
The predicted RNA-binding protein regulome of axonal mRNAs
- 1Idiap Research Institute, Martigny 1920, Switzerland;
- 2SIB Swiss Institute of Bioinformatics, Lausanne 1015, Switzerland;
- 3UCL Laboratory for Molecular Cell Biology, University College London, London WC1E 6BT, United Kingdom
-
↵4 These authors contributed equally to this work.
Abstract
Neurons are morphologically complex cells that rely on the compartmentalization of protein expression to develop and maintain their cytoarchitecture. The targeting of RNA transcripts to axons is one of the mechanisms that allows rapid local translation of proteins in response to extracellular signals. 3′ Untranslated regions (UTRs) of mRNA are noncoding sequences that play a critical role in determining transcript localization and translation by interacting with specific RNA-binding proteins (RBPs). However, how 3′ UTRs contribute to mRNA metabolism and the nature of RBP complexes responsible for these functions remains elusive. We performed 3′ end sequencing of RNA isolated from cell bodies and axons of sympathetic neurons exposed to either nerve growth factor (NGF) or neurotrophin 3 (NTF3, also known as NT-3). NGF and NTF3 are growth factors essential for sympathetic neuron development through distinct signaling mechanisms. Whereas NTF3 acts mostly locally, NGF signal is retrogradely transported from axons to cell bodies. We discovered that both NGF and NTF3 affect transcription and alternative polyadenylation in the nucleus and induce the localization of specific 3′ UTR isoforms to axons, including short 3′ UTR isoforms found exclusively in axons. The integration of our data with CLIP sequencing data supports a model whereby long 3′ UTR isoforms associate with RBP complexes in the nucleus and, upon reaching the axons, are remodeled locally into shorter isoforms. Our findings shed new light into the complex relationship between nuclear polyadenylation, mRNA localization, and local 3′ UTR remodeling in developing neurons.
Axonal RNA transport and local translation are a widespread phenomenon observed in most neuronal cell types and across species (Holt et al. 2019). In developing axons, local translation plays a crucial role in mediating cell survival and axon growth in response to extracellular cues. It enables axon extension, guides growth cone turning, and promotes nerve regeneration after injury (Dalla Costa et al. 2020).
During neuronal development, axons extend over long distances to reach their targets, driven by their growth cone, which responds rapidly to local cues along its migratory path (Dorskind and Kolodkin 2021). At early developmental stages, sympathetic neurons respond to neurotrophin 3 (NTF3, also known as NT-3) released from the vasculature surrounding the axons (Makita et al. 2008), eliciting a local signal that supports initial axon growth and cell survival (Elshamy and Ernfors 1996; Kuruvilla et al. 2004). At later stages, when axons have reached a considerable length and approach their final targets, nerve growth factor (NGF) is released, internalized within signaling endosomes, and transported back to the cell bodies, where it activates transcription (Kuruvilla et al. 2004; Ascano et al. 2012; Scott-Solomon and Kuruvilla 2018; Scott-Solomon et al. 2021). In sympathetic neurons, both neurotrophins bind to the same tyrosine kinase receptor, NTRK1 (also known as TrkA), although their ability to signal to the nucleus depends on the internalization into signaling endosomes and transport to cell bodies. In contrast to NGF, NTF3/NTRK1 complexes are not found in signaling endosomes and are not retrogradely transported. Thus, NTF3 in axons is thought to act mostly locally and to be a poor activator of gene expression (Kuruvilla et al. 2004).
In addition to conveying the genetic information from the chromatin to the translational machinery, mRNA transcripts also carry information stored in their untranslated regions (UTRs). The 3′ UTRs regulate many aspects of RNA metabolism, including transcript localization, mRNA stability, and translation by interacting with RNA-binding proteins (RBPs) (Andreassi and Riccio 2009; Mayr 2017; Andreassi et al. 2018; Andreassi et al. 2021). RBP complexes are initially assembled cotranscriptionally and are essential for regulating RNA splicing, polyadenylation site (PAS) choice, alternative polyadenylation (APA), and mRNA nuclear export (Hentze et al. 2018; Van Nostrand et al. 2020). Most RBPs are multifunctional, and the complexes undergo extensive remodeling in the cytoplasm (Hentze et al. 2018). RBP interaction with elements in the 3′ UTR mediates mRNA targeting to dendrites and axons, and this event is necessary for the establishment of synapses, axon growth, and nerve repair after injury (Doyle and Kiebler 2011; Allen et al. 2013; Holt and Schuman 2013; Cosker et al. 2016; Terenzio et al. 2017; Dalla Costa et al. 2020; Thelen and Kye 2020).
Here, we performed a comprehensive analysis of transcription, APA, mRNA transport, and putative RBP binding in cell bodies and axons of developing rat sympathetic neurons in response to neurotrophins. We aim to further characterize the shift in the transcriptional landscape that takes place in developing sympathetic neuron axons in response to extracellular cues.
Results
NGF and NTF3 applied to distal axons induce distinct transcriptional programs
To identify mRNAs localized in cell bodies and axons of sympathetic neurons in response to neurotrophins, we integrated previously published 3′ UTR sequencing data obtained from compartmentalized cultures of sympathetic neurons exposed to NGF (Andreassi et al. 2021), with data simultaneously obtained from axons exposed to NTF3. Compartmentalized chambers allow the physical separation of cell bodies from distal axons and are especially suited for sympathetic neurons because they grow in culture as a highly homogeneous population without glial cells. Neurons were seeded in the central compartment with NGF (100 ng/mL), and after 5 d, either NGF or NTF3 was added to the lateral compartments. A low concentration of NGF (10 ng/mL) was maintained in the cell bodies to preserve viability. After seven additional days necessary to achieve robust axon growth, RNA was isolated from cell bodies or axons, subject to two rounds of linear amplification, and sequenced using 3′ end sequencing. This technique allowed the sequencing of transcripts 3′ ends independently of the length of the transcript (for a detailed description of the protocol used, see Methods) (Fig. 1A; Andreassi et al. 2021).

NGF and NTF3 are equally capable of regulating transcriptional changes when applied to distal axons. (A) Schematic of the experimental set-up. (B) Volcano plot representing log2 fold-change (log2FC) in gene expression values between neurons whose axons were exposed to NGF or NTF3, as well as corresponding P-values (−log10). Blue dots indicate genes significantly up-regulated in NGF; purple dots, genes significantly up-regulated in NTF3 (FC > 1.5 and P-value < 0.01). (C) Enrichment scores of GO biological pathways associated with up-regulated genes in NGF-treated (top) and NTF3-treated (bottom) neurons. (D) Fisher's enrichments in predicted binding sites motifs for 21 transcription factors in the 1000-nucleotide (nt) promoter region of the up-regulated genes in NGF- or NTF3-treated neurons. (E,F) Sequence logos of predicted motifs bound by transcription factors (top) and fractions of promoter regions with these motifs (bottom) in the total pool of expressed genes (gray bar), the genes up-regulated in NGF (blue bar), and the genes up-regulated in NTF3 (purple bar). Fisher's enrichment test. (G, left) Number of transcripts showing significant promoter distal-to-proximal shifts in NGF compared with NTF3 (filled-in bar) alongside their composition in terms of proximal shift in NGF versus distal shift in NTF3 (outlined bars). (Right) Legend of the color outline schematizing the relative usage of the promoter-proximal (Ip) and promoter-distal (Id) 3′ UTR isoforms underlying differential 3′ UTR isoform usage between NGF and NTF3. (H) Same as G for changes in NTF3 compared with NGF. (I) Enrichment scores of GO biological pathways associated with significant increase in distal-to-proximal promoter 3′ UTR usage in NGF compared with NTF3 (top) and vice versa (bottom).
Analyses of the reads obtained from two independent biological replicates indicated the high reliability of the RNA sequencing in both compartments (Supplemental Fig. S1A). The lower correlation between the replicates from axonal compartments is likely owing to the very low amount of RNA isolated in axons. Unsupervised analysis of the sequencing reads indicated the dominant effect of the compartments over the treatment condition. Indeed, despite axon-only exposure to different neurotrophins, we primarily observed the effects of the treatments in the cell bodies (Supplemental Fig. S1B,C). Thus, we first aimed to examine the transcriptional changes in the cell bodies upon exposure of distal axons to NGF or NTF3. Because a control condition using neurons grown without neurotrophins is not possible, the experimental setting does not distinguish between changes owing to the direct effect of each neurotrophin or the indirect effect owing to the lack of thereof. Differential gene expression analysis of neurons whose axons were treated with NGF or NTF3 identified 232 genes up-regulated in NGF-treated neurons compared with NTF3 (FC > 1.5 and P-value < 0.01) (Fig. 1B; Supplemental Table S1), enriched in terms related to cell adhesion and glucose metabolism (Fig. 1C, top). Conversely, the expression of 119 genes was increased in the cell bodies of neurons whose axons were exposed to NTF3, and they were enriched for neuron projection and synaptic transmission terms (Fig. 1C, bottom; Supplemental Table S2). Transcription factor binding site (TFBS) enrichment analysis indicated a neurotrophin-specific transcriptional regulation of the differentially expressed genes (Fig. 1D; Supplemental Table S3). The 1000-nucleotide (nt) promoter regions of genes up-regulated in NGF compared with NTF3 were enriched in 14 TFBS motifs, including motifs bound by early growth response 1 (EGR1), early growth response 2 (EGR2), early growth response 3 (EGR3; P-value = 3.46 × 10−4, 43% of the promoter regions of NGF up-regulated genes), Fos proto-oncogene, AP-1 transcription factor subunit (FOS), FosB proto-oncogene, AP-1 transcription factor subunit (FOSB), FOS like 1, AP-1 transcription factor subunit (FOSL1), JunB proto-oncogene, AP-1 transcription factor subunit (JUNB), and JunD proto-oncogene, AP-1 transcription factor subunit (JUND; P-value = 3.91 × 10−3, 16% the promoter regions of NGF up-regulated genes) (Fig. 1E), all belonging to the reactome biological pathway of NGF-stimulated transcription (HSA-9031628). Fewer motifs were enriched within the promoter regions of transcripts up-regulated in neurons whose axons were exposed to NTF3, and they all related to embryonic development. Such motifs were bound by the dimer of transcription factors SOX2 and POU class 5 homeobox 1 (POU5F1; 2.26 × 10−2, 8% of the promoter regions of NTF3 up-regulated genes) and transcription factor SOX17 (P-value = 2.39 × 10−2, 23% of the promoter regions of NTF3 up-regulated genes) (Fig. 1F).
APA takes place principally cotranscriptionally, generating transcripts expressing identical coding regions and 3′ UTRs of different length (Tian and Manley 2013). To ask whether neurotrophins induce distinct APA, we investigated the shifts of the relative 3′ UTR usage of transcripts expressed in cell bodies of neurons whose axons were exposed to NGF or NTF3. Twenty-seven transcripts showed distal-to-proximal promoter 3′ UTR shift in NGF compared with NTF3 (Fig. 1G; Supplemental Fig. S1D; Supplemental Table S4), and 45 transcripts showed distal-to-proximal promoter 3′ UTR shifts in response to NTF3 (Fig. 1H; Supplemental Fig. S1D; Supplemental Table S5). Differential relative 3′ UTR isoform usage between the two conditions can be owing to selective increase of promoter-proximal 3′ UTR isoforms, promoter-distal isoforms, or both. The 27 transcripts showing significant promoter distal-to-proximal 3′ UTR shifts in NGF included 20 promoter-proximal shifts in NGF and 17 promoter-distal shifts in NTF3 (Fig. 1G). The 45 transcripts showing significant promoter distal-to-proximal 3′ UTR shifts in NTF3 included 34 promoter-proximal shifts in NTF3 and 26 promoter-distal shifts in NGF (Fig. 1H). In both conditions, some transcripts showed changes in both directions. Thus, although neurons exposed to NGF showed a similar number of promoter-proximal and promoter-distal 3′ UTR shifts (20/26), NTF3 induced twice as many promoter-proximal shifts as promoter-distal shifts (34/17), indicating a preferential usage of isoforms with short 3′ UTR in NTF3-treated condition. Depending on the neurotrophin, 3′ UTR shifts targeted genes associated with distinct biological pathways: Whereas target genes related to cellular stress and DNA damage were overrepresented in response to NGF (Fig. 1I, top), target genes in neurons treated with NTF3 were enriched in cell migration and differentiation biological pathways (Fig. 1I, bottom). When comparing changes in gene expression and APA between the two conditions, we found that the genes affected by APA were distinct from those undergoing transcriptional changes under NTF3 and NGF conditions (Supplemental Fig. S1E). We also observed a lack of correlation between changes in 3′ UTR usage and overall gene abundance (Supplemental Fig. S1F). These results are in line with previous studies (Tian and Manley 2017) showing the role of APA in global mRNA metabolism independent of mRNA expression. Thus, both NTF3 and NGF propagate signals from distal axons to the nucleus to regulate gene expression and cotranscriptional APA.
Identification of RBPs that regulate APA in response to NGF and NTF3
RBPs regulate mRNA processing and metabolism, including APA (Erson-Bensan 2016; Hentze et al. 2018). To identify putative regulators of neurotrophin-specific 3′ UTR APA, we interrogated publicly available cross-linking and immunoprecipitation (CLIP) sequencing data for 126 RBPs assayed in human cell lines (Supplemental Table S6). Visual inspection of the distribution of RBP cross-link events along the 3′ UTR isoforms revealed that the 500-nt region preceding the 3′ ends showed the highest percentage of bound RBPs (70%) (Supplemental Fig. S2A). This region is expected to serve a regulatory function given its high conservation score (Supplemental Fig. S2B). We first searched for associations between cross-linking events mapping to defined regions along the promoter-proximal and promoter-distal 3′ UTRs, as well as the relative usage in cell bodies of the short 3′ UTR isoform. One hundred twenty-six RBPs were tested individually both in NGF and NTF3 conditions (for details, see Methods). A positive association between RBP binding and the expression of 3′ UTR isoforms predominantly occurred at the 3′ end of both the promoter-proximal (Ip) and the promoter-distal (Id) 3′ UTR, irrespective of the neurotrophin used (Fig. 2A). We identified 17 RBPs preferentially bound within the [−350:+150] region surrounding the 3′ end, acting as positive regulators of polyadenylation (Supplemental Fig. S2C,E; Supplemental Table S7). These included the cleavage and polyadenylation specific factor 1 (CPSF1, also known as CPSF160 in human) (Fig. 2B) and cleavage stimulation factor subunit 2 (CSTF2) (Fig. 2C), which are known to reside in the [−50:0]- and [0:50]-nt regions around the 3′ end (Mitschka and Mayr 2022). Thus, the binding of RBPs to specific 3′ end terminal regions was positively associated to both short and long 3′ UTR isoforms, which is consistent with the fact that these factors may promote 3′ end processing irrespective of 3′ UTR length (Laishram and Anderson 2010).
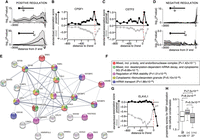
Predicted RBP regulome underlying alternative polyadenylation (APA). (A) Distribution of significant positive association between the binding of 126 RBPs in defined regions along the 3′ UTR and the relative usage of the promoter-proximal (top) or promoter-distal (bottom) 3′ UTR isoform. Dark lines display the median significance; shaded areas, lower and upper quartiles. (B,C) Scatterplot of the extent of standardized significant association between CPSF1 (B) or CSTF2 (C) cross-link events in defined regions along the 3′ UTR and the relative usage of the promoter-proximal (black) or promoter-distal (gray) 3′ UTR isoform in the axons. (D) Negative association between RBP binding and the relative usage of the promoter-proximal (top) or promoter-distal (bottom) 3′ UTR isoform. (E) Network of protein–protein interactions for 27 candidate regulators of APA predicted to prevent promoter-proximal usage when bound to the [0:150]-nt region downstream from the 3′ end. Edges represent experimentally determined protein–protein interactions annotated in the STRING database (Szklarczyk et al. 2017). Nodes indicate proteins colored according to the biological pathways they are enriched in. (F) Top five biological pathways and associated P-values overrepresented in the 27 candidate negative regulators of short 3′ UTR isoform. Fisher's enrichment test. (G) Same as B,C for ELAVL1. (H) Distributions of the propensity scores of the 126 RBPs, the 17 positive regulators of APA (short and long 3′ UTR isoform), and the 27 negative regulators of the short 3′ UTR isoform to localize into cellular condensates as predicted by GraPES (Kuechler et al. 2022). Welch's t-test assessing the significant difference between the mean propensity scores.
Negative association between 3′ UTR isoform usage and RBP binding revealed that negative regulators of polyadenylation are uniquely detected in the [0:150]-nt region downstream from the promoter-proximal 3′ end (Fig. 2D). Long 3′ UTR isoforms were not associated with negative regulators. The 27 negative regulators of the short 3′ UTR form a densely connected network of experimentally validated interacting proteins that are enriched in biological processes related to mRNA stability (Fig. 2E,F; Supplemental Fig. S2D,F; Supplemental Table S8). ELAV like RNA binding protein 1 (ELAVL1; also known as HuR) is a well-characterized RBP that competes with the CSTF factors downstream from poly(A) sites to block polyadenylation (Zhu et al. 2007). ELAVL1 binding downstream from the 3′ end of the promoter-proximal 3′ UTRs was significantly associated with decreased promoter-proximal 3′ UTR usage, promoting the transcription of long 3′ UTR isoforms. In contrast, ELAVL1 binding upstream of the 3′ end was not predicted to affect polyadenylation (Fig. 2G). Therefore, our analysis supports a model by which the selection between short and long 3′ UTR isoforms primarily depends on negative factors bound within the 150-nt region downstream from the promoter-proximal 3′ end. Analysis of the potential to localize into cellular condensates revealed significant differences between the 17 positive and the 27 negative regulators of polyadenylation, with the former showing higher probability to localize into P-bodies and cytoplasmic granules (Fig. 2H). Our analysis identifies new candidates and known regulators of polyadenylation along with their preferential location around the 3′ end, with positive regulators more likely to concentrate into condensates in which they may operate to promote the cleavage of the 3′ end (Andreassi et al. 2021).
Next, we tested whether the subtle but significant differences in APA observed in dozens of transcripts upon exposure of distal axons to NGF or NTF3 (Fig. 1G) were associated with specific RBPs. 3′ UTR position-dependent Fisher's enrichment in RBP cross-link events was studied for the four groups of APA isoforms previously identified (Fig. 1G,H). The distributions of the enrichment scores (−log10(P-value)) along the 3′ UTR revealed that the regulatory regions with significant enrichment of RBPs were restricted to the region downstream from the promoter-proximal 3′ end, irrespective of the grouping (Fig. 3A–D; Supplemental Fig. S3), which aligns with the negative regulators of polyadenylation identified through the Welch's t-test (Fig. 2D; Supplemental Fig. S2F). Using this approach, four groups of RBPs that serve as candidate regulators of preferential promoter-proximal or promoter-distal usage between the two conditions were identified (Fig. 3E–H). Although this analysis does not distinguish between positive and negative regulators of differential APA between each condition, several of these factors, including heterogeneous nuclear ribonucleoprotein C (HNRNPC), Y box binding protein 3 (YBX3), and KH-type splicing regulatory protein (KHSRP), were previously identified as global negative regulators of APA (Fig. 2E). Notably, these RBPs bind within the 150-nt region downstream from the promoter-proximal 3′ end (Fig. 3A–D), similar to the predicted negative regulators of APA (Fig. 2D). These findings collectively suggest that each neurotrophic treatment leads to a condition-specific change in the negative regulatory activity of these factors (Fig. 3E–H). Specifically, the negative regulatory activity on the promoter-proximal 3′ UTR isoform is diminished in one condition compared with the other when there is a shift toward the usage of short 3′ UTR isoforms in the former (Fig. 3E,F,I,J). Conversely, it is enhanced when there is a preference for long 3′ UTR isoforms in the former (Fig. 3G,H,K,L). For example, HNRNPC is enriched in the regions downstream from the 3′ end of the promoter-proximal 3′ UTR isoforms whose related distal isoforms are up-regulated in the NGF condition compared with the NTF3 condition (Fig. 3D,H). Furthermore, HNRNPC is enriched in the regions downstream from the 3′ end of the promoter-proximal 3′ UTR isoforms that are up-regulated in the NTF3 condition compared with the NGF condition (Fig. 3A,E). These results support a stronger repressor activity of HNRNPC in response to NGF compared with NTF3. Tracks of representative transcripts expressing long or short 3′ UTR in cell bodies of neurons treated with NGF or NTF3 along with CLIP-seq data are shown in Figure 3, I through L.
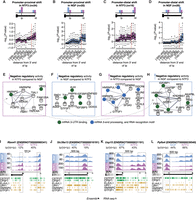
Reduction of negative regulatory activity is predicted to underlie neurotrophin-driven APA. (A–D) Distribution of Fisher's enrichment scores (−log10(P-value)) in cross-link events of 126 RBPs along the short 3′ UTR isoforms of the pairs of isoforms showing significant promoter-proximal shifts in NTF3 (A) or NGF (B) and promoter-distal shifts in NTF3 (C) or NGF (D). (E–H) Networks of protein–protein interactions for the RBPs showing significant enrichment in cross-link events in the [0:+150]-nt regions downstream from the 3′ end of the short 3′ UTR isoforms associated with significant promoter-proximal shifts in NTF3 (A) or NGF (B) and promoter-distal shifts in NTF3 (C) or NGF (D). Edges represent experimentally determined protein–protein interactions annotated in the STRING database. Nodes indicate proteins colored according to the biological pathways they are enriched in. Arrows indicate the predicted directions in changes in activity of the RBPs. (I–L) Genome browser views of 3′ end sequencing profiles and CLIP cross-linking events for predicted positive (green) and negative (gold) regulators of APA for 3′ UTR isoforms showing a marked shift toward increase in promoter-proximal 3′ UTR usage (Rbm41 and Slc39a13) or in promoter-distal 3′ UTR usage (Usp15 and Ppfia4) in either condition. Gray boxes highlight the location of promoter-proximal and promoter-distal 3′ UTR isoforms.
NGF and NTF3 induce the localization of distinct mRNA transcripts in axons
We next investigated whether NGF and NTF3 prompted the transport of different mRNAs in axons. A similar number of transcripts and 3′ UTR isoforms were found in cell bodies and axons independently of neurotrophin treatment (Fig. 4A; Supplemental Fig. S4A). However, a large number of axonal mRNAs with distinct cellular functions (Supplemental Fig. S4B) was uniquely detected in response to either NGF (n = 1962) or NTF3 (n = 1089; Fig. 4B). In line with previous studies (Tushev et al. 2018; Andreassi et al. 2021), a larger percentage of axonal transcripts expressed multiple (Fig. 4C) and longer (Fig. 4D) 3′ UTR isoforms compared with cell bodies in both conditions.

Axons exposed to NGF or NTF3 contain distinct 3′ UTR isoforms. (A) Number of Ensembl transcripts ID detected in cell bodies only (filled-in bars), and cell bodies and distal axons (empty bars) in NGF (blue) and NTF3 (purple) culture conditions. (B) Overlap between the transcripts detected in the distal axons of neurons exposed to either NGF or NTF3. (C) Cell body (filled-in bars) and axonal (empty bars) transcript IDs showing multiple 3′ UTRs in either NGF or NTF3 culture conditions. Two-sided Fisher's exact count test. (D) Maximum 3′ UTR lengths for existing annotations in Ensembl Rn5 (Ensembl) and for those newly identified by 3′ end RNA sequencing (extended annotation) in either NGF or NTF3. Two-sided Wilcoxon rank-sum test. (E) Top five GO biological pathways with associated transcripts showing the most significant increase in axonal localization in NGF (left) compared with NTF3 (right), as quantified with standardized scores comparing GO-annotated transcript with the full pool of detected transcripts (Z-score). (F) Distributions of the localization scores (LSs) for the background genes and the genes belonging to the collagen catabolic biological pathway in NGF (left) and in NTF3 (center) and the differences in LSs between the two conditions (right). Welch's t-test assessing the significant difference between the mean LSs. Boxplots display the five number summary of median, lower and upper quartiles, minimum and maximum values. (G) Same as E for the top five GO biological pathways showing excess in axonal localization in NTF3 compared with NGF. (H) Same as F for genes belonging to the regulation of VEGF production. (I) Average mRNA abundance across the four cell bodies samples and the difference in LSs between NGF and NTF3. Four hundred eighty-two transcripts significantly more localized in NGF compared with NTF3 (blue dots; Z-score > 1.96) and 348 transcripts significantly more localized in NTF3 compared with NGF (purple dots; Z-score < −1.96). (J, top) Genome browser view of a representative transcript with significant detection in the axonal compartment in NTF3-treated culture condition and residual detection in NGF condition (Atf3). (Bottom) eCLIP cross-linking events along the gene for NTF3-specific pairs of positive regulators of axonal transport (DDX24:SLTM and EFTUD2:SRSF1; green) and negative pairs regulators of axonal transport specific to NGF or NTF3 (PUM2:HNRNPC and CELF4:CELF2, respectively; orange). (K) Lower levels of Atf3 localized to NGF-treated axons (top) compared with NTF3-treated axons (bottom). Black arrows point to cell body signal; white arrowheads, axonal signal. mRNA puncta were not subject to pixel dilation. (Insets) Magnification (2×) of the boxed areas. Scale bar, 10 μm.
The abundance of axonal mRNAs mostly correlated with both expression levels in the cell bodies and transcript length (Supplemental Fig. S4C,D), suggesting that diffusion and anchoring are important mechanisms regulating mRNA localization (St. Johnston 2005). To study the mechanisms responsible for active mRNA transport, we developed a statistical model and a novel metric that we named localization score (LS). LS quantifies the efficiency of transcripts localization in axons irrespective of mRNA length and abundance in the cell bodies (see Methods) (Supplemental Table S9; Supplemental Material). A positive LS indicates higher axonal mRNA abundance than expected for transcripts with similar size and expression levels, and correlates with active transport and stabilization. Conversely, negative LS values are indicative of either restricted transcript diffusion from the cell bodies or higher mRNA degradation. Because LS enables the identification of over- and undertransported mRNA irrespective of the expression levels (Supplemental Fig. S4E–G), it provides a better statistical tool than the ratio of the gene abundance between axons and cell bodies commonly used (Supplemental Fig. S4H), as the latter is more prone to identify extreme values for highly expressed transcripts given the larger dynamic ranges in expression of this class of transcripts. Analysis of GO enrichment per range of abundance ratios versus LS shows that terms associated with overtransported transcripts using LS scores better reflect the biological system (axon development, cell adhesion) compared with transcripts showing excess in abundance ratios (Supplemental Results; Supplemental Fig. S7).
Using LS, thousands of over- and undertransported 3′ UTR isoforms were identified in both of the NGF and NTF3 conditions (Supplemental Fig. S4I,J). To validate the statistical model, real-time quantitative PCR (RT-qPCR) was performed on transcripts predicted to be restricted to cell bodies. Analysis of Eid2 and Rab22a indicated that these mRNAs were virtually absent in NGF- and NTF3-treated axons, despite being highly expressed in the cell bodies (Supplemental Fig. S4K). LS analysis revealed that NGF and NTF3 promote axonal localization of transcripts associated with largely similar GO biological pathways, such as vesicular localization and axo-dendritic transport (Supplemental Fig. S4L). Subtle but significant differences in axonal transcripts associated with specific biological pathways were detected in both conditions. Transcripts related to the collagen catabolic pathway showed a higher LS in NGF axons compared with NTF3 axons (Fig. 4E,F), whereas those related to the vascular endothelial-derived growth factor–related pathway had a higher LS in NTF3 axons (Fig. 4G,H). The latter finding is especially interesting considering that when sympathetic neurons are exposed to NTF3, axons grow in close contact with blood vessels (Scott-Solomon et al. 2021), possibly inducing the transport of transcripts that mediate cross-signaling between neurons and endothelial cells. Analysis performed on individual transcripts identified 482 3′ UTR isoforms with a significantly higher LS in NGF-treated axons and 348 with a higher LS in NTF3-treated axons (Fig. 4I; Supplemental Tables S10, S11). Single-molecule RNA fluorescence in situ hybridization (smFISH) of Atf3, a transcript predicted to localize in NTF3-treated but not NGF-treated axons, confirmed that neurotrophins induce mRNA axonal localization in a highly specific manner (Fig. 4J,K; Willis et al. 2007). We next investigated the interplay between mRNA regulation in the cell body (transcription and APA) and axonal localization. As differences in LSs were not correlated with differences in gene expression (Supplemental Fig. S4M), we found limited overlap between the significantly differentially expressed genes (Supplemental Tables S1, S2) and the genes differentially localized between the two conditions (Supplemental Fig. S4N; Supplemental Tables S10, S11). However, comparison of the LSs of genes targeted by APA in response to NGF and NTF3 (Supplemental Tables S5, S6) revealed that genes showing significant distal-to-proximal shifts in the NGF condition displayed lower axonal localization compared with that of NTF3 (Supplemental Fig. 4O). Although the difference in LS between NGF and NTF3 is not statistically significant (P-value = 0.06), these findings suggest a specific correlation between axonal localization and APA, especially in the NGF condition.
Computational prediction of the RBP regulome for axonal mRNA localization
To study whether putative RBP binding accounted for the distinct axonal localization observed among 3′ UTR isoforms in response to neurotrophins, we conducted statistical tests comparing the LS of transcripts showing a cross-link event for individual RBPs in the 50-nt regions along the 3′ UTR with those that did not show such events (see Methods). A [−200:−50]-nt region preceding the 3′ end showed the highest regulatory potential for axonal localization in both of the NGF and NTF3 conditions (median P-value = 10 × 10−25 across the 126 RBPs) (Fig. 5A). The preferential binding of trans-acting factors to this specific region is in line with previous findings showing the presence of NGF-dependent localization elements within −150 nt from the 3′ end (Andreassi et al. 2010). Indeed, the number of cross-link events (331 experiments for 126 RBPs) within the [−150:−100]-nt region preceding the 3′ end correlated with the LSs (Fig. 5B). The 32 RBPs showing the highest positive association with localized transcripts were enriched in the mRNA transport biological pathway (P-value = 8.74 × 10−5) (Fig. 5C; Supplemental Table S12). Regulators of mRNA transport, such as fragile X messenger ribonucleoprotein 1 (FMR1) (Fig. 5D; Antar et al. 2004; Dictenberg et al. 2008), insulin-like growth factor 2 mRNA binding protein 1 (IGF2BP1) (Supplemental Fig. S5A; Kislauskis et al. 1994; Ross et al. 1997), and TAR DNA binding protein (TARDBP, also known as TDP43) (Supplemental Fig. S5B; Štalekar et al. 2015; Nagano et al. 2020), also showed significantly higher cross-linking events in transcripts overtransported in response to NGF and NTF3 (Fisher's count test). Although the 126 studied RBPs showed similar positive association with axonal localization in both NGF and NTF3 (Supplemental Fig. S5C), eukaryotic translation initiation factor 4, gamma 2 (EIF4G2) (Supplemental Fig. S5D) and SNRPB (Supplemental Fig. S5E) showed significant enrichment in overtransported 3′ UTR isoforms in either the NGF or NTF3 condition, respectively. Interestingly, EIF4G2 is locally translated in rat sympathetic neuron axons, where it supports axon growth (Kar et al. 2013). Finally, 12 RBPs showed significantly higher binding occurrence (Fisher's count test) in the [−250:−50]-nt region preceding the 3′ end of the undertransported isoforms compared with either all transcripts or overtransported isoforms (Supplemental Table S13).
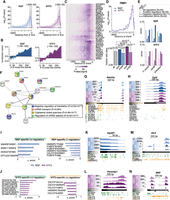
The RBP regulome underlying axonal transcript localization in NGF and NTF3. (A) Distribution of the significance in the difference between the mean LSs in NGF (left) and NTF3 (right) of the isoforms showing or not a cross-link event in 50-nt-long regions along the 3′ UTR for 126 RBPs. Dark line displays the median; shaded areas, lower and upper quartiles. (B) Increasing LSs in NGF (left) and NTF3 (right) conditions as a function of the running average of detected number of cross-link events across the 332 CLIP-seq data in the [−150:−100]-nt upstream of the 3′ end. (C) Heatmap showing the significance in the difference between the mean LSs of the 3′ UTR isoforms showing or not a cross-link event in 50-nt-long regions along the 3′ UTR for the top 32 RBPs positively associated with axonal localization. (D) Scatterplot of the extent of significant association between FMR1 cross-link events and the axonal localization along the 3′ UTR (top). Blue indicates NGF; purple, NTF3. (Bottom) Barplots showing the fraction of [−250:−50]-nt regions upstream of the 3′ ends showing a FMR1 cross-link event in the full set of detected transcripts (gray bar) and the pools of over- and undertransported transcripts in NGF (blue bars) and NTF3 (purple bars), respectively. Fisher's enrichment test. (E) Logistic regression classifier performances trained with different feature sets: all regulators (43 RBPs; M1), positive regulators only (32 RBPs; M2), negative regulators only (11 RBPs; M3), and interaction terms (pairs of RBPs; M4) for the NGF data set (top) and NTF3 data set (bottom). (F) Protein–protein interactions for 16 predicted negative RBPs showing significant enrichment in cross-link events in the [−250:−50]-nt regions upstream of the 3′ end of the undertransported transcripts. Edges represent experimentally determined protein–protein interactions annotated in the STRING database. Nodes indicate proteins colored according to biological pathways they are enriched in. (Inset) Top four biological pathways and associated enrichment P-values as obtained from a Fisher's enrichment test. (G,H) Genome browser view of representative transcripts either restricted (G) or with significant detection in the axonal compartment (H) in NTF3-treated and NGF-treated culture conditions and CLIP cross-linking events for the most-negative (orange) and most-positive (green) RBP regulators of axonal transport common to NGF and NTF3 conditions. (I,J) Barplot showing the significance in features importance to logistic regression classifier performance of pairs of RBPs that are specific to NGF (I) or NTF3 (J). (K–N) Similar to G for transcripts over- and undertransported in the NGF condition (K,M) and over- and undertransported in the NTF3 condition (L,N) (see also Supplemental Fig. S5J).
Logistic regression was used to investigate the collective contribution of 43 RBPs (consisting of 32 positive and 12 negative candidate regulators) that were individually associated with mRNA axonal localization. For discrimination between over- and undertransported 3′ UTR isoforms (Supplemental Fig. S5F), we trained three types of classifiers: (1) Model M1 used the complete set of 32 positive and 12 negative regulators of axonal localization (refer to Supplemental Tables S12, S13); (2) model M2 used exclusively the 32 positive regulators; and (3) model M3 used exclusively the 11 negative regulators. Forty-two interaction terms were also generated from a subset of positive and negative regulators to explore their potential synergistic regulatory effects (model M4; see Methods). Using these models, groups of RBPs were identified that enabled the classification of localized versus cell body–restricted 3′ UTR isoforms. RBPs either were positive regulators of axonal localization (Supplemental Fig. S5G) or were linked to decreased localization of specific axonal mRNAs in response to either NGF or NTF3 (Supplemental Fig. S5H). Although models using a higher number of predicted features (M1 and M4) showed superior performance (Fig. 5E), the classifiers based on the 11 candidate negative regulators showed superior performance in discriminating between over- and undertransported transcripts than the classifiers based on 32 positive regulators in both NGF and NTF3 conditions (i.e., higher model performance of M2 compared with M3) (Fig. 5E). These 11 negative RBPs form a network of interacting proteins enriched in regulators of translation and mRNA stability (Fig. 5F), including ELAVL1, which reduces neuronal cytoplasmic mRNA; CUGBP Elav-like family member 4 (CELF4); and KHSRP (Supplemental Fig. S5I; Engel et al. 2022; Olguin et al. 2022; Patel et al. 2022). KHSRP and CELF4 regulate mRNA abundance in axons and dendrites (Snee et al. 2002), whereas pumilio RNA binding family member 2 (PUM2) shapes the transcriptome in developing axons by retaining mRNAs in the cell body (Martínez et al. 2019). The visualization of positive and negative regulators shared by NGF and NTF3 along the 3′ UTRs of overtransported (Fig. 5G) and cell body–restricted transcripts (Fig. 5H) revealed a pronounced increase in the density of cross-linking events involving negative regulators of axonal localization. These findings suggest that mRNA destabilization and compartment-specific restriction play crucial roles in regulating the axonal transcriptome (Shav-Tal and Singer 2005; Wagnon et al. 2012; Patel et al. 2022; Loedige et al. 2023).
Further comparison of model performances revealed that classifiers trained with data from the NTF3 condition showed superior performance compared with the model trained with data from the NGF condition (Fig. 5E), particularly in classifying a lower proportion of cell body–restricted 3′ UTR isoforms as axonally localized (resulting in a reduced false-positive rate). Thus, transcripts with restricted axonal localization in the NTF3 condition display a more coherent RBP code and a better-defined regulation than those in NGF.
The enhanced performance of the classifiers trained with 42 interaction terms derived from 14 pairs of RBPs compared with those trained with the 43 individual RBPs suggests a combined and dependent regulation among these RBPs (Fig. 5E). This finding also highlights the identification of RBP combinations that show greater specificity for each cue (Fig. 5I,J). Furthermore, a systematic increase in the density of cross-linking events was observed for pairs of negative regulators in both of the NGF and NTF3 conditions (Fig. 5K–N; Supplemental Fig. S5J), highlighting the contribution of combined rather than individual RBPs.
The RBPome for 3′ UTR remodeling in axons
We recently discovered that the 3′ UTR of transcripts localized in axons may undergo local remodeling, resulting in the generation of polyadenylated isoforms expressing a shorter 3′ UTR that is stable and efficiently translated (Andreassi et al. 2021). Analysis of isoforms localized in NGF-treated and NTF3-treated axons revealed a similar number of 3′ UTR isoforms with different usage of long and short 3′ UTRs in both cell bodies and axons (665 and 458, respectively) (Supplemental Fig. S6A,B; Supplemental Tables S14–S17). Candidate transcripts of axonal remodeling were identified in both NGF and NTF3 conditions as isoforms expressing short 3′ UTR in axons but virtually absent in cell bodies (Fig. 6A, left; Supplemental Tables S18, S19). This unique expression pattern suggests that they are not the product of cotranscriptional APA and are cleaved in axons. At least some transcripts expressing short 3′ UTR only in axons are not transported (Andreassi et al. 2021); however, we cannot exclude that their localization may be owing to a very efficient and fast translocation from cell bodies to axons. Isoforms expressing short 3′ UTRs only in axons were largely not overlapping between NGF and NTF3 (Fig. 6A, right). Tracks of representative transcripts expressing unique short 3′ UTR in axons treated with either NGF or NTF3 are shown in Figure 6, B and C.
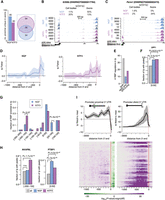
Negative regulators of APA in the cell body are candidate 3′ UTR cleavage factors in the axons. (A, left) Number of 3′ UTR isoforms with proximal shift uniquely detected in axons in NGF (blue) and NTF3 (purple) culture condition. (Right) Venn diagram showing the overlap between the candidate of axonal remodeling in NGF and NTF3. (B,C) Representative transcripts with a marked shift toward axonal increase in promoter-proximal 3′ UTR uniquely detected either in axons treated with NGF (Kif26b; B) or in axons treated with NTF3 (Parm1; C). (D) Distribution of the extent of significant enrichment in 126 RBP cross-link events in defined regions along the 3′ UTR of the 80 (left) and 60 (right) candidate isoforms of axonal remodeling in NGF and NTF3 condition, respectively. Dark lines display the median significance; shaded areas, lower and upper quartiles. (E) Number of candidate RBPs regulators of axonal remodeling identified in the [−250:+150]-nt region surrounding the 3′ end of the promoter-proximal isoform in both conditions, in NGF only and NTF3 only (gray, blue, and purple bars, respectively). (F) Fraction of promoter-proximal 3′ UTR isoforms showing cross-link events for UPF1 in the [0:50]-nt region downstream from the cleavage site. Gray bar indicates all promoter-proximal 3′ UTR; blue bars, 80 candidates of axonal remodeling in NGF; and purple bars, 60 candidates of axonal remodeling in NTF3. (G) Fraction of candidate RBPs regulators of axonal remodeling in NGF and NTF3 at specific positions along the 3′ UTR where they show the most significant enrichment. Fisher's enrichment test. (H) Same as G for AKAP8L and PTBP1. (I, top) Distribution of the extent of significant positive or negative association between the cross-link events in defined regions along the short (left) and long (right) 3′ UTR isoforms of 65 candidate regulators for axonal remodeling, as well as their relative usage of the short and the long isoform in the cell body. Dark lines display the median significance; shaded areas, lower and upper quartiles. (Bottom) Heatmaps showing the individual significance association between cross-link events of individual RBPs along the short (left) or long (right) 3′ UTR isoform and their relative usage in the axons.
We identified argonaute RISC catalytic component 2 protein (AGO2) as the endonuclease responsible for 3′ UTR cleavage (Andreassi et al. 2021). Fisher's enrichment in RBP cross-link events along the 3′ UTR of the remodeled isoforms (n = 80 in NGF and n = 60 in NTF3) revealed that although NGF-related predicted regulators of axonal remodeling preferentially localized to the [0:+50]-nt region downstream from the 3′ end, NTF3-related predicted regulators were found on both sides of the 3′ end (Fig. 6D). Thirty-five enriched RBPs were observed in both NGF and NTF3 remodeled isoforms, whereas 10 and 18 were unique for NGF or NTF3, respectively (Fig. 6E; Supplemental Tables S20, S21). Common RBPs included ELAVL1, which is known to inhibit polyadenylation when bound to the region downstream from the 3′ end (Supplemental Fig. S6C). The RNA helicase and ATPase UPF1, which belongs to the protein complex that we previously showed to mediate the 3′ UTR cleavage (Andreassi et al. 2021), was enriched in the 50-nt region downstream from the 3′ end of remodeled isoforms in both NGF-treated and NTF3-treated axons (Fig. 6F; Supplemental Fig S6D). Analysis of the positional preference of the top predicted regulators of axonal cleavage revealed that for NTF3-remodeled isoforms, binding to the [−200:−150]-nt window upstream of the 3′ end is favored, whereas regulatory regions of NGF-remodeled isoforms reside in the [50:100]-nt region downstream from the 3′ end (Fig. 6G). Neurotrophin-specific predicted regulators of axonal remodeling include A-kinase anchoring protein 8 like (AKAP8L), which is enriched in the [−200:−150]-nt region of 3′ UTR short isoforms and is uniquely detected in NTF3 condition, and polypyrimidine tract binding protein 1 (PTBP1), which is enriched in the [0:50]-nt region downstream from the pool of short 3′ UTR isoforms detected only in NGF-treated axons (Fig. 6H). Analysis of the association of the RBPs predicted to regulate 3′ cleavage in axons with APA in the nucleus revealed that these factors behaved as negative regulators of polyadenylation when bound downstream from the 3′ end of short 3′ UTR isoforms (Fig. 6I). Together, these findings indicate that RBPs predicted to regulate 3′ cleavage in axons may determine nuclear APA and 3′ UTR isoform expression.
Discussion
NGF and NTF3 are required for axon growth and neuron survival (Dorskind and Kolodkin 2021). In developing sympathetic neurons, NGF and NTF3 elicit distinct intracellular signaling pathways despite acting through the same receptor, NTRK1. NGF–NTRK1 complexes are internalized into signaling endosomes that travel back long distances to the cell bodies, where they activate transcription (Bhattacharyya et al. 1997; Riccio et al. 1997, 1999). In contrast, NTF3 cannot be endocytosed within signaling endosomes and is thought to act mostly locally (Kuruvilla et al. 2004; Harrington et al. 2011; Ascano et al. 2012; Scott-Solomon and Kuruvilla 2018; Scott-Solomon et al. 2021). It should be noted that sympathetic neurons do not express NTRK3 (also known as TrkC) (Scott-Solomon et al. 2021), the higher affinity receptor for NTF3 (Barbacid 1994), and therefore, any NTF3-dependent effect in these cells is likely owing to NTF3 binding to NTRK1. Our data show that both neurotrophins, when applied to distal axons, induce a robust transcriptional response of 3′ UTR isoforms that is only partially overlapping (Fig. 1). Our findings challenge the current understanding that NTF3 does not signal retrogradely to the cell body compartment, and open the possibility that it may initiate a signaling cascade that is faithfully propagated to the nucleus.
RBPs are key regulators of mRNA metabolism, including mRNA transport and translation, and are essential in determining when and where specific proteins are expressed. By integrating our 3′ end sequencing data with CLIP data generated in human cell lines, we discovered novel candidates and known regulators of APA (Fig. 2), corroborating the suitability of human data to study RBP regulomes underlying mRNA metabolism in rodent neurons. RNA transport in axons is known to be bidirectional, and transcripts complexed with RBPs can move both anterogradely and retrogradely (Holt and Schuman 2013; Terenzio et al. 2017; Das et al. 2019; Dalla Costa et al. 2020). We identified a large repertoire of transcripts that are localized and stored in sympathetic neuron axons in response to either NGF or NTF3 (Fig. 3), as well as the putative RBPome responsible for mRNA localization. We also showed that restricted axonal localization in response to NTF3 is associated with a more-defined RBP regulome compared with that of NGF (Fig. 5). Axonal localization is positively regulated by RBPs that facilitate mRNA transport and is negatively modulated by RBPs that regulate mRNA stability. Albeit universal axon-targeting motifs have not been identified so far, 32 RBPs were positively associated with axonal localization. Using a logistic regression method, groups of RBPs were identified that influenced mRNA localization to axons in response to either NGF or NTF3. Our analysis strongly suggests that mRNA stability and restriction to cell bodies play a pivotal regulatory role in axonal mRNA localization, aligning with recent research (Loedige et al. 2023). Future studies will clarify whether a similar combination of RBPs drives transcript localization to other neuronal compartments, such as dendrites and dendritic spines, or in other cell types.
We recently discovered that some axonal transcripts including inositol monophosphatase 1 (Impa1) undergo 3′ UTR remodeling in sympathetic neuron axons (Andreassi et al. 2021). Importantly 3′ UTR cleavage is necessary for triggering the translation of Impa1 mRNA isoforms expressing a short, cleaved 3′ UTR (Andreassi et al. 2021). Hundreds of transcripts with a proximal-to-distal shift of the 3′ UTR are detected in axons. Several short 3′ UTR isoforms are expressed exclusively in axons and, in some cases, specifically in response to either NGF and NTF3 (Fig. 5; Supplemental Fig. S5). Analysis of the RBP complexes interacting with the remodeled isoforms revealed an enrichment of UPF1 and PTBP1 previously identified as binding partners of the cleavage complex (Andreassi et al. 2021). It should be noted that predicted regulators of 3′ UTR cleavage behaved as negative regulators of nuclear polyadenylation when bound downstream from the 3′ end of short 3′ UTR isoforms. Thus, we propose that RBPs responsible for 3′ UTR cleavage are recruited cotranscriptionally to downstream regions of the promoter-proximal 3′ end in the nucleus. They favor the expression of the long 3′ UTRs required for axonal localization (Cosker et al. 2016; Terenzio et al. 2018; Andreassi et al. 2021), possibly by competing with cleavage factors and suppressing the use of the proximal PAS. The factors may remain bound to the 3′ UTR within RBP granules and hitchhike along the axons, eventually colocalizing in the axonal compartment with the long 3′ UTR isoform. Upon deassembly of the transport granules in axons, some factors, including UPF1, promote the cleavage of the long 3′ UTR (Fig. 7). Transport granules have been considered as “translation factories” that contain RBPs, mRNAs, ribosomes, and translation factors, regulating local protein synthesis (Krichevsky and Kosik 2001; Kanai et al. 2004). Here, we propose a similar mechanism by which APA, RNA localization, and neurotrophin-dependent translation are coupled and coregulated. Release of the 3′ UTR isoform remodeling factors from granules may serve as the final step that allows the translational activation in axons of transcripts expressing shorter 3′ UTRs.
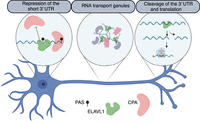
Proposed model in which the local translation and neurotrophin-mediated axon growth are linked to cotranscriptional APA, RNA localization, and axonal 3′ UTR remodeling through release of mRNAs and cleavage factors from transport granules.
Although further mechanistic studies will be necessary to validate the integration of compartmentalized 3′ end RNA-seq and CLIP sequencing data, our study sheds new light on the nature of axonal mRNA and the RBPs that regulate the transport and 3′ UTR remodeling. Moreover, given that most neurological diseases are considered as disorders of the RNA (Wang et al. 2007; Nussbacher et al. 2019), our data provide new targets potentially amenable for the cure of degenerative disorders of the nervous system.
Methods
Reagents
Cell culture reagents, molecular biology reagents, and kits were purchased from Thermo Fisher Scientific, and all other chemicals were from Sigma-Aldrich, unless stated otherwise.
Compartmentalized cultures of rat sympathetic neurons
All animal studies were approved by the institutional animal care and use committees at University College London. Superior cervical ganglia were dissected from postnatal day 1–2 Sprague Dawley rats, enzymatically dissociated, and plated on glass coverslips or in compartmentalized chambers precoated with homemade collagen and laminin (5 μg/mL), as previously described (Riccio et al. 1997). Undifferentiated cells seeded in the central compartment of the chambers were initially exposed to 100 ng/mL NGF to support survival and cell differentiation. Five to six days after plating, NGF was reduced to 10 ng/mL in the central compartment, and neurons were maintained with either NGF (100 ng/mL) or NTF3 (1 μg/mL) in the lateral compartment only where they promoted extensive axon growth. The concentration of NTF3 used was thoroughly tested to ensure a rate of axonal extension, similar and often even higher than that of NGF. The medium was changed every 48–72 h with a fresh amount of growth factors added. Cytosine arabinoside (ARA-C; 10 μM) was added 24 h after plating to block the proliferation of nonneuronal cells.
RNA isolation, reverse transcription, linear amplification, and 3′ end RNA-seq
3′ End sequencing was performed as previously (Andreassi et al. 2021) and as described in the Supplemental Methods. Primer sequences and PCR conditions are provided in Supplemental Table S23.
3′ UTR isoform quantification and identification of transcripts localized to axons
As previously described (Andreassi et al. 2021), the last 500-nt portion of each transcript contains >70% of the reads originating from that transcript irrespective of their length. We thus used the number of reads mapped to the −500-nt terminal region of each 3′ UTR isoform as a proxy for the 3′ UTR isoform expression levels. Because 3′ end sequencing amplifies the 3′ end of the transcript, it is not, in principle, influenced by the transcript length as is the case for classic RNA-seq, and therefore, no further normalization is performed to correct for transcript length as usually performed with RPKM. The density of mapped reads in the −500-nt terminal region of 3′ UTR isoforms is bimodal, with a low-density peak probably corresponding to background transcription, that is, 3′ UTR isoforms of low abundance or 3′ UTR isoforms to which reads were spuriously mapped, and a high-density peak corresponding to expressed 3′ UTR isoforms (Supplemental Fig. S4A). To identify 3′ UTR isoforms expressed in axons and cell body, a two-component Gaussian mixture was fitted to the data using the R package mclust (Fraley and Raftery 2002). An isoform was called expressed if there was a <5% chance of belonging to the background category in both replicates or if there was a >10% chance of belonging to the expressed category in at least one replicate.
Analysis of alternative cleavage and polyadenylation
The analysis of APA was performed as previously described (Andreassi et al. 2021) and as detailed in the Supplemental Methods. In brief, we first extracted the log2 ratio of promoter-proximal and promoter-distal 3′ UTR isoform expression levels, hereafter called RUD, in each sample. We also calculated the ratios between the read count in the promoter-proximal and the sum of the read counts in the promoter-proximal and the promoter-distal 3′ UTR isoforms, hereafter called PUD. In the case of multiple promoter-distal 3′ UTR isoforms per transcript ID, a value was computed for each individual promoter-distal 3′ UTR isoform. To identify transcripts that show a marked change in the 3′ UTR isoform between conditions, we scored the differences in proximal-to-distal poly(A) site usage using the differences in both RUD and PUD between conditions of interest. The statistical significance of the changes in proximal-to-distal poly(A) site ratio between conditions was assessed by a Fisher's exact count test. We adjusted the P-value controlling for a false-discovery rate (FDR) of 0.01.
Axonal localization analysis
The ratio between the genes’ abundance in the axons and in the cell body is often used to quantify mRNA axonal localization (Martínez et al. 2019; Olguin et al. 2022); however, this metric correlates with the mRNA abundance in the cell body and the transcript length (Supplemental Fig. 3C,D). Consequently, such metrics fail to identify highly transported but lowly expressed transcripts, and similarly, they are more likely to associate highly expressed transcripts with high mRNA axonal LSs despite low transport efficiency compared with transcripts of a similar expression level. Here, we aimed to develop a novel metric that accurately infers the axonal transport efficiency and stability, irrespective of the transcript length and the expression in the cell body, using a hierarchical Bayesian model procedure.
First, we looked at the global relationship between the normalized read count per the 3′ UTR isoforms in the axonal compartment
and their corresponding normalized read counts in the cell body compartment. We found that the average 3′ UTR isoform abundance
level—average log2 expression—in the axonal compartments of either the NGF or NTF3 condition is best approximated by a combination of polynomial
regression model of degree four of the abundance in the cell body and a linear model of the transcript length as revealed
by the Akaike's an information criterion from an ANOVA analysis (Supplemental Table S22; Supplemental Fig. S4E). Given our goal to set out a metric of axonal localization independent of the cell body read counts and the transcript length,
we created 102 groups of 3′ UTR isoforms of fixed expression ranges in the cell body (18 bins from 23 to 220 nt) and fixed ranges of transcript lengths (10 bins from 102 to 104.5 nt). For each of these 102 groups, we generated a matrix of 104 simulated draws of axonal read count values predicted using the fitted polynomial regression model of degree four, obtained
from the total pool of transcripts, over a regularly interspaced grid of 100 possible transcript lengths, ranging from the
minimal to the maximal transcript length of this specific group, and 100 possible cell body read count values, again ranging
from the minimal to the maximal cell body read count of this specific group. We then extracted the average and variance over
the predicted 104 axonal read count values for each of these 102 groups. Using these 102 pairs of averages and variances, we next fitted 102
negative binomial distributions to the 102 groups of axonal read counts by maximum likelihood (mle) using the fitdist function
from the fitdistrplus R package (Dutang 2015). These 102 posterior predictive distributions of axonal read counts could then serve to assess whether the observed axonal
abundance of each individual 3′ UTR isoform 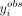 is consistent with the fitted models given their associated transcript length and cell body abundance. Here we propose that
the more extreme the
is consistent with the fitted models given their associated transcript length and cell body abundance. Here we propose that
the more extreme the 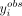 , for a given 3′ UTR isoform of specific transcript length and cell body abundance, is on the histogram of simulated values,
, for a given 3′ UTR isoform of specific transcript length and cell body abundance, is on the histogram of simulated values,
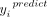 , from the corresponding predictive distribution, the more likely the axonal localization of its corresponding 3′ UTR isoform
has been actively regulated as opposed to the unspecific active transport, which is expected to affect most transcripts detected
in the axons. If
, from the corresponding predictive distribution, the more likely the axonal localization of its corresponding 3′ UTR isoform
has been actively regulated as opposed to the unspecific active transport, which is expected to affect most transcripts detected
in the axons. If 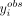 is in the lower tail, we expect this 3′ UTR isoform to be either restricted to the cell body or actively degraded in the
axonal compartment; conversely, if
is in the lower tail, we expect this 3′ UTR isoform to be either restricted to the cell body or actively degraded in the
axonal compartment; conversely, if 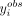 is in the higher tail of the histogram, we expect the 3′ UTR isoform to be either actively transported or stabilized in the
axonal compartment. Thus, for each 3′ UTR isoform i, we next computed the proportion of 104 values, randomly generated from the posterior negative distribution associated with its corresponding transcript length and
abundance in the cell body, that were smaller or larger than the observed axonal read count. To get a single value per transcript,
hereafter called the localization score (LS), we selected the smallest of these two values (probabilities to observe smaller
or larger axonal read count for a given isoform), transformed it using the log10, and multiplied it by −1 when the latter was selected. Hence, positive values are associated with active transport or stabilization,
whereas negative values are associated with cell body restriction or degradation. Notably, although the ratios between gene
abundance in the axons and in the cell body depend on cell body abundance (Supplemental Fig. S4F, top) and transcript length (Supplemental Fig. S4G, bottom), this is not the case anymore with the novel LS metric (Supplemental Fig. S4F,G, bottom). This analysis has been restricted on the 30,450 3′ UTR isoforms detected in the axonal compartments of neurons
exposed to either NGF or NTF3. The axonal LSs for these 3′ UTR isoforms have been computed for the NGF and NTF3 conditions
independently and are reported in Supplemental Table S9.
is in the higher tail of the histogram, we expect the 3′ UTR isoform to be either actively transported or stabilized in the
axonal compartment. Thus, for each 3′ UTR isoform i, we next computed the proportion of 104 values, randomly generated from the posterior negative distribution associated with its corresponding transcript length and
abundance in the cell body, that were smaller or larger than the observed axonal read count. To get a single value per transcript,
hereafter called the localization score (LS), we selected the smallest of these two values (probabilities to observe smaller
or larger axonal read count for a given isoform), transformed it using the log10, and multiplied it by −1 when the latter was selected. Hence, positive values are associated with active transport or stabilization,
whereas negative values are associated with cell body restriction or degradation. Notably, although the ratios between gene
abundance in the axons and in the cell body depend on cell body abundance (Supplemental Fig. S4F, top) and transcript length (Supplemental Fig. S4G, bottom), this is not the case anymore with the novel LS metric (Supplemental Fig. S4F,G, bottom). This analysis has been restricted on the 30,450 3′ UTR isoforms detected in the axonal compartments of neurons
exposed to either NGF or NTF3. The axonal LSs for these 3′ UTR isoforms have been computed for the NGF and NTF3 conditions
independently and are reported in Supplemental Table S9.
Mapping and analysis of CLIP data
To identify RBPs that bind to 3′ UTR regions, we examined iCLIP data for 18 RBPs (Attig et al. 2018) and eCLIP data from K562 and HepG2 cells for 89 and 70 RBPs, respectively, available from ENCODE (Sloan et al. 2016; Van Nostrand et al. 2020). In total, we analyzed CLIP-seq data for 126 RBPs (for complete list of CLIP sequencing data, see Supplemental Table S6). Before mapping the reads, adapter sequences were removed using cutadapt v1.9.dev1 (Martin 2011), and reads <18 nt were dropped from the analysis. Reads were mapped with STAR v2.4.0i (Dobin et al. 2013) to the UCSC hg19/GRCh37 genome assembly. To quantify binding to individual loci, only uniquely mapping reads were used. The results were lifted to rn5 using liftOver (Hinrichs et al. 2006). The mapping of the CLIP data was performed in 2018. Although updated versions of the human genome assembly have been released since then (GRCh38, T2T-CHM13), mapping the CLIP to a newer version of the genome assembly is not expected to significantly change the results given that novel annotated regions are not biased toward specific regions (3′ UTR long vs. short isoform) or to specific groups of genes related to neuronal functions. Indeed, the statistics based on these data capitalize on the large variety of transcripts and regions where these are mapped.
RBP regulome underlying APA
To identify positive and negative RBPs regulators of APA in developing rat sympathetic neurons, we tested the association between RBP binding in defined regions along the 3′ UTR and the relative usage of the promoter-proximal 3′ UTR isoforms (PUDs) in the cell body compartments of either NGF or NTF3 conditions. In particular, we used the Welch's t-test to compare the distributions of the PUD between groups of isoforms showing or not cross-link events in the following 29 defined regions surrounding the 3′ ends of the promoter-proximal 3′ UTR isoforms: [−3000:−2950], [−2750:−2700], [−2500:−2450], [−2250:−2200], [−2000:−1950], [−1750:−1700], [−1500:−1450], [−1400:−1350], [−1300:−1250], [−1200:−1150], [−1100:−1050], [−1000:950], [−900:−850], [−800:−750], [−700:−650], [−600:−550], [−500:−450], [−450:−400], [−400:−350], [−350:−300], [−300:−250], [−250:−200], [−200:−150], [−150:−100], [−100:−50], [−50,0], [0,+50], [+50:+100], and [+100:+150]. There were 126 RBPs with available CLIP-seq data in human cell lines; thereby we obtained 30 P-values per RBPs. These were then −log10-transformed and multiplied by the sign of the difference in PUD between the group of isoforms showing or not a cross-link event in the defined region along the 3′ UTR. Thus, a positive value indicates that RBP binding to regions surrounding the 3′ end of the promoter-proximal 3′ UTR promotes the usage of the promoter-proximal 3′ UTR isoforms, hence acting as positive regulators of the promoter-proximal and negative regulators of promoter-distal 3′ UTR isoforms, whereas a negative value indicates that RBP binding to regions surrounding the 3′ end of the promoter-proximal 3′ UTR promotes the usage of the promoter-distal isoforms, hence acting as negative regulators of the promoter-proximal and positive regulators of promoter-distal 3′ UTR isoforms. We repeated this analysis to compare the distributions of the PUD between groups of isoforms showing or not cross-link events in the same 30 defined regions surrounding the 3′ ends of the promoter-distal 3′ UTR isoforms, therefore inspecting the regulatory potential of RBP through the binding of the distal 3′ UTR isoforms. Thus, we obtained a map of regulatory potential of the relative 3′ UTR usage along the short and long 3′ UTR for each of the 126 RBPs.
RBP regulome underlying APA between NGF and NTF3 and axonal remodeling
To identify RBPs regulators of APA between NGF and NTF3, we performed a Fisher's count enrichment analysis to test for significant enrichment in RBP cross-link events in defined regions (see previous paragraph) around the 3′ end of either the promoter-proximal or the promoter-distal 3′ UTR isoform between the groups of isoforms showing significant shifts in either group and the total pool of 3′ UTR isoforms. This analysis is indeed more efficient in recovering candidate RBP regulators compared with the Welch's t-test comparing the distributions in ΔPUD between groups of isoforms showing or not a cross-link event given the relatively low number of pairs of isoforms showing promoter-proximal or promoter-distal shifts in the cell bodies of NGF-treated and NTF3-treated neurons compared with the full set of 3′ UTR isoforms. We used a similar approach to identify candidate regulators of axonal remodeling. Specifically, we used the Fisher's count test to assess for significant enrichment in RBP cross-link events in defined regions along the 3′ UTRs of predicted remodeled promoter-proximal 3′ UTR isoforms (n = 80 in NGF and n = 60 in NTF3) compared with the total pool of promoter-proximal 3′ UTR.
RBP regulome underlying axonal transport
To identify positive and negative RBPs regulators of axonal 3′ UTR isoform localization and stability in developing rat sympathetic neurons, we tested the association between RBP binding in defined regions along the 3′ UTR and the LS in NGF or NTF3 conditions. We used the Welch's t-test to compare the distributions of the LSs between groups of isoforms showing or not cross-link events in 30 defined regions surrounding their 3′ ends (see above for detailed regions) for 126 RBPs with available CLIP-seq data in human cell lines, thereby obtaining 30 P-values per RBPs. These were then −log10-transformed and multiplied by the sign of the difference in LSs between the group of isoforms showing or not a cross-link event in the defined region along the 3′ UTR. Thus, positive values indicate that RBP binding to regions surrounding the 3′ end of 3′ UTR isoform promotes axonal transport and/or stability, whereas negative values indicate lower LSs for the group of isoforms showing cross-link events compared with the group that does not show a cross-link event, hence indicating that that RBP binding to regions surrounding the 3′ end of 3′ UTR isoforms prevents axonal transport and/or promotes mRNA degradation.
Combinatorial regulatory potential of RBPs in axonal localization
To investigate potential combinatorial effects of RBPs in regulating axonal mRNA localization, we used a binary logistic regression classifier trained to distinguish between over- and undertransported 3′ UTR isoforms. For this analysis, we focused on the 5% isoforms showing the highest (n = 1356) and lowest (n = 1356) LSs in each treatment condition to have a sufficiently high number of training data (Supplemental Fig. S5F), ensuring a balanced representation of both classes. We split the total pool of 3′ UTR isoforms into a training set (n = 2168) and a testing set (n = 542). The training set was used to perform a randomized search on hyperparameters, in which a cross-validation strategy with 20 folds was used to assess the performance of the different hyperparameter combinations.
A combination of four classifiers was trained and tested, in which the predictor variables were, respectively, (1) evidence of cross-link events for the 32 positive and 11 negative regulators of axonal transport identified using Welch's t-test (M1), (2) evidence of cross-link events for the 32 positive regulators of axonal transport identified using statistical testing (M2; Supplemental Table S12), (3) evidence of cross-link events for the 11 negative regulators of axonal transport identified using statistical testing (M3; Supplemental Table S13), and (4) combinations of cross-link events involving 42 selected pairs of the 43 RBPs identified as potential positive and negative regulators of axonal transport in the region preceding the 3′ end, specifically the [−250, −50] region (M4). An isoform was considered bound by an RBP if there was evidence of a cross-link event in the region [−250:−50] nt preceding the 3′ end in at least one of the multiple CLIP sequencing data generated in different cell lines (HepG2, K652) and with different techniques (eClip and iClip).
The development of the M4 model aimed to study the synergistic potential of RBP was limited by the size of the training set (n = 2000 3′ UTR isoforms), making it challenging to learn the coefficients for the total number of possible pairwise interaction terms (ntot = 583; n = 528 interactions terms for 32 positive candidate regulators of axonal localization and n = 55 interactions terms for the negative regulators). We therefore selected the four RBPs showing most positive weights and the four RBPs showing most negative weights in the M1 regression models trained with the total set of 43 positive and negative regulators in either the NGF (top positive regulators: SF3B4, SRSF1, SNRPB, EFTUD2; top negative regulators: CELF4, KHDRBS1, PUM2, FUBP3) or NTF3 (top positive regulators: SF3B4, DDX24, SLTM, SRSF9; top negative regulators: CELF4, KHSRP, CELF2, HNRNPC; M1) (Supplemental Fig. S5G,H) condition. We next created 42 pairwise interaction predictor variables (21 positive and 21 negative interaction terms) by identifying 3′ UTR isoforms showing evidence of cross-link events for pairs of RBPs.
All logistic regressions have been fitted using the sklearn library in Python (Pedregosa et al. 2011). For each set-up, we performed a randomized search to optimize the hyperparameters, namely, the solver among [“lbfgs,” “newton-cg,” “liblinear,” “sag,” “saga”] and the regularization technique among [“l2,” “l1,” “elasticnet”]. We fixed the inverse regularization term to C = 0.01 for all models to make it easier to compare and interpret the performance between the different models, and the maximum number of iterations taken for the solvers to converge has been set to max_iter = 10,000. The selected regularization term for both NGF and NTF3 sets is the L2 penalty term for all four models, whereas the solver depends on the model (lbfgs for M1, M3, and M4 and liblinear for M2 in the NGF condition; sag for M1, liblinear for M2, and lbfgs for M3 and M4 in the NTF3 condition). The predictive power of this set of models was determined using the recall and precision and F1-score.
To identify the statistically significant predictors in each model (RBPs or interaction of RBPs) in each culture condition,
we compared the observed predictor coefficients to their respective null distributions. To generate a null distribution for
each predictor, we shuffled the response variable label randomly, retrained the model, and recalculated the predictor coefficients.
We repeated this process 2000 times. To compare the observed predictor coefficients with the null distributions generated,
we computed their Z-scores as
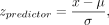 where x is the observed predictor coefficient, and µ and σ are the mean and standard deviation of the null distribution for this predictor. If |zpredictor| > 1.645 (95% confidence; one-tailed), then the predictor is a significant regulator of localization. In particular, if zpredictor < 0, the predictor is a negative regulator of localization, whereas if zpredictor > 0, the predictor is a positive regulator of localization. To compare regulators between the NGF and NTF3 conditions, we
applied the following rule: If a predictor is significant in both the NGF and the NTF3 model, then that predictor is a common
regulator of localization. However, if a predictor is significant in only one of the two models, that predictor is a specific
regulator of this model.
where x is the observed predictor coefficient, and µ and σ are the mean and standard deviation of the null distribution for this predictor. If |zpredictor| > 1.645 (95% confidence; one-tailed), then the predictor is a significant regulator of localization. In particular, if zpredictor < 0, the predictor is a negative regulator of localization, whereas if zpredictor > 0, the predictor is a positive regulator of localization. To compare regulators between the NGF and NTF3 conditions, we
applied the following rule: If a predictor is significant in both the NGF and the NTF3 model, then that predictor is a common
regulator of localization. However, if a predictor is significant in only one of the two models, that predictor is a specific
regulator of this model.
Statistical analyses
Data are expressed as averages ± SEM. A t-test was used as indicated to test for statistical significance, which was placed at least P < 0.05 unless otherwise noted. Statistical analysis was performed with the R statistical package version 4.2.2 (2022) and Bioconductor library version 3.16 (R Core Team 2013).
Data access
The processed data required to implement the code and generate figures, as well as Supplemental Tables, are available at Zenodo (https://doi.org/10.5281/zenodo.8047412). Code and scripts used to make the results are available at GitHub (https://github.com/RLuisier/AxonLoc) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank all members of the Riccio and Luisier laboratories, as well as Pierre Klein for stimulating discussions and critical review of the data. The work was supported by the Wellcome Trust Investigator awards 103717/Z/14/Z and 217213/Z/19/Z (to A.R.), the Medical Research Council LMCB Core grant MC/U12266B (to A.R), a Wellcome Trust Institutional Strategic Support Fund (to C.A.), and Idiap Research Institute (to R.L. and L.F.).
Authors contributions: R.L., C.A., and A.R. conceived and designed the study. C.A. performed the screen and all experiments presented in the study. R.L. designed the computational framework, performed data analysis and interpretation of results, designed the figures, and derived the model. L.F. developed machine learning frameworks for combinatorial analysis of RBPome underlying axonal localization. R.L. and A.R. wrote the manuscript with the critical input from C.A. and L.F. All authors discussed the results and contributed to the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277804.123.
-
Freely available online through the Genome Research Open Access option.
- Received February 14, 2023.
- Accepted August 10, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








