable adaptation to changing conditions and have contributed evolution, possibly to the acquisition and spread of genes linked to the origin of cellular complexity. The availability of MGEs that could be adapted for delivery of genome editing tools in a community context (Rubin et al. 2022) may pave the way for genetic manipulation of these archaea.

Phylogenetic placement of Asgard archaeal genomes and distribution of associated MGEs. The phylogeny shows the evolutionary relationships among different Asgard archaeal lineages, with symbols indicating the presence and types of published MGEs in various candidate groups. Descriptions of the phylogenomic analyses have been previously described in detail (Valentin-Alvarado et al. 2024). Our work on Atabeyarchaeia and Freyarchaeia is highlighted, demonstrating the novel MGEs identified in this study and the presence of two novel defense systems in Freyarchaeia. This figure provides a comprehensive overview of the current state of knowledge regarding MGEs across the Asgardarchaeota phylum.
Our study opens up avenues for future research. The first relates to comparative genomics of MGEs across the Asgard archaea and beyond, including investigations across diverse environments and temporal studies. Using enrichments of Asgard archaea or, if they become available, pure cultures, experimental studies could test the adaptive significance of specific MGEs. With enrichments, genetic manipulation methods designed for community editing may be used to inactivate prophages or eliminate the function of specific genes (e.g., methylases). Methylases are of particular interest, given that novel methylation patterns and restriction-modification systems were identified in Atabeyarchaeia and Freyarchaeia and are likely involved in epigenetic regulation and host–MGE interactions. Specifically, the unassigned methylase genes for unassigned motifs reported here per genome, are immediate targets for biochemical studies via heterologous expression. Of particular interest would be the functional characterization of MGE-encoded genes, including methylases, potentially linked to the development of multicellularity.
Methods
Sample acquisition, nucleic acid extraction, and sequencing
Metagenomic data were generated from deep wetland soil in Lake County, California (Al-Shayeb et al. 2022; Valentin-Alvarado et al. 2024). Briefly, we collected soil cores from a seasonally flooded wetland (SRVP) in Lake County, California, in October 2018, October 2019, November 2020, and October 2021 (38°41′39″N 122°31′36″W 571 m). Samples were frozen in the field using dry ice and kept at −80 C until extraction. The Qiagen PowerSoil max DNA extraction kit was used to extract DNA from 5–10 g of soil, and the Qiagen AllPrep DNA/RNA extraction kit was used to extract RNA from 2 g of soil. Samples were sequenced by the QB3 sequencing facility at the University of California, Berkeley on a NovaSeq 6000. Read lengths for the 2018 DNA samples and the RNA samples were 2 × 150 bp and 2 × 250 bp for the 2019–2021 DNA samples. A sequencing depth of 10 Gb was targeted for each of 2018, 2020, and 2021 samples and 20 Gbp for each of the 2019 samples. PacBio sequencing was obtained from a subset of deep soil samples from 2021 via the University of Maryland sequencing facility. Samples from September 9, 2021, from 140 cm and 75 cm were sequenced using a Sequel II to generate PacBio HiFi reads. Reads were quality trimmed using BBDuk (bbduk.sh minavgquality = 20 qtrim = rl trimq = 20) (Bushnell 2014) and assembled with hifiasm-meta (Feng et al. 2022).
Discovery of integrated genetic elements using closed complete genomes
Our manual approach to identifying integrated MGEs was based on three criteria:
-
Anomalous coverage: We evaluated regions with unusually low or high coverage compared with the host chromosome. Coverage changes occurring in localized regions often indicate MGE boundaries. We used the following rule to determine candidate integrated genetic elements: If a region's coverage was significantly different from the overall host chromosome (e.g., 3× higher or lower), we delineated it as a candidate MGE (see section Coverage Calculation of Integrated MGEs and Host-Chromosome).
-
Short reads supporting excision: We examined reads that indicated potential excision events. This included paired reads with large separations that mapped to locations before and after the integration boundaries, or partially discrepant reads in which the discrepant portion matched the sequence on the opposite side of the integration region (see Fig. 1).
-
Gene annotations: We analyzed the functional annotations of genes within candidate regions (see section Functional Annotation of Candidate MGEs). Although these often included functions associated with plasmids or viruses, we also frequently observed an unusually high proportion of hypothetical proteins in these elements.
To implement this strategy, metagenome reads were aligned to the reference genome sequences and coverage anomalies and read discrepancies identified. Read coverage was employed to determine the relative abundance and distribution of each MGE across the different samples. Additionally, in-depth examination of reads sequence discrepancies allowed for the identification of integration sites and mini elements within other integrated elements. To further characterize the MGEs, we compared genomes lacking the MGE with those containing them. This combination of approaches enabled us to accurately determine the length of the MGE and pinpoint the specific sites of integration within the plasmid.
Coverage calculation of integrated MGEs and host-chromosome
We aligned metagenome reads to reference genomes using the BBMap's short-read aligner. Using minimum identity threshold of 0.95, ambiguously mapping reads were discarded. The coverage values of the MGEs and total genome coverage values were calculated from the alignment/map (BAM) files. The positions of integrated elements were defined. For the genome coverage calculation, the entire genome was divided into two sections: the region before the start of the larger integrated element and the region after the end of the larger integrated element. This approach enabled systematic, efficient calculation of the coverage of integrated elements within genomes.
Functional annotation of candidate MGEs
We functionally annotated putative MGEs using multiple databases and tools. Protein families and domains were identified using TIGRFAMs 15.0 and Pfam v31.0 (both downloaded September 2018; Haft et al. 2013; Paladin et al. 2021). Carbohydrate-active enzymes were annotated using CAZymes (dbCAN-HMMdb-V7, dbCAN v2, downloaded September 2018; Drula et al. 2022). Peptidases were identified using MEROPS (November 2018) (Mistry et al. 2007). Hydrogenases were annotated using HydDB (November 2018) (Søndergaard et al. 2016). Clusters of orthologous groups (COGs) were assigned using the NCBI COG database (https://www.ncbi.nlm.nih.gov/research/cog; November 2018). Membrane transporters were identified using TransportDB (http://www.membranetransport.org/transportDB2/index.html; November 2018; Galperin et al. 2021). We employed InterProScan (Jones et al. 2014; v5.50–84.0) for comprehensive protein annotation. All annotations were performed using the pipeline described by Dombrowski et al. (2020) with default parameters. Detailed results are provided in Supplemental Table S6.
For MGE-specific annotations, we utilized several manually curated databases: Nucleo-Cytoplasmic Virus Orthologous Groups (NCVOGs) (Yutin et al. 2009), prokaryotic Virus Orthologous Groups (pVOGs) (Grazziotin et al. 2017), Prokaryotic Virus Remote Homologous Groups (PHROGs) (Terzian et al. 2021), Phage Artificial Neural Networks (PhANNs) (Cantu et al. 2020), Virus Orthologous Groups (VOGs) (Bao et al. 2004), and Giant virus metagenome-assembled genomes (GVMAG) (Schulz et al. 2020; Rambo et al. 2022). Viral and plasmid protein hallmarks were classified using geNomad v. 2.16 (Camargo et al. 2024). Given the challenges in annotating archaeal MGEs owing to limited reference databases and validated features, we conducted manual inspections supplemented with phylogenetic and gene synteny analyses for these sequences. Detailed results are provided in Supplemental Table S5.
Identification and genome curation of Atabeyarchaeia-associated exogenous MGEs
We used metagenomic data sets to search for candidate MGEs associated with Atabeyarchaeia and Freyarchaeia. Screening was based on taxonomic profile, GC contents, and CRISPR-based targeting. All the candidate contigs were manually curated using Geneious Prime-2023.1.2 (https://www.geneious.com). The manually curated genomes were de novo reconstructed from high-quality Illumina metagenomic data. Manual genome curation methods generally follow the method of Chen et al. (2020). Long-read PacBio data were used to verify and expand the sequence data set. Replichores of complete genomes were predicted according to the GC skew, and cumulative GC skew was calculated by the iRep package gc_skew.py (https://github.com/christophertbrown/iRep). We classified complete MGE genomes as viruses if they contained viral structural genes. Genomes lacking viral structural genes were categorized as plasmids or other MGEs (such as transposons or conjugative elements) based on length, the presence of transposases (classified using ISFinder) (Siguier et al. 2006), genomic composition, and the presence of circularized and excised forms. Genomes not fitting these categories were left unclassified. We further validated our viral and/or plasmid candidates using geNomad v. 2.16 (Supplemental Table S7; Camargo et al. 2024).
CRISPR-Cas systems and classification of soil Asgard-associated viruses
CRISPR-Cas systems in Atabeyarchaeia and Freyarchaeia genomes were identified using CRISPRCasTyper v1.8.0 (Russel et al. 2020). Spacers were extracted from reads by mapping reads to the corresponding CRISPR arrays via BBMap (https://sourceforge.net/projects/bbmap/). Recruited spacers were matched against all assembled scaffolds with one or fewer mismatch using Bowtie v1.3.1 (Langmead et al. 2009). Scaffolds that are targeted by CRISPR spacers and not affiliated with microbial genomes were curated manually to completion. The phylogenetic classification was predicted based on genome-wide similarities using ViPTree whole-proteome-based similarity of MGE from Atabeyarchaeia and other Asgardarchaeota (Supplemental Table S8).
Comparative analysis of archaeal MGEs sizes
To contextualize the sizes of the identified MGEs in Atabeyarchaeia, we compiled a comprehensive data set of archaeal MGEs from various sources. We downloaded all complete reference plasmids from the NCBI Plasmid Browser (https://www.ncbi.nlm.nih.gov/genome/browse#!/plasmids/) and incorporated MGEs previously published (Al-Shayeb et al. 2022; Medvedeva et al. 2022; Rambo et al. 2022; Wu et al. 2022). The data set included various types of MGEs such as plasmids, viruses, iMGEs, mini-Borgs, Aloposons, and unclassified MGEs from diverse archaeal hosts including ANME-1, Asgard archaea, DPANN, Euryarchaeota, and Thermoproteota. We filtered the data set to include only MGEs with total genome sizes ≤ 100,000 bp to focus on elements comparable in size to those found in Atabeyarchaeia. The resulting data were visualized using a scatter plot, with MGE types represented by different shapes and host lineages distinguished by colors, allowing for a comprehensive comparison of MGE sizes across archaeal taxa (Supplemental Fig. S1).
Methylation analysis via REBASE and single-molecule, real-time
Methylation patterns within the genomes of Atabeyarchaeia and Freyarchaeia were investigated by mapping PacBio circular-consensus reads metagenomic reads to each of the three curated circular reference genomes for Atabeyarchaeia-1, Atabeyarchaeia-2, and Freyarchaeia using minimap2. The resulting BAM files were then processed and analyzed to identify methylation patterns using the ipdSummary and motifMaker commands in the single-molecule, real-time (SMRT) link analysis software package (v11.0, PacBio). To annotate MTase activities and restriction enzyme sites, the sequenced genomes and identified methylated motifs were compared against REBASE (Roberts et al. 2015; Blow et al. 2016). This comparison enabled the annotation of methylation sites and the determination of specific motifs associated with MTase activity and restriction-modification systems within these archaeal genomes.
Methylation motif validation with nanopore sequencing
Reads were base-called by dorado using the 4 kHz v.4.0.1 5 mC and 6 mA Rerio models (https://github.com/nanoporetech/rerio). Reads were aligned to the genomes with minimap2 (Li 2018). To exclude partial mappings and reads from other organisms, alignments with <80% alignment or >5% SNPs were excluded. Modifications were aggregated with “modkit pileup,” and motifs were called with “modkit find-motifs” (https://github.com/nanoporetech/modkit). Motifs were refined by inspecting the methylation distribution of related or off-by-one motifs to see if methylation was increased or decreased by more specific or more generic motifs.
Identification and network analysis of archaeal phage integrases
Phage integrase sequences were extracted from a custom database of NCBI archaeal genomes and archaeal-associated MGEs using HMMER (v3.3.2; Finn et al. 2011). The Pfam-established E-value cutoff for the Phage_integrase family (PF00589) was used as the threshold for sequence identification. The resulting integrase sequences were then analyzed using the Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST; Gerlt et al. 2015) to generate a sequence similarity network. The network was visualized and further analyzed using Cytoscape (v3.9.1; Shannon et al. 2003).
Phylogenetic analyses of proteins present in MGEs
Hallmark and poorly characterized proteins
For the terminase, PCNA, viral capsid, CAAX, and Type IIG RM fusion protein, we compiled the top 25 to 50 best matches for candidate proteins from the NCBI database, ggKbase, and UniProt (E-value cutoff of 1 × 10−5, 70% query coverage). The sequences were aligned using MAFFT (v7.310) (Katoh and Standley 2013) with the parameters ‐‐localpair ‐‐maxiterate 1000 ‐‐reorder. Following alignment, each was trimmed using TrimAl, applying a gap threshold of 0.7. The final alignments underwent manual inspection using Geneious (v11.0.20.1 + 1; https://www.geneious.com; see above). Maximum likelihood trees were inferred using IQ-TREE v1.6.12 (v1.4), (Nguyen et al. 2015), employing the auto option for model selection and a bootstrap value of 1000 and identifying the best-fit model for constructing the final trees. The details of all models used are included in the description of each figure. Trees were visualized using iTOL (Letunic and Bork 2021). All hallmark MGEs protein alignments and trees have been provided in the Supplemental Material for further reference.
GTPases
A curated set of proteins for the eukaryotic Arf family described by Vargová et al. (2021) was used to search in both soil-associated Atabeyarchaeia and Freyarchaeia genomes and MGEs. A nonredundant subset of the references and Asgardarchaeota hits with >25% protein identity were aligned and trimmed with MAFFT auto (v7.310) (Katoh and Standley 2013) and TrimAl -gt 0.5 (v1.4.rev15) (Capella-Gutiérrez et al. 2009). An initial phylogeny was produced with IQ-TREE (v.1.6.1) (Nguyen et al. 2015), and the LG + R9 model was chosen according to BIC.
By blasting the MGE GTPases against the NCBI database (June 19, 2024), we added sequences with percentage identity ≥35% with >71% query coverage. Our expanded reference set includes 10 Asgardarchaeota (one Baldrarchaeia [previously misidentified as Odindarchaeia], six Lokiarchaeia, and three Heimdallarchaeia) and two grassland soil-associated Deferrimicrobium sp. sequences. We also added one eukaryotic sequence from Hericium alpestre collected from forest deadwood that had high query coverage (70%–71%) and percentage identity (33.33%–34.92%) to both putative MGE GTPases. Sequences were aligned with MAFFT (v7.490) (Katoh and Standley 2013), trimmed with TrimAl -gt 0.5 (v1.4.rev22) (Capella-Gutiérrez et al. 2009), and used to create a maximum likelihood tree with IQ-TREE (v1.6.12) (Nguyen et al. 2015). LG + R7 was the best-fit model chosen according to BIC (Supplemental Fig. S10). The phylogeny was visualized with iTOL (Letunic and Bork 2021).
Hallmark viral proteins structural analysis
Protein structures for Opia virus hallmark proteins (capsid, terminase large subunit, and PCNA) were predicted using AlphaFold 3 (Abramson et al. 2024). The resulting models were compared to known structures using Foldseek (van Kempen et al. 2024) to identify structural homologs. Reference structures from the Protein Data Bank (PDB; https://www.rcsb.org) were used for comparison: bacteriophage capsid (PDB: 3BJQ), thermophilic bacteriophage D6E large terminase (PDB: 5OEB), and P. abyssi PCNA (PDB: 6T8H). Structural alignments and visualizations were performed using UCSF Chimera X v1.8 (Meng et al. 2023). Confidence scores from AlphaFold 3 predictions were mapped onto the structures using a blue-to-red color gradient, representing high to low confidence, respectively.
Structural analyses and structural phylogeny of the ESP
The protein sequences of small GTPases found were analyzed along with eukaryotic small GTPases identified as sequence homologues (Supplemental Figs. S10–S12). These sequences were submitted for structural modeling using ColabFold v1.5.240 (Mirdita et al. 2022). Multisequence alignments were performed using the MMseqs2 (Steinegger and Söding 2017) mode and the AlphaFold2_ptm models. Two recycling steps were employed to improve model prediction. The structural models were used as queries to search for structural homologues in the RCSB Protein Data Bank using FoldSeek easy-search feature, with a cutoff of >15% identity and <1 × 10−5 E-value.
Protein structures identified by FoldSeek were integrated with the models generated in ColabFold (V1.5.240; Mirdita et al. 2022). A multistructural alignment (MSTA) of these structures was carried out using the default parameters of mTM-align (Supplemental Fig. S12; Dong et al. 2018). The resulting MSTA was further analyzed using IQ-TREE v2.0.3 (model LG + I + G4 chosen according to BIC), yielding the dendrogram in Supplemental Figure S11A. The pairwise matrix obtained from the mTM-align process (Supplemental Table S12) was utilized to select proteins suitable for 3D reconstruction of their alignments (Supplemental Fig. S11B). This was done using the Needleman–Wunsch algorithm and the BLOSUM-62 matrix within the ChimeraX software (Meng et al. 2023)
Data access
The metagenomic data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1050611. The Asgard archaea genomes can be accessed via ggKbase (https://ggkbase.berkeley.edu/SRVP_asgard/organisms). The complete viral genome sequences generated in this study have been submitted to the NCBI BioSample database (https://www.ncbi.nlm.nih.gov/biosample/) under accession numbers SAMN43308743, SAMN43308744, and SAMN43308745. The methylation data and candidate MTases identified in this study are available via REBASE (http://rebase.neb.com/rebase/private/pacbio_Banfield10.html). All the read mapping files, protein sequence alignments, and phylogenetic tree and modeled structures are available via Zenodo (https://zenodo.org/records/12617226).
Competing interest statement
J.F.B. is a cofounder of Metagenomi. D.F.S. is a cofounder and scientific advisory board member of Scribe Therapeutics. R.J.R. works for New England Biolabs, a company that sells research reagents including restriction enzymes and DNA MTases to the scientific community. L.L. is an employee of Oxford Nanopore Technologies and is a stock or stock option holder of Oxford Nanopore Technologies. The other authors declare that they have no competing interests.
Acknowledgments
This research was funded by the Bill & Melinda Gates Foundation (grant number INV-037174 to J.F.B.), a University of California Dissertation-Year Fellowship (to L.E.V.A.), a Stengl-Wyer Graduate Fellowship and University of Texas Graduate Continuing Fellowship (to K.E.A.), and the Innovative Genomics Institute at UC Berkeley. This work was also supported by the Moore-Simons Project on the Origin of the Eukaryotic Cell, Simons Foundation 73592LPI (https://doi.org/10.46714/735925LPI to B.J.B.) and Simons Foundation early career award 687165 (to B.J.B.). The findings and conclusions contained within are those of the authors and do not necessarily reflect positions or policies of the Bill & Melinda Gates Foundation. D.F.S. is an Investigator of the Howard Hughes Medical Institute, and the work was supported by the U. S. Department of Energy under Award number DE-SC0016240 to D.F.S. We thank Dr. Basem Al-Shayeb and Dr. Marie C. Schoelmerich for their contribution to the field work and generation of sequence data sets; Dr. Jacob West-Roberts, Shufei Lei, and Jordan Hoff for bioinformatics support; Dr. Mart Krupovic and Dr. Gavin Knott for helpful discussion; and Dr. Lin LinXing Chen for critical reading over the manuscript.
Author contributions: L.E.V.A. and J.F.B. designed the study. L.E.V.A., J.F.B., A.C.C., and L.-D.S. collected samples and extracted nucleic acids. L.E.V.A., R.S., A.C.C., and J.F.B. generated metagenomic data. L.E.V.A. and J.F.B. performed genome binning, and J.F.B., L.E.V.A., and L.-D.S. conducted genome curation. L.E.V.A. and J.F.B. analyzed the MGE genomes. L.E.V.A., K.E.A., and P.L. analyzed the functionally annotated MGEs. A.C.C. and L.E.V.A. performed correlation analyses. L.E.V.A. conducted phylogenetic analyses of hallmark MGE proteins. L.E.V.A., A.C.C., and J.F.B. analyzed MGE integration and excision events. L.E.V.A. and L.-D.S. analyzed whole viral proteomes. B.A.A. provided guidance on transposase and MGE classification. K.E.A., P.L., V.D.A., and B.J.B. analyzed eukaryotic signature proteins. A.C.C., L.E.V.A., and R.J.R. performed methylation analyses. R.S. contributed to bioinformatics analyses. B.J.B. and D.F.S. provided feedback on study design, methodology, and financial support. L.E.V.A. and J.F.B. wrote the manuscript, with contributions from all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279480.124.
-
Freely available online through the Genome Research Open Access option.
- Received April 17, 2024.
- Accepted August 27, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








