
The rate of de novo structural variation is increased in in vitro–produced offspring and preferentially affects the paternal genome
- Young-Lim Lee1,2,
- Aniek C. Bouwman2,
- Chad Harland1,3,
- Mirte Bosse2,
- Gabriel Costa Monteiro Moreira1,
- Roel F. Veerkamp2,
- Erik Mullaart4,
- Nadine Cambisano5,
- Martien A.M. Groenen2,
- Latifa Karim5,
- Wouter Coppieters1,5,
- Michel Georges1 and
- Carole Charlier1
- 1Unit of Animal Genomics, GIGA-R, Faculty of Veterinary Medicine, University of Liège, B-4000 Liège, Belgium;
- 2Wageningen University and Research, Animal Breeding, and Genomics, 6708 WG Wageningen, The Netherlands;
- 3Livestock Improvement Corporation, Hamilton 3240, New Zealand;
- 4CRV B.V., 6842 BD Arnhem, The Netherlands;
- 5GIGA Genomics Platform, GIGA Institute, University of Liège, B-4000 Liège, Belgium
Abstract
Assisted reproductive technologies (ARTs), including in vitro maturation and fertilization (IVF), are increasingly used in human and animal reproduction. Whether these technologies directly affect the rate of de novo mutation (DNM), and to what extent, has been a matter of debate. Here we take advantage of domestic cattle, characterized by complex pedigrees that are ideally suited to detect DNMs and by the systematic use of ART, to study the rate of de novo structural variation (dnSV) in this species and how it is impacted by IVF. By exploiting features of associated de novo point mutations (dnPMs) and dnSVs in clustered DNMs, we provide strong evidence that (1) IVF increases the rate of dnSV approximately fivefold, and (2) the corresponding mutations occur during the very early stages of embryonic development (one- and two-cell stage), yet primarily affect the paternal genome.
The genetic polymorphism observed in populations results from a balance between the birth of new variants by the process of de novo mutation (DNM) in the germline, the loss of variants by genetic drift, and selective forces that increase or decrease the frequency of variants in the population. Until recently, the rate of DNM in the germline was estimated indirectly by phenotypic mutation screening (including the incidence of inherited diseases), from between-species sequence divergence, or from within-species levels of polymorphism (Kondrashov and Kondrashov 2010; Ségurel et al. 2014). Advances in massively parallel sequencing now allow estimating the rate of de novo mutation directly. This is mostly performed by sequencing father–mother–offspring trios. Alternatively, the rate of DNM can be estimated from the number of sequence differences detected by massively parallel sequencing between identical-by-descent chromosome segments in relatives separated by a known number of generations (Palamara et al. 2015).
Massively parallel sequencing–based approaches have first been used to estimate the rate of DNMs for the most abundant class of variants; that is, point mutations or single-nucleotide variants. We refer to this category as dnPMs. In humans, the average number of dnPMs detected in a newborn is of the order of 45 (e.g., Goldmann et al. 2016). The rate of dnPMs in the male germline is estimated on average at about 20 per gamete at puberty with an extra ∼1.3 dnPMs per year after puberty (Goldmann et al. 2016; Sasani et al. 2019). This paternal age effect is usually attributed to the increased number of cell divisions separating the zygote from sperm cells with increasing age of the father, although the validity of this explanation has been questioned (Wu et al. 2020). The rate of dnPM in the female germline is estimated at approximately five per oocyte on average at puberty (Goldmann et al. 2016). As oogonia enter meiosis before birth, the number of cell divisions separating zygote and oocyte is independent of maternal age, yet a small effect of maternal age on the number of dnPMs per oocyte (∼0.4 dnPM per year) has also been observed (Sasani et al. 2019), which may in part arise from reduced DNA repair capacity of aging oocytes (Goldmann et al. 2018; Gao et al. 2019). In addition to studies in humans, the rate of dnPMs has been estimated from massively parallel sequencing data in approximately 10 vertebrate species including primates, birds, fish, and cattle (Bergeron et al. 2022). These revealed large differences in mutation rate across species (e.g., 1.66 × 10−8 in orangutans [Besenbacher et al. 2019] and 0.2 × 10−8 in herrings [Feng et al. 2017]), which may be owing to differences in effective population size and, hence, the effectiveness of purifying selection against mutations affecting DNA maintenance (Lynch et al. 2016).
Massively parallel sequencing–based methods have also been used to study the generation of other types of genetic variants, particularly structural variants. Structural variants are operationally defined as variants that affect ≥50 bp at once (Sudmant et al. 2015). They include deletions, duplications, inversions, and combinations thereof. We refer to this category as dnSV. dnSVs are of particular interest as they have a higher probability than dnPMs to perturb the function of genes and to have phenotypic consequences including diseases (e.g., Belyeu et al. 2021). In humans, the rate of dnSV has been estimated at one dnSV per six to 13 births (Brandler et al. 2018; Werling et al. 2018; Collins et al. 2020), with an approximately twofold higher paternal than maternal contribution (Kloosterman et al. 2015; Belyeu et al. 2021).
Germline DNMs are accurately defined as genetic variants that were not present in the gametes that fused to generate a zygote but are observed in one or more gametes produced by the individual that developed from that zygote (Moorjani et al. 2016). The mutational process that gave rise to the DNM must have occurred in the germline of that individual. It can have occurred as early as in the zygote itself, as late as in the final stages of meiosis, or at any time between these two extremes. If the DNM occurred early during development, the individual may show detectable “mosaicism”; that is, the DNM will be detectable in the DNA of the individual. If the DNM occurred in the zygote (before S phase), its allelic dosage will be 50% in all cells. The later the DNM occurred during development, the lower its allelic dosage will be. For instance, allelic dosages of 25%, 12.5%, or 6.25% suggest that the DNM occurred after the first, second, or third cell divisions. However, as the organism's cell lineage is not a simple exponentially growing bifurcating tree, these extrapolations have to be considered with caution: Bottlenecks in the cell lineage may increase the allelic dosage of DNM that occurred later during development (Park et al. 2021). Nevertheless, DNMs that are detectably mosaic are likely to have occurred before segregation of primordial germ cells and are therefore often detected in both soma and the germline. On the other hand, DNMs that occur during the final stages of gametogenesis are typically not detected in DNA samples from the individual in whom they occurred (including sperm DNA). The individual is not detectably mosaic for such “late” DNMs, which can be detected only via their transmission to offspring.
DNMs are thought to arise from errors during the repair of lesions undergone by the genomic DNA. These lesions can be mismatches in the double helix owing to the incorporation of erroneous bases by the DNA polymerase during DNA replication or the deamination of methylated cytosines. But they can also be single- and double-stranded DNA breaks, abnormal bases such as uracil produced by deamination of unmethylated cytosines, pyrimidine dimers produced by UV irradiation, 8-oxoguanine produced by oxidation of guanine, or ribonucleotides incorporated during lagging-strand replication (Reijns et al. 2015; Gao et al. 2016). For clarity, we consider that the DNM exists once the new sequence is present on both the Crick and Watson strands of the DNA. Before that, we refer to the alterations that induce the DNM as “lesions.”
The rate of germline DNM is thought to be potentially affected by intrinsic as well as extrinsic factors. There is evidence that the rate of DNM varies between individuals, and part of these differences may be inherited (Sasani et al. 2019). There is also evidence that not only the rate but also the spectrum of DNM changed during human evolution (Harris 2015; Harris and Pritchard 2017; Mathieson and Reich 2017; Narasimhan et al. 2017). Additionally, the rate of DNM varies along the genome at different scales, involving factors such as local base pair composition, transcription, and replication timing (for review, see Ségurel et al. 2014). Exposure to chemical and physical mutagens in the environment is likely to affect the DNM rate as well (Adewoye et al. 2015; Holtgrewe et al. 2018; Kaplanis et al. 2022). An important consideration regarding extrinsic factors is the growing utilization of assisted reproductive technologies (ARTs) in humans and domestic animals. ART include artificial insemination (the use of frozen semen to inseminate females [AI]), transfer of in vivo–fertilized embryos (usually after inducing multiple ovulations in the females [MOET]), in vitro fertilization (with fresh or frozen semen) of in vitro–matured oocytes obtained by oocyte pickup from ovaries (in vivo or postmortem [IVF]), and intracytoplasmic sperm injection (ICSI) (Duranthon and Chavatte-Palmer 2018). To what extent and how ARTs affect the rate of DNM in humans and animals remain controversial owing to limited sample sizes and possible confounding factors (e.g., in humans, ART is primarily used for couples with fertility issues, which may confound the study of the direct effect of ART on DNM) (Wong et al. 2016; Tšuiko et al. 2017; Esteki et al. 2019; Wang et al. 2021; Smits et al. 2022). Domestic cattle genealogies have been accurately recorded over many generations and are characterized by large paternal and maternal half-sib pedigrees. This provides unique opportunities to assemble large pedigrees that are ideally suited for the detection of DNM. Moreover, the reproduction of domestic cattle makes extensive use of ARTs ranging from AI to MOET to IVF, facilitating the study of the effect of ART on DNM. In this work, we exploited a multigenerational bovine pedigree designed for the detection of DNM (referred to as the Damona pedigree) (Harland et al. 2016) to explore the rate and mechanisms that generate dnSVs.
Results
Identifying DNM in the Damona pedigree
DNMs are most often detected using DNA samples from father–mother–offspring trios. In these studies, DNMs are variants for which both parents are called homozygous wild type (or “reference”), whereas the offspring is called heterozygous by standard genotyping software including GATK (Van der Auwera and O'Connor 2020). In addition, to qualify as a DNM in most studies, the variant must not have been previously reported in a variant database; that is, it must be a novel variant. This filtering step eliminates lots of false DNMs but excludes recurrent DNMs, which are known to exist and constitute an interesting research topic on their own (Seplyarskiy et al. 2021). It is important to realize that the trio-based experimental design primarily detects DNMs that (1) occurred late during gametogenesis of the father or mother and were transmitted to the offspring, who is “constitutively” heterozygous (50% allelic dosage) or (2) occurred very early during the development of the offspring such that the allelic dosage in the offspring is high enough for it to be called heterozygous by genotyping software. In these experiments, the DNM can be assigned to either the paternal or maternal homolog of the offspring if a parent-specific variant can be captured by the same sequence reads as the DNM. An important limitation of the trio-based design is that one cannot always be 100% sure that the DNM detected in the offspring transmits to the next generation; that is, that it is truly a germline DNM. Also, most DNMs do not have a close variant that will allow assignment to either the paternal or maternal homolog. Having multiple samples from an additional generation (i.e., grand-offspring) allows confirmation of transmission and allows assignment to the paternal or maternal homolog by linkage analysis in the grand-offspring (Harland et al. 2016).
It is with these considerations in mind that we assembled the Damona pedigree for the detection of DNM (Fig. 1A). The Damona pedigree comprises 743 Dutch Holstein Friesian cattle that can be assigned to 127 three-generation pedigrees comprising a sire, a dam, an offspring, an average of eight sibs of the offspring (range: 0–17), and an average of five grand-offspring (range: 1–11). Moreover, an average of 1.4 grandparents per pedigree were available as well (five pedigrees with four grandparents, 16 with three, 38 with two, 34 with one1, 34 with zero) (Fig. 1B).
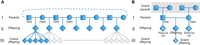
The Damona pedigree structure. (A) A sire is bred with multiple dams and vice versa. Using this feature, we minimized the number of animals to be sequenced while increasing the number of independent three-generation pedigrees. Squares and circles mark sires and dams, respectively (first generation). Diamonds mark offspring (second generation) and grand-offspring (third generation) of unspecified sex. (B) Example of a Damona pedigree. Each pedigree consists of a minimum of three generations (parents, offspring, and grand-offspring). Some pedigrees have grandparents available, making four-generation pedigrees (marked with a dotted gray box). The numbers indicate the average number of animals per pedigree (six paternal half-sibs [HS], two maternal HS, and five grand-offspring per offspring).
The 127 pedigrees overlap: For instance, an animal can be an offspring in one pedigree and a parent in another. All animals from the Damona pedigree had their whole genome sequenced (females: blood DNA; males: 24% blood, 76% sperm DNA). The average sequence depth was 26× for sire–dam–offspring trios, 17× for sibs, 10× for grand-offspring, and 27.3× for grandparents. This pedigree is well suited for the detection of germline DNM and their reliable assignment to sire, dam, or offspring.
The first type of DNM that is detected in the Damona pedigree are DNMs that occurred late during gametogenesis of either sire or dam and were transmitted to the offspring. Accordingly, both parents will be called homozygous reference and the offspring heterozygous by standard genotyping algorithms. To qualify as “late parental DNM,” we further require the DNM (1) to be transmitted to at least one grand-offspring in perfect linkage (i.e., r2 = 1) with either the paternal (in which case it is assigned to the sire) or the maternal (in which case it is assigned to the dam) homolog of the offspring, (2) to have an allelic dosage in the offspring that is not significantly inferior to that in the grand-offspring (who are known for sure to be heterozygotes; this controls for allelic imbalance of technical origin), (3) to be absent—even at very low dosage—in parental sequence reads (if the dosage is very low, the parent may have been called homozygous reference by standard genotyping software despite the DNM being clearly present in the sequence reads), and (4) to not to be transmitted to any of the offspring's siblings. The second type of DNM that is detected in the Damona pedigree are DNMs that occurred early during the development of the offspring: “early offspring DNM.” These DNMs differ from the first category (late parental DNMs) by the fact that (1) the allelic dosage in the offspring is significantly inferior to that in the grand-offspring and/or (2) it may be transmitted to grand-offspring in imperfect yet complete linkage (r2 < 1, D′ = 1) with one of the parental homologs of the offspring and/or (3) if there is a nearby parental variant, three haplotypes are observed among the sequence reads of the offspring (i.e., for differences 2 and 3, the haplotype upon which the DNM occurred is observed with and without the DNM). It is noteworthy that if an “early offspring DNM” occurred in the zygote (i.e., before the S phase of the first embryonic cell division), it can a priori not be distinguished from a “late parental DNM” (i.e., in the absence of a new source of information; see hereafter). The third type of DNM that are detected in the Damona pedigree is “early parental DNMs.” These differ from the first category (late parental DNM) by the fact that (1) sequence reads with the DNM are found at low dosage in a parent (despite it being called homozygous reference by genotyping software) and/or (2) the DNM is also transmitted to one or more sibs of the offspring. We further require for the DNM of either category that none of the ascendants of the offspring in the pedigree be called heterozygous.
The Damona pedigree was first used to detect dnPMs using the rules defined above. The results of a pilot experiment were reported (Harland et al. 2016). We herein report on the use of the same data set and rules for the detection of dnSVs.
Extreme paternal bias of dnSV in IVF-produced offspring
To detect dnSV in the Damona pedigree, we analyzed the whole-genome sequence data for the 127 three-generation pedigrees using Smoove (https://github.com/brentp/smoove), followed by extensive manual curation and application of the rules defined in the previous section. This resulted in the identification of 19 dnSVs, consisting of 14 deletions, three duplications, one inversion, and one complex dnSV (Del/Dup), ranging from 58 bp to 1.2 Mb in size (Table 1; Supplemental Figs. S1–S24). This corresponds to one dnSV per 6.7 births, which is in line with recent estimates in human studies (vide supra) (Supplemental Fig. S25; Supplemental Table S1). Three of the 19 dnSVs encompassed protein-coding exons, including one that disrupted the centromere protein C (CENPC) gene, likely causing a loss-of-function mutation of this essential gene (Kalitsis et al. 1998). Examination of the breakpoint sequences revealed microhomologies (1–14 bp) suggestive of microhomology-mediated break-induced replication (MMBIR) for 10 dnSVs, the absence of microhomologies suggestive of nonhomologous end-joining (NHEJ) for six dnSVs (with small insertions for two), interspersed repeat elements spanning the breakpoints suggestive of nonallelic homologous recombination (NAHR) for two (SINE: NM-2, LINE: M-4), and a variable number of tandem repeat (VNTR) structures for another (A3) (Table 1; Supplemental Fig. S26; Hastings et al. 2009a,b). Eleven of the 19 dnSVs were categorized as “late parental,” three as “early offspring,” two as “early parental” DNMs, and three as “ambiguous” (most likely “late parental,” yet possibly “early offspring”) as defined in the previous section. Of note, the three “early offspring” dnSVs were detected in blood DNA of three female offspring (and hence present in the soma) yet were transmitted to grand-offspring (and hence present in the germline), testifying of the fact that they indeed occurred during early development.
dnSVs detected in the Damona pedigree
The 19 dnSVs revealed two noteworthy features. The first is that 10 of the 11 “late parental” events were assigned to the germline of the sire. This 10:1 paternal bias is statistically significant (P = 1.2 × 10−2) and considerably higher than the approximately 2:1 paternal bias reported in humans (Kloosterman et al. 2015; Belyeu et al. 2021). The second is that 14 of the 19 events (74%) were detected in pedigrees in which the offspring was produced by IVF. Of note, 27% (34/127) of Damona offspring were produced by AI, 37% (47/127) by MOET, and 36% (46/127) by IVF (Supplemental Table S2). The enrichment of dnSV in IVF-produced offspring is statistically significant (P = 1.6 × 10−3). These numbers indicate that the rate of dnSVs in offspring produced by AI or MOET is of the order of one in 16 births, whereas the corresponding rate in offspring produced by IVF is of the order of one in three births; that is, about five times higher (95% CIbootstrap: 1.6–31.7 times higher) (Supplemental Fig. S27). Of note, two offspring produced by IVF carried two distinct dnSVs (ID-59: A-2 and NM-3; ID-248: M-3 and NM-1). Although this may seem to further support the effect of IVF on the rate of dnSV, the departure from Poisson distribution was not statistically significant (P = 0.55).
We fitted a linear model to jointly test the effect of sample type of the offspring (sperm or blood), age of the sire and dam at reproduction, sequencing depth (of sire, dam, and offspring), and ART used to produce the offspring. Only IVF (P = 0.016) and sequencing depth of the sire (P = 0.028) were nominally significant. IVF increased the dnSV rate (number of dnSV per newborn animal) by 0.218 compared with AI, whereas an extra 1× coverage of the sire's genome increased the dnSV rate by 0.012. There was no evidence of an effect of paternal or maternal age (P ≥ 0.50), reminiscent of recent reports in human (e.g., Belyeu et al. 2021). It is noteworthy, in this regard, that the age range for the sires was 2.1–11.1 yr and for the dams 1.8–6.7 yr, hence much more contracted than in human studies (Supplemental Table S2; Supplemental Fig. S27).
Clustered dnSVs and dnPMs reveal the common occurrence of complex postfertilization events
It has been shown in human studies that dnSVs are accompanied by nearby dnPMs (including small indels) more often than expected by chance, generating so-called clustered DNMs. Clustered DNMs are thought to result from one and the same mutational event (Michaelson et al. 2012). Estimates of the proportion of dnSVs with associated dnPMs in humans range from 11%–16% at <20-kb intermutational distance (Brandler et al. 2016; Goldmann et al. 2018). In light of these findings, we scanned the genomic vicinity of the 19 detected dnSVs for associated dnPMs. We found associated dnPM at ≤3 kb for six of the 19 dnSVs (31%) (Table 2; Supplemental Figs. S28–S33). The overall validation rate of dnPMs detected in the Damona data using an independent method (Allegro, Tecan) was 80%. Four of the six dnPMs associated with a dnSV were included in the validation and were all confirmed. Of note, all six affected the homolog transmitted by the sire, and all clustered DNMs were detected in pedigrees in which the offspring was produced by IVF (Table 2).
Clustered dnSVs and dnPMs in the Damona pedigree
The probability to observe dnPM at ≤3-kb for six of the 19 dnSVs by chance alone, given the individual rates of dnPM in the Damona pedigree, was estimated to be <10−7. The closest next dnPM outside of the clustered DNM was ≥22 Mb away. Thus, we can safely assume that the associated dnSVs and dnPMs arose as a result of the same mutational event, in agreement with human studies. In further support of the fact that clustered DNMs arise from a distinct mutational process, four of the six associated dnPMs were indels, whereas the proportion of indels only accounts for ∼10% among the collection of dnPMs detected in the Damona pedigree (P = 1.7 × 10−4) (Harland et al. 2016). Also, for all six associated dnPMs, we showed (using inherited SNPs in the same reads) that the dnPM affected the same homolog as the corresponding dnSV (Supplemental Figs. S28–S33).
As a consequence, one would naturally expect associated dnSVs and dnPMs to be of the same mutational category, whether late parental, early offspring, or early parental (see previous sections). For three of the six clustered DNMs, the dnSV was a “late parental” event, for two an “early offspring” event, and for one an “ambiguous” event. To our surprise, for two of the six clustered DNMs for which the dnSV was of a “late parental” type, the associated dnPM was of an “early offspring” type (NM-1 and NM-8). For both, the classification of the dnSV as a “late sire” event and of the dnPM as an “early offspring” event was based on multiple independent lines of evidence. In particular, linkage with the paternal chromosome in the grand-offspring was perfect for the dnSV yet imperfect for the dnPM (NM-1 and NM-8) (Table 2). For both associated dnPMs, the allelic dosage in the offspring was ∼≥20%, strongly suggesting that they occurred after the S phase of the first cell stage of the offspring's development (Supplemental Table S3).
Discussion
Two initial key observations of this work, namely, (1) that a majority of dnSVs appeared to have occurred during the late stages of gametogenesis in the sire and (2) that the use of IVF increases the rate of dnSV by a factor of five, jointly create a conundrum. Indeed, why would the IVF procedure increase the incidence of dnSV in the germline of the sire that produced the used sperm (often collected a long time before the IVF procedure)? The discordant behavior of at least two of the six associated dnSVs and dnPMs gave us a hint. We mentioned before that it is a priori impossible to distinguish a “late paternal” event from the earliest possible “early offspring” event; that is, a zygotic DNM that occurs before the S phase of the first embryonic cell division. The evidence in support of (1) clustered DNMs resulting from unique mutational events and (2) associated dnPMs (5/6) being postfertilization events strongly suggests that the corresponding dnSVs—rather than being late paternal events as initially surmised for at least two—are most likely postfertilization “early offspring” events that occurred before the S phase of the first cell division (and are therefore “on their own” indistinguishable from late parental events; see above).
Assuming that these clustered DNMs indeed occurred after fertilization, why would they preferentially occur in embryos produced by IVF, and why would they nearly exclusively affect the paternal homolog? The impact of IVF could be because DNA repair mechanisms are less effective (more error prone) in in vitro– than in vivo–produced embryos. The predilection for the paternal homolog could be related to known biological differences between the paternal and maternal homologs during gametogenesis and early embryonic development. For example, the haploid spermatid genome is known to undergo active DNA fragmentation as a result of postmeiotic chromatin remodeling (nucleosomes-to-protamines), resulting in clustered (∼4-kb) DNA breaks (i.e., lesions) that could remain unrepaired until after fertilization (Grégoire et al. 2018). Alternatively, DNA lesions are known to be introduced in the paternal genome during the active DNA demethylation that it undergoes during zygotic reprogramming (Ladstätter and Tachibana-Konwalski 2016). It is interesting to note in this regard that data in humans suggest that the aging of oocytes may cause an increase in the rate of postfertilization DNM specifically on the paternal chromosome (Gao et al. 2019).
The distinct segregation pattern in the grand-offspring of the clustered dnPMs and dnSVs indicates that the conversion of the corresponding clustered lesions into actual DNM does not necessarily occur exactly at the same time point during early development. In the case of NM-1 and NM-8, for instance, the data suggest that the corresponding dnSVs were likely established before the first cell division (dosage = 0.5), whereas the associated dnPMs were established after the first cell division (dosage ≤ 0.25). As stated before and to be more precise, one should say “prior and after the S phase of the first cell division.” Indeed, for the dosage of a postfertilization DNM to be 0.5, it needs to have occurred on one of the homologs when the cell is diploid (2C amount of DNA). After the S phase, the cell is in essence tetraploid (4C amount of DNA). If a DNM occurs at that stage, its dosage will be 0.25. It is interesting to note in this regard that 10 of the 19 detected dnSVs were characterized by microhomologies at the breakpoints (Table 1). This is typically regarded as evidence in support of MMBIR being the molecular mechanism underpinning the occurrence of dnSV. Yet MMBIR is assumed to occur during S phase, as it results from replication fork stalling/collapse (Lee et al. 2007; Hastings et al. 2009a,b). Thus, postfertilization dnSVs that are caused by MMBIR should have a dosage ≤ 0.25. We have at least two examples of dnSVs (NM-1 and NM-8) with microhomologies of, respectively, 3 and 4 bp and with strong support for being postfertilization events (based on associated dnPM), for which the allelic dosage in the offspring does not depart from 0.5 (Tables 1, 2). This suggests that as-of-yet-undefined nonreplicative, nonhomologous repair mechanisms may be accompanied by microhomologies at the breakpoints as well (Hastings et al. 2009b).
ART are increasingly used, not only in animals but in humans as well. Knowing whether these technologies are increasing the DNM rate and to what extent is therefore of the utmost importance. A study conducted with bovine cleavage-stage embryos showed that IVF increases the proportion of blastomeres with large (>100-kb) chromosomal anomalies from ∼20% in in vivo–derived embryos to ∼≥70% in in vitro–derived embryos (Tšuiko et al. 2017). Yet when applying the same methods to human postpartum fetal and placental tissues, de novo numerical aberrations and large structural imbalances appeared to occur at similar rates in IVF and naturally conceived infants (Esteki et al. 2019). At least three studies examined the effect of IVF on the number of dnPMs in the offspring. Two reported a modest increase of dnPMs (of the order of five) per offspring (Wong et al. 2016; Wang et al. 2021), whereas the other did not see any effect or even a trend (Smits et al. 2022). It is noteworthy that the excess dnPMs observed by Wang et al. (2021) were primarily owing to paternal mutations, and this was considered an argument against a direct effect of IVF per se. This interpretation may have to be reconsidered in light of our own results, as these may be postfertilization events preferentially affecting the paternal genome. In any case, these findings appear in sharp contrast with the results reported in this study, as we found a 4.9-fold ((14 / 46) / (5 / 81)) increase of the number of dnSVs in IVF-derived cattle compared with AI- or MOET-derived animals. Two of the dnSVs considered in these calculations are early parental events and could therefore be ignored to obtain better estimates of the effect of IVF. When doing so, the effect of IVF increases to 5.7-fold: (13 / 46) / (4 / 81). One possible explanation is that IVF specifically increases the incidence of small-scale structural variants, which were not studied in any of the above-mentioned publications.
This study obviously has its limitations. Although the Damona pedigree comprises 743 animals, the number of gametes that can be studied for the presence of DNM is only 254. As a consequence, the number of dnSV events that are available for analysis (i.e., 19) remains rather limited. Thus, the estimates of the effects, although significant, have large confidence intervals (i.e., 1.6 to 31.7 for the effect of IVF). Follow-up studies are therefore needed to reach more definitive conclusions. Also, one may argue that the short-read technologies that have been used to sequence the Damona pedigree are not optimal for the detection of dnSV. More modern, long-read technologies may have uncovered additional events. We observed a modest (yet nominally significant) effect of the sequence depth of the sire on the number of detected dnSV in the offspring: 0.01 extra dnSV per additional fold coverage. As a matter of fact, the linear model pointed toward a similar (yet nonsignificant) effect of the sequence depth of the dam. The significance of these observations, if any, remains unclear. We expected the detection power to increase with the sequence depth of the offspring but did not see any such effect (P = 0.2, β = −0.009) (Supplemental Fig. S27) despite the fact that the sequence depth of offspring ranged from 18× to 48×. We expected the sequence depth of the parents to have a negative effect on the number of detected dnSVs, as increased depth would facilitate the detection of SV in the parents. SVs transmitted from parents to offspring could erroneously be declared as dnSVs (false positives) if missed in the parent. Other studies combine multiple dnSV detection algorithms as a way to increase the sensitivity and specificity of dnSV detection (e.g., Belyeu et al. 2021). We believe that the unique features of the Damona pedigree, as well as the extensive manual curation that we conducted on the Smoove output, will have at least in part compensated for the use of a more limited suite of algorithms. Finally, we should note that the developmental biologies of cattle and humans are obviously different and that our observations may therefore not directly extrapolate to humans. However, our findings will urge the scientific community to look at the effect of ART on the rate of dnSVs in humans, which—to the best of our knowledge—has not yet been conducted systematically. Distinct yet interesting questions are whether freezing sperm has an impact on the DNM rate and whether frozen sperm accumulates extra DNM as storage time increases. Unfortunately, this information was not available for the studied material.
To conclude, the simultaneous analysis of associated dnSVs and dnPMs in clustered DNMs suggests that the rate of dnSVs is increased approximately fivefold during the early development of IVF-produced embryos and that the corresponding DNMs primarily affect the paternal homolog.
Methods
Damona massively parallel sequencing data
Trio animals in the 127 pedigrees amounted to 235 animals. Their DNA was extracted from blood (143 females and 22 males) or sperm samples (70 males; all sperm samples were obtained as frozen straws) using standard procedures. Briefly, DNA extractions were performed with a KingFisher flex 96 (Thermo Fisher Scientific) robot. For semen samples, the Macherey-Nagel NucleoMag tissue kit was used. Lysates from semen (straws; 10 µL) were prepared in the T1 lysis buffer from the Macherey-Nagel NucleoMag tissue kit with addition of Proteinase K (2.7 mg/mL) and DTT (230 mM) and digested overnight at 56°C, followed by extraction on the KingFisher flex robot as described by the kit's manufacturer. For blood samples, the Macherey-Nagel NucleoMag blood kit was used for 200 µL of blood by adding 75 µL of MBL1 lysis buffer from the Macherey-Nagel NucleoMag blood kit and 20 µL of Proteinase K (50 mg/mL), with a subsequent short 10-min digestion at room temperature, followed by the extraction on the KingFisher flex robot as described by the kit's manufacturer.
Familial relationships were confirmed by genotyping all samples with the 10-K Illumina SNP chip. We constructed 550-bp insert size whole-genome Illumina Nextera PCR free libraries following the protocols recommended by the manufacturer. All samples were then sequenced on Illumina HiSeq 2000 instruments, using the 2 × 100-bp paired-end protocol, by the GIGA Genomics platform (University of Liège). The sequence data were mapped using BWA-MEM 0.7.5a (Li 2013) to bovine reference genome ARS-UCD1.2. Afterward, SAMtools 1.9 (Li and Durbin 2009) was used to convert SAM files into BAM files. Subsequently, the BAM files were sorted with sambamba 0.6.6 (Tarasov et al. 2015), and the PCR duplicates were removed using Picard Tools 2.7.1 (https://github.com/broadinstitute/picard). The 235 trio animals were sequenced at a target coverage of ∼26×, and the rest (grand-offspring and half-sibs) were sequenced at target coverage of 8×.
dnSV discovery
We discovered SVs using the population calling mode in Smoove (https://github.com/brentp/smoove). First, Lumpy used evidence from split and discordant reads to detect population-wide SVs in 127 trios (Layer et al. 2014). Lumpy was designed to detect deletions, duplications, inversions, and breakends. The latter are junctions that could not be classified into canonical forms of SVs (Abel et al. 2020). In our study, the scope of dnSV was limited to deletions, duplications, and inversions. Afterward, the population-wide SVs were merged using SVtools (Larson et al. 2019), generating a nonredundant SV call set. Subsequently, the full cohort of 743 animals was genotyped (235 animals forming 127 trios and 508 animals either half-sibs or grand-offspring) using SVtyper (https://github.com/hall-lab/svtyper). The fold-coverage change in the read depth of copy number variants (deletions and duplications) was annotated using duphold (Pedersen and Quinlan 2019).
To detect late parental dnSVs, we scanned the SV call set for the following criteria: (1) Both parents are homozygous reference with evidence in the massively parallel sequencing data that the number of alternative allele supporting reads (ALT) ≤ 1, (2) offspring is heterozygous with evidence in the massively parallel sequencing data that the number of ALT ≥ 6 and ratio of ALT over the reference allele supporting reads (REF) ≥ 0.2, (3) all of the HS of the offspring are homozygous reference, and (4) the dnSV is transmitted to at least one GO, and the dnSV and the haplotype on which the dnSV arose are in perfect linkage in GO. We made an exception for NM-10 (a 1.2-Mb inversion; the offspring has only one GO to which NM-10 was not transmitted).
Additionally, our three-generational pedigree structure enables detection of dnSVs that occurred early during development of the parents (early parental) or offspring (early offspring). The individuals in which an early dnSV occurs (1) show imperfect linkage between the dnSV and the haplotype of origin in the subsequent generation (thus, the haplotype of origin can be transmitted, with or without the dnSV) and/or (2) carry three haplotypes in the sequence reads—paternal haplotype, maternal haplotype, and either of the two with the dnSV (Supplemental Figs. S20–S24)—and/or (3) show allelic imbalance for the early dnSV (e.g., including for SNPs within de novo deletions). We asked “early” events to meet at least two of the forementioned conditions. Of these early events, early offspring events are required to have the same evidence in the sequencing reads as the late parental events (see above). Early parental events required one parent in which dnSV occurred to have four or more ALT reads and an ALT/REF ratio ≥0.03. dnSVs with conflicting evidence were called ambiguous (see legend to Table 1). All candidate sites that passed these filters were manually inspected using the Integrative Genomics Viewer (IGV) (Robinson et al. 2011) in all members of the pedigree.
dnPM discovery
We scanned the 20-kb flanking regions of dnSVs for dnPMs. dnPMs were detected using GATK (McKenna et al. 2010) and filtered subsequently using custom scripts (for detailed explanation, see Harland et al. 2016). Overall, six dnPMs are identified in the 20-kb flanking bins, and four of them were orthogonally validated using Allegro amplicon sequencing (Tecan).
Determining parent of origin using parental variants
The parental origin of dnPM can be inferred if sequence reads with the dnPM also encompass constitutive variants that can be traced back unambiguously to one of the parents. For instance, if a read with the dnPM harbors a G at a variant site for which the sire is AG and the dam is AA, the dnPM was transmitted by the sire. In the case of dnSV, the use of constitutive variants to determine the parental origin of dnSV is slightly different. In the case of de novo deletions and duplications, we used constitutive variants that map within the structural variant. As an example, for a de novo deletion, if the offspring is A− (hemizygous), the sire GG, and the dam AA, the deletion is assumed to have been transmitted by the sire. As an example of a de novo duplication, if the offspring is AG with allelic proportions of 2A:1G, the sire is GG, and the dam AA, the duplication is assumed to have been transmitted by the dam.
Detecting mosaicism: quantifying and testing the statistical significance of allelic imbalance
Mutations occurring early during the development of an individual may generate detectable mosaicism, which manifests by an allelic ratio inferior to 50% (expected for a variant for which an animal is constitutively [i.e., in all its cells] heterozygous). In the case of dnPM, the allelic ratio is estimated as the fraction of sequence reads overlapping the dnPM site that carry the DNM as opposed to the reference allele. To test whether the observed allelic ratio differs significantly from that expected under the null hypothesis of constitutive heterozygosity, we compared—for candidate late offspring dnPMs constituting clustered DNMs (NM-1/8/9, A-2, M-3/5)—the allelic ratio observed in the offspring with that observed in the grand-offspring (assumed to be constitutively heterozygous). If a dnPM was transmitted to multiple grand-offspring, the sequence reads of these multiple grand-offspring were pooled and treated as if they originated from a single individual. The significance of the observed difference (offspring minus grand-offspring) was estimated using a permutation test. All reads (offspring and grand-offspring) were merged, and a number of reads corresponding to the real number of reads of the offspring was sampled at random from the pool. The difference in the allelic ratio between the pseudo-offspring and pseudo-grand-offspring (rest of the reads) was computed, and this operation was repeated 10,000 times, generating a list of 10,000 pseudodifferences. The statistical significance of the difference in allelic ratio observed with the real data was estimated as the proportion of pseudodifferences that would be as small or smaller (i.e., we performed one-tailed tests). The reads used for these tests were either the reads from the massively parallel sequencing data alone (NM-1 and NM-8) or the reads from the whole-genome sequencing and the reads from targeted sequencing experiments conducted using the Allegro amplicon sequencing (Tecan) to confirm the veracity of the dnPM (NM-9, A-2, M-3, and M-5). The average sequence depth with the Allegro amplicon sequencing was ∼150×.
In the case of deletion- and duplication-type dnSVs, we computed the ratio in average sequence depth within the dnSV and average sequence depth in the two 10-kb windows flanking the dnSV for the offspring and grand-offspring (early offspring dnSV). Allelic imbalance in the offspring was estimated as the difference between these two values (offspring minus grand-offspring for deletions, grand-offspring minus offspring for duplications). To estimate the statistical significance of the observed difference, we pooled the reads of the offspring and grand-offspring (dnSV plus flanking windows), randomly sampled a number of reads equal to the actual number of reads in the offspring from the pool to create a pseudo-offspring and pseudo-grand-offspring (remaining reads), and computed a pseudodifference as described above. The operation was repeated 10,000 times, yielding a list of 10,000 pseudodifferences. The statistical significance of the true differences was estimates as the proportion of pseudodifferences that would be as small or smaller than that observed with the real (unpermuted) data. One dnSV (M-1) was a candidate “early sire” mutation. In that case, we compared the allelic ratio between the sire and the offspring (assumed to be constitutively heterozygous).
Data access
The sequencing data generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number PRJEB53518/ERA15565221. The code to reproduce the analysis is available as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work, and in particular the generation of the “Damona” Dutch Holstein Friesian whole-genome sequence data set, was funded by the DAMONA ERC advanced grant to M.G. (ERC AdG-GA323030). C.C. is senior research associate from the Fonds de la Recherche Scientifique-FNRS (F.R.S.-FNRS). Y-L.L., A.C.B., M.B., M.A.M.G., and R.F.V. are financially supported by the Dutch Ministry of Economic Affairs (TKI Agri and Food project 16022) and the Breed4Food partners Cobb Europe, CRV, Hendrix Genetics, and Topigs Norsvin. Y-L.L. is postdoctoral fellow of the Dutch Research Council (NWO) Rubicon grant (019.222EN.017). G.C.M.M. is postdoctoral fellow of the H2020 EU project BovReg (grant agreement number 815668).
Authors’ contributions: M.G. and C.C. designed the study. Y.-L.L. performed the data analyses. E.M. collected the cattle pedigree data. W.C., L.K., and N.C. generated the sequencing data. C.H. processed the Damona sequencing data. G.C.M.M. mapped the Damona sequencing data to the ARS-UCD1.2 reference genome. A.C.B., M.B., R.F.V., and M.A.M.G. assisted in interpreting the results and editing of the manuscript. M.G., C.C., and Y.-L.L. wrote the manuscript. All authors contributed to the final version of the paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277884.123.
- Received March 23, 2023.
- Accepted August 8, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








